


Abstract
The Arabidopsis thaliana decapping enzyme (AtDcp2) was characterized by bioinformatics analysis and by biochemical studies of the enzyme and mutants produced by recombinant expression. Three functionally significant regions were detected: (i) a highly disordered C-terminal region with a putative PSD-95, Discs-large, ZO-1 (PDZ) domain-binding motif, (ii) a conserved Nudix box constituting the putative active site and (iii) a putative RNA binding domain consisting of the conserved Box B and a preceding loop region. Mutation of the putative PDZ domain-binding motif improved the stability of recombinant AtDcp2 and secondary mutants expressed in Escherichia coli. Such recombinant AtDcp2 specifically hydrolysed capped mRNA to produce 7-methyl GDP and decapped RNA. AtDcp2 activity was Mn2+- or Mg2+-dependent and was inhibited by the product 7-methyl GDP. Mutation of the conserved glutamate-154 and glutamate-158 in the Nudix box reduced AtDcp2 activity up to 400-fold and showed that AtDcp2 employs the catalytic mechanism conserved amongst Nudix hydrolases. Unlike many Nudix hydrolases, AtDcp2 is refractory to inhibition by fluoride ions. Decapping was dependent on binding to the mRNA moiety rather than to the 7-methyl diguanosine triphosphate cap of the substrate. Mutational analysis of the putative RNA-binding domain confirmed the functional significance of an 11-residue loop region and the conserved Box B.
INTRODUCTION
One of the key processes in mRNA turnover is decapping, that is, the removal of the 7-methyl guanosine cap from the 5′ end of the mRNA strand. Capped mRNA is estimated to constitute 95–97% of cellular mRNA and it has been proposed that the availability of the 7-methyl guanosine cap in mRNA is a dominant feature in competition for limited translation initiation factors, primarily eIF4E (1). Cap-independent translation mechanisms such as re-initiation and internal initiation generally lack the efficiency associated with cap-dependent translation (1). Since capping is co-transcriptional and occurs in the nucleus (2), while decapping occurs in granules known as the mRNA processing bodies (P bodies) (3,4), decapped mRNAs are unlikely to be recapped for cap-dependent translation. Rather, deadenylation followed by removal of 7-methyl guanosine diphosphate from the 5′ end of mRNA appears to initiate degradation of mRNA by exoribonucleases in the 5′ to 3′ direction (5). There are four primary pathways of mRNA decay in eukaryotes: two deadenylation-dependent mechanisms classified in accordance to the orientation of decay—5′→3′ or 3′→5′, endonucleolytic digestion and quality control pathways (6). Of these, deadenylation followed by decapping is the pathway gaining importance in yeast, humans and now in plants as a mechanism of controlling both gene expression and protein synthesis (5,7,8).
In yeast, the major mRNA decay pathway involves a deadenylation-dependent decapping mechanism, where the mRNA is initially deadenylated, then decapped by an enzyme named Dcp2p and ultimately degraded by a highly processive exoribonuclease in the 5′→3′ direction (5,9–11). A similar enzyme in humans, hDcp2, is active on capped mRNA and is again deadenylation-dependent (7,12). Although decapping by Dcp2 enzymes is a mandatory step in the degradation of capped mRNA in the 5′→3′ direction, the mechanism of action of this enzyme is still poorly understood and this enzyme is yet to be characterized in plants. Annotation of the Arabidopsis thaliana genome in the NCBI database includes one gene, At5g13570, with strong sequence homology to the Dcp2-type mRNA decapping enzymes. In this article, the protein encoded by gene At5g13570 is studied in detail.
Dcp2-type decapping enzymes are members of the Nudix superfamily of enzymes (13). Nearly 1800 open reading frames coding for Nudix hydrolases have been identified by bioinformatics searches in over 360 species (14). Nudix enzymes catalyse the hydrolysis of a wide range of small nucleotide substrates composed of a nucleoside diphosphate linked to another moiety X, hence the name Nudix (14). All members of this superfamily share a conserved amino acid sequence (GX5EX7REUXEEXGU) termed the Nudix motif/box, forming the catalytic site of these enzymes (15,16). In the Dcp2-type decapping enzymes from yeast and humans, the Nudix box has been shown to be essential for the hydrolysis of the pyrophosphate bond linking the cap to the 5′ extremity of mRNA (7,11).
It is proposed that hDcp2, the decapping enzyme from humans, binds to the RNA body first and subsequently positions the active site against the cap structure for decapping (17). An intact cap structure on an RNA strand of 23 nucleotides is mandatory for decapping by hDcp2 (7). The hDcp2 enzyme is unable to bind to cap analog [m7G(5′)ppp(5′)G] alone (17) and decapping by hDcp2 is not regulated by the addition of cap analog competitor (7), again suggesting the importance of a short RNA region for preliminary binding. In addition, uncapped transcripts are efficient competitors for capped RNA, suggesting that binding of mRNA to hDcp2 is independent of the cap itself (17). A region essential for RNA binding by the hDcp2 protein was demonstrated by deletion mutagenesis. The deleted region was C-terminal to the Nudix motif in a region comprising an α-helical structure and a preceding loop region (17,18). The region corresponding to the above α-helix was designated as Box B (17,18). The contributions of structural motifs and individual residues to mRNA binding within this region have not been established.
In vivo studies in plants suggest an important role for the Dcp2 type decapping enzyme in post-embryonic development (8,19). Heterozygous knockouts of the dcp2 allele coding for the AtDcp2 protein, which resulted in small leaves and short hypocotyls and roots, and homozygous T-DNA insertions, which resulted in a lethal phenotype, demonstrate the significance of the AtDcp2 decapping enzyme for growth and elongation of plants (8). In this article, we present studies on the enzymatic properties and structural motifs central to the mechanism of action of this Dcp2-type decapping enzyme from the plant A. thaliana and confirm that gene At5g13570 does indeed encode an active Dcp2-type decapping enzyme. We conduct in addition a mutational analysis on AtDcp2 as a full length recombinant protein. Previous biochemical studies largely used truncated Dcp2 proteins with greater inherent stability (11,17). Indeed, we detected significant proteolysis of the recombinant full length AtDcp2 expressed in E. coli. To minimize the proteolysis of the full length AtDcp2 protein in a bacterial expression system, we first conducted bioinformatics analyses to identify the structurally disordered regions that are susceptible to proteolysis in the AtDcp2 sequence. In addition to the regions with high propensity of structural disorder, we also discovered a putative PSD-95, Discs-large, ZO-1 (PDZ) domain-binding motif in the AtDcp2 sequence. This motif could potentially target the recombinant AtDcp2 protein to the PDZ domain-containing proteases in Escherichia coli. Thus, its mutation to abolish such interaction could minimize degradation of the recombinant AtDcp2 protein in E. coli. We found that mutation of the putative PDZ domain-binding motif did minimize degradation of recombinant AtDcp2 protein in E. coli. The adoption of such a strategy permits us to express full length AtDcp2 protein and its mutants for biochemical analysis.
Bioinformatics analysis was used to identify further structural motifs within the sequence of the full length AtDcp2 protein. The functional significance of these motifs was investigated by biochemical characterization of the recombinant AtDcp2 mutants carrying mutations of key residues in these motifs. The mechanism of hydrolysis was investigated by mutation of key residues in the consensus REXXEE found within the conserved Nudix box of this enzyme superfamily. In addition, biochemical analyses to decipher the molecular basis of substrate recognition by AtDcp2 and the ability of the reaction products to selectively suppress AtDcp2 decapping activity were conducted. Our results demonstrated the involvement of a putative loop region and the neighbouring Box B region in decapping.
MATERIALS AND METHODS
Bioinformatics analysis
Structural disorder prediction was performed with the DISOPRED (20) and DisEMBL (21) disorder prediction servers. Information on introns/exons and intron phases was downloaded from the Xpro database (22). Homology modelling of the 3D structures of proteins was performed using SWISS-MODEL (23) and the structures further analysed using PyMOL™ version 0.97.
Cloning of cDNAs
A cDNA library in the pSPORT-P vector prepared from mRNA from 42-day-old seedlings of A. thaliana cv Columbia was obtained from Invitrogen, USA. The cDNA corresponding to the At5g13570 gene was amplified by PCR from the Arabidopsis cDNA library with primers introducing a BamH I site at the 5′ end and an EcoR I site at the 3′ end. PCR reactions were performed in 50 μl reaction volumes containing the PCR amplification buffer, 400 nM of each dNTP, 100 ng of the 5′ and 3′ gene-specific primers, 2 μl of the cDNA template and 1–5 U of Taq DNA polymerase (Invitrogen, USA). The cycling conditions were 95°C for 30 s, 45–55°C for 30–45 s and 72°C for 1 min/kb of the expected products. The amplification conditions consisted of 25–40 cycles, with final 5 min incubation at the end of the amplification cycles. PCR products were ligated into digested vectors using the T4 DNA ligase (Roche, Switzerland). Reactions were carried out with 10–100 ng of the vector with 3- to 20-fold molar excess of the insert in the ligation buffer supplied by the manufacturer. The ligation reactions were incubated at 14°C overnight and immediately used for transformation of competent bacterial cells. Plasmid purification was performed using the PerfectPrep mini kit (Eppendorf Scientific Inc., USA). The plasmid clones containing the entire At5g13570 coding region were sequenced to confirm authenticity of the insert and that the insert was in frame for expression as a glutathione S-transferase (GST)-fusion in E. coli.
Site-directed mutagenesis
The QuikChange™ site-directed mutagenesis kit (Stratagene, USA) was used to create the sequences coding for the mutant proteins. PCR reactions comprised of 40–180 ng of plasmid DNA, 100 ng of each mutagenic primer, 200 μM dNTPs, 10 mM KCl, 6 mM (NH4)2SO4, 20 mM Tris–HCl and 2.5 U of Pfu Turbo DNA polymerase in a total volume of 50 μl. The reaction was subjected to an initial heating step of 30 s at 95°C and 21 cycles of 95°C for 30 s, 55°C for 1 min, 68°C for 14 min in a GeneampR PCR system (Perkin-Elmer, USA). A 5 μl aliquot of the reaction was obtained for analysis by agarose gel electrophoresis and the remaining 45 μl of the reaction was subjected to digestion, with Dpn I endonuclease for 1–2 h at 37°C. A total of 1–10 μl of the reaction was used to transform 60 μl of E. coli XL1-Blue cells for amplification of the mutant plasmids. Mutant cDNAs isolated from bacterial cells were sequenced to verify the mutations.
In vitro transcription of mRNA
All transcripts were synthesized by in vitro transcription using an SP6 polymerase. Omp (Outer membrane protein) gene (Saccharomyces cerevisiae YBR230C) cloned into the pSP73 vector, for SP6 driven transcription, was donated by Dr Lena Burri. The Omp transcripts were synthesized by SP6 polymerase driven transcription on linearized plasmids using the MEGAscript® in vitro transcription kit (Ambion, USA) as described by the manufacturer.
Capping reactions
The capping reaction mixture contained 50 mM Tris–HCl, pH 8, 6 mM KCl, 2.5 mM DTT, 1.25 mM MgCl2, 0.1 mg/ml BSA, 20 U RNaseOUT™ ribonuclease inhibitor, 0.4 mM SAM, 3.33 pmol (3000 Ci/mmol) [α-32P] GTP and 10 U of vaccinia virus capping enzyme (Ambion, USA) in a final volume of 200 μl. The reactions were incubated at 37°C for 3 h and the total RNA precipitated with 3.25 M LiCl. The precipitated RNA was resuspended and dissolved in 200 μl of H2O prior to its use as the substrate for the decapping activity assay.
Expression and purification of the AtDcp2 protein
Escherichia coli BL-21(DE3)pLysS transformed with the wild-type/mutant plasmids was grown with orbital shaking (180–200 r.p.m.) at 37°C to an A600nm of 0.3–0.6 and induced with 0.5–1 mM IPTG. Expression was induced for 3–40 h, depending on the post-induction temperature. Post-induction temperatures ranging from 15°C to 37°C were used. Cells were harvested, sonicated to disrupt the cells and the soluble and insoluble fractions separated by centrifugation at 13 000g for 15 min. The expressed GST-AtDcp2 and GST-AtDcp2-PD fusion proteins were purified by affinity chromatography on a glutathione agarose column. The GST-AtDcp2-PD-His10 protein was purified by affinity chromatography using a Co-IDA agarose column. The histidine-tagged fusion proteins were supplemented with Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) to a final concentration of 5.7 mM.
Decapping reaction and separation of the components of the decapping reaction by thin layer chromatography
Decapping of the capped mRNA by the AtDcp2 protein was assayed in reactions containing 50 mM Tris–HCl, pH 8, 5 mM MnCl2 or 5 mM MgCl2, 0.0167 pmol (final concentration 0.14 nM) capped mRNA containing 32P at the α-phosphate group ([α-32P]-7-methyl-Gppp-RNA) and up to 8 μg of the decapping enzyme or its mutants in a final volume of 120 μl. For comparative mutational analyses, 8 μg of the recombinant enzyme was used for each reaction. All assays were conducted using the capped Omp mRNA transcripts of 548 nucleotides. Reactions were incubated at 25°C for up to 3 h. Aliquots (2 μl) were directly blotted onto PEI-Cellulose (Merck & Co. Inc., USA) and TLC performed using 0.75 M LiCl or 0.45 M (NH)4SO4 as the mobile phase. The TLC plates were dried and the distribution of radioactivity determined using a Typhoon 8600 phosphorimager and ImageQuant 5.2 software (Amersham International, UK). The extent of decapping at the respective time points was calculated using the following formula.
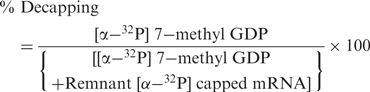 |
For competition studies of mRNA decapping, reactions were run with a fixed amount (0.14 nM) of radiolabelled capped Omp mRNA and 164 μM to 2 mM of each non-radioactive competitor. The competitors were uncapped Omp mRNA, cap analog dinucleotide (7-methyl diguanosine triphosphate) and 7-methyl GDP. Inhibition of decapping by fluoride ions was assayed under standard conditions in the presence of 50 μM to 25 mM NaF.
Identification of the major reaction product of the AtDcp2-catalysed decapping of capped mRNA as 7-methyl-GDP
The radioactive capped mRNA ([α-32P]-m7Gppp-RNA) was incubated with GST-AtDcp2-PD-His10 in the presence of Mn2+ for a prolonged period of time (>3 h) to allow complete decapping of [α-32P]-m7Gppp-RNA by the enzyme. An aliquot of the reaction mixture was mixed with up to 1 μg of 7-methyl-GDP standard prior to application onto the TLC plate. After chromatography, the 7-methyl-GDP standard on the TLC plate was detected by Abs254nM and its location was marked and the mobilities of the radioactive reaction products of the decapping reaction were detected by autoradiography. Co-migration of a radioactive spot with the 7-methyl-GDP standard indicates that the radioactive spot corresponds to 7-methyl-GDP generated in the AtDcp2-catalysed decapping of [α-32P]-m7Gppp-RNA.
RESULTS
The recombinant AtDcp2 protein preparation from expression in E. coli contains a significant amount of degradation products
The cDNA corresponding to gene At5g13570, annotated in the NCBI database as a putative Dcp2 type mRNA decapping enzyme from the genome of A. thaliana, was amplified by PCR from a cDNA library prepared from seedlings of A. thaliana cv Columbia. The encoded protein, a putative Dcp2-type mRNA decapping enzyme, was expressed in E. coli as a GST fusion protein, GST-AtDcp2. Expression at post-induction temperatures of 23°C and 37°C did not produce any soluble GST-AtDcp2 protein. Co-expression with chaperones GroES and GroEL did not significantly increase the yield of soluble GST-AtDcp2. When the expression temperature was lowered to 15°C, a significant amount of soluble GST-AtDcp2 protein was obtained. As shown in Figure 1A, purification by glutathione-agarose affinity chromatography generated an enzyme preparation that contains the intact recombinant GST-AtDcp2 of ∼69 kDa and a significant amount of lower molecular weight contaminating proteins. As all the proteins bound to glutathione agarose, we therefore conclude that the contaminating proteins retained an intact N-terminal GST moiety and were degradation products of the recombinant protein with a portion of the C-terminus end truncated. The most likely source of these contaminating proteins was either proteolysis of the C-terminal end of the proteins following expression or premature termination of translation. In agreement with our observation, extensive proteolysis of the decapping enzymes from yeast (Dcp2p) and human (hDcp2) was observed when these proteins were expressed as recombinant proteins in bacterial expression systems (7,11).
Figure 1.

SDS–PAGE analysis under non-reducing conditions of the purified variants of recombinant AtDcp2 enzyme expressed in E. coli. (A) GST-AtDcp2. (B) GST-AtDcp2-PD. (C) GST-AtDcp2-PD-His10. The arrow indicates the mobility of the intact recombinant protein in each case.
Bioinformatics analysis revealed structural disorder and identified a putative PDZ domain-binding motif at the C-terminus
Since the purified GST-AtDcp2 preparation contained a significant amount of proteolytic products, we conducted a bioinformatics analysis to identify regions in the AtDcp2 sequence that are prone to proteolysis. The rationale is that mutation or deletion of the putative proteolysis-prone regions may allow generation of intact recombinant AtDcp2 proteins for biochemical analysis. Our analysis revealed a region of extensive structural disorder at the C-terminus of the AtDcp2 protein. Seventy-six percent of the C-terminus from residues 251–373 was predicted by DISOPRED (20) to be disordered, while only 8% of the N-terminal region spanning residues 1–250 was disordered. The C-terminus was similarly predicted by DisEMBL (21) to contain a high percentage of structural disorder in the form of loops and coils, hot loops that constitute a subset of loops comprising inherent high mobility and missing coordinates in X-ray structures (Figure 2A). In comparison to other segments in the AtDcp2 sequence, the C-terminal extremity was predicted to exhibit the highest degree of structural disorder. From this analysis, it was concluded that there are two topologically different regions present in the AtDcp2 protein, a structured globular N-terminus and a highly disordered C-terminus of 123 amino acids.
Figure 2.

Bioinformatics analysis predicts region of structural disorder and existence of a putative PDZ binding domain in the AtDcp2 sequence. (A) Prediction of disordered regions using the DisEMBL server—in the graphical output from DisEMBL, the disorder prediction is calculated using the three criteria: (i) secondary structural types of loops/coils (blue), (ii) hot loops which constitute a subset of loops comprising inherent high mobility (red) and (iii) missing coordinates in X-ray structures (green). (B) Schematic representation of the protein fragments encoded by each exon of the At5g13570 gene coding for AtDcp2 mRNA decapping enzyme—labels indicate the size of proteins fragments encoded by each exon (below) and the intron phases (above) of the At5g13570 gene. The positions of the Nudix fold and the disordered tail are shown. (C) The sequence corresponding to the disordered region of the AtDcp2 protein (underlined) with emphasis on the putative PDZ domain-binding motif. (D) The putative PDZ domain-binding motif in the wild-type AtDcp2 protein and mutations introduced to generate the recombinant GST-AtDcp2-PD and GST-AtDcp2-PD-His10 proteins.
The presence of such a significant region of structural disorder is highly unusual for Nudix hydrolases. Most members of the Nudix superfamily are compact globular enzymes. When the structural disorder profile of each of the 25 members of the Nudix hydrolase superfamily in A. thaliana was assessed, it was apparent that the AtDcp2 protein was the only Nudix protein in Arabidopsis to possess a region with a high degree of structural disorder (data not shown). To assess the genetic basis for the presence of unique structural features at the protein level, the intron–exon architecture of the At5g13570 gene, coding for AtDcp2, was investigated. The majority of the disordered C-terminal extremity of AtDcp2 was encoded by a longer exon (coding for 111 residues) comprising of a phase 1 intron at the 5′ end and a minor segment encoded by a smaller exon containing a phase 2 intron at the 5′ end (Figure 2B). In contrast, the Nudix fold contained phase 0 introns between exons (Figure 2B). Whereas the presence of phase 0 introns in the Nudix fold is consistent with its more ancient origins—it is found in eukarya, prokarya and archaea—the phase 1 and 2 introns found between exons forming the disordered C-terminus of the decapping enzyme are consistent with its more recent formation in the eukaryotic lineage alone.
The structural disorder within the AtDcp2 protein was consistent with its putative role as an RNA chaperone that binds to the unwound 5′ RNA backbone prior to cap hydrolysis. Large disordered regions are a characteristic feature of RNA and protein chaperones (24). In the AtDcp2 protein, this region of disorder maps outside of the region inclusive of an α-helix named Box B and an adjacent loop region that had previously been demonstrated to be essential for RNA binding by the human hDcp2 enzyme (17) and may function separately in RNA binding. Many unfolded and misfolded regions of proteins undergo proteolysis more readily than regions that adopt well-defined secondary and tertiary structures (25–27). Thus, the presence of such a long C-terminal segment with high propensity of structural disorder may contribute to its susceptibility to proteolysis by the endogenous proteases when expressed as a recombinant protein in E. coli.
In addition to this, we have also identified another structural feature that could potentially contribute to its susceptibility to proteolysis in E. coli—the C-terminal GNSA motif, which is a putative PDZ domain-binding motif (Figure 2C). The GNSA motif could further enhance proteolysis of recombinant AtDcp2 by directing the recombinant protein to bind the PDZ domain-containing proteases. PDZ domains are protein–protein interaction domains, chiefly responsible for the binding of partner proteins (28). The protein–protein interactions mediated by PDZ domains rely on the low-binding affinity of PDZ domains to apolar PDZ domain-binding motifs of partner proteins, in particular, those which contain a hydrophobic residue at the C-terminal extremity (28). Several of the cytoplasmic and periplasmic proteases in E. coli responsible for the degradation of disordered or misfolded proteins, contain PDZ or PDZ-like domains. For example, both the tail-specific protease (Tsp) and the isolated PDZ domain from the Tsp protease bind to the apolar residues of substrates with a dissociation constant KD of 1.8–1.9 μM (29). Further, the periplasmic PDZ domain-containing protease DegS preferentially recognizes the sequence motif YYF-COOH at the C-terminal extremity of protein substrates (30,31).
Taken together, the susceptibility of recombinant AtDcp2 to proteolysis is likely due to the presence of (i) a long segment with high propensity of structural disorder and (ii) a putative PDZ domain-binding motif at the C-terminus.
Mutation of the putative PDZ domain-binding motif significantly reduces proteolysis of the recombinant AtDcp2 protein expressed in E. coli
The inclusion of broad-specificity proteases inhibitors in the extraction buffer did not prevent proteolysis of the recombinant GST-AtDcp2 protein (data not shown). We decided to mutate the PDZ domain-binding motif of AtDcp2 to minimize proteolysis of the recombinant enzyme by the endogenous proteases in E. coli. The C-terminal residues Ser-Ala were mutated to Pro-Asp to replace the apolar tail with an acidic residue to abolish any possible interaction of the recombinant enzyme with the PDZ domain-containing proteases. Indeed, the resultant mutant protein GST-AtDcp2-PD was less susceptible to degradation than the parent molecule with an intact PDZ domain-binding motif at the C-terminus. The purified GST-AtDcp2-PD preparation (Figure 1B) contains predominantly the intact recombinant enzyme of ∼69 kDa and a much smaller amount of degradation products than the GST-AtDcp2 preparation (Figure 1A). Consistent with our observation, similar approaches (i.e. insertion of a polar tail motif) was adopted by other researchers to prevent proteolysis of the recombinant proteins by proteases containing PDZ or PDZ-like domains. For example, the sequence KNQHE was attached to the C-terminal extremity of the Arc repressor to abolish its rapid proteolysis by the Tsp (32). Similarly, the presence of a polar C-terminal extremity in the λ repressor N-terminal domain prevented its degradation by Tsp (32).
The inclusion of a poly-histidine tag and the addition of tris-(2-carboxyethyl) phosphine hydrochloride (TCEP) facilitates purification and prevents aggregation of recombinant AtDcp2 proteins
A poly-histidine tag (His10) was engineered at the C-terminus of GST-AtDcp2-PD to facilitate purification of the intact recombinant AtDcp2 protein. The resultant recombinant enzyme GST-AtDcp2-PD-His10 contains (i) a GST moiety at the N-terminus, (ii) mutations of the last two residues at the C-terminus of the AtDcp2 sequence and (iii) a poly-histidine tag at the C-terminus. The presence of the poly-histidine tag permitted efficient purification of the intact recombinant GST-AtDcp2-PD-His10 proteins from crude bacterial lysate (Figure 1C). We also included 5.7 mM of TCEP in the buffer for storage of the final purified enzyme preparation to prevent aggregation of the recombinant enzyme. A summary of the further mutations carried out on GST-AtDcp2-PD-His10 is shown in Figure 3
Figure 3.

Summary of the mutations and purity of the purified preparations of GST-AtDcp2-PD-His10 and its mutants. (A) The location of the functional motifs and mutated residues in the AtDcp2 protein. All proteins also contained an additional 10 histidine residues C-terminal to residue 373. (B) SDS–PAGE analysis of the preparations of GST-AtDcp2-PD-His10 protein (His10) and mutant variants purified with buffers containing TCEP to minimize aggregation. The single amino acid substitutions are presented using the conventional mutational nomenclature. Del 11 indicates the mutant AtDcp2Δ222–232. The arrow indicates the mobility of the intact recombinant proteins (molecular masses ranging from ∼ 67 to 69 kDa).
7-methyl GDP is a major product of the divalent cation-dependent decapping reaction catalysed by GST-AtDcp2-PD-His10
The activity of expressed GST-AtDcp2-PD-His10 protein as a decapping enzyme was assessed with an assay procedure adapted from the method developed by Zhang et al. (33). This method involves the use of radioactive [α-32P]-labelled capped mRNA (referred to as [α-32P]-m7Gppp-RNA) as the substrate and separation of the reaction products from the substrate by TLC. Since longer capped transcripts (those of more than 99 bases) were preferred over shorter capped transcripts (20–29 bases) by the yeast decapping enzyme Dcp2p (11), we chose the yeast outer membrane protein (Omp) transcript of 548 bases as the substrate for all decapping reactions described in this article. To generate the capped RNA, in vitro transcribed Omp mRNA was radiolabelled at the α-phosphate group of the cap with [α-32P] GTP using a capping enzyme from vaccinia virus.
As shown in Figure 4A, GST-AtDcp2-PD-His10 catalysed formation of several radioactive reaction products from [α-32P]-m7Gppp-RNA in the presence of Mg2+ or Mn2+. These reaction products were separated by TLC and depicted in the figure as spots a, b and c. Since [α-32P]-m7Gppp-RNA could not migrate during chromatography, it remained at the origin. Thus, the radioactive spot at the origin corresponded to residual [α-32P]-m7Gppp-RNA left at the end of the reaction. Among spots a, b and c, spot b represents the major reaction product. To identify reaction product in spot b, [α-32P]-m7Gppp-RNA was incubated with GST-AtDcp2-PD-His10 in the presence of Mn2+ for a prolonged period of time (>3 h) to allow complete decapping of [α-32P]-m7Gppp-RNA. An aliquot of the reaction mixture was mixed with up to 1 μg of 7-methyl-GDP standard prior to application onto the TLC plate. After chromatography, the 7-methyl-GDP standard on the TLC plate was detected by Abs254nM and its location was marked (data not shown). When the location of radioactive spots a, b and c on the TLC plate was monitored by autoradiography, we found that spot b co-localized with 7-methyl GDP (The key in Figure 4A), suggesting that 7-methyl-GDP was generated in the decapping reaction catalysed by GST-AtDcp2-PD-His10 in vitro.
Figure 4.

Characterization of the mRNA decapping activity of AtDcp2. (A) TLC identification of the products of hydrolysis by GST-AtDcp2-PD-His10 protein of capped mRNA labelled with 32P at the α-phosphate of the cap. Reactions were carried out in the presence of 5 mM Mg2+ or 5 mM Mn2+ and in the absence of both ions. The ‘key’ shows the mobilities of the standard 7-methyl GDP (corresponding to spot b) and capped mRNA on the TLC plate. According to the report by Bergman et al. (34), spots a and c likely correspond to [32P]7-methyl-GMP and [α-32P]7-methyl-GTP, respectively. (B) TLC profile of the time course of decapping of [32P]-labelled capped RNA by the GST-AtDcp2-PD-His10 mutant carrying the catalytic site mutation (E154A) in the presence of 5 mM Mn2+ ions. (C) Twenty percent SDS/Urea polyacrylamide gel electrophoresis of the products of the AtDcp2-catalysed decapping of [32P]-labelled capped RNA before (−) and after (+) treatment with alkaline phosphatase. The multiple radioactive bands in the lane labelled (−) correspond to [α-32P]7-methyl-GDP and other nucleotides including [32P]7-methyl-GMP and [α-32P]7-methyl-GTP generated in this extended (3 h) decapping reaction. (D) Time course of decapping by the GST-AtDcp2-PD-His10 protein in the presence of Mg2+ or Mn2+ determined by TLC as in panel A. In the absence of Mn2+ and Mg2+ ions, production of 7-methyl GDP was undetectable (data not shown).
When the conserved glutamate-154 essential for decapping activity was replaced by alanine (the rationale of this mutation is presented in the next section), the mutant could only generate a negligible amount of spot b after prolonged incubation (120 min) (Figure 4B). Thus, the generation of radioactive reaction products in spots a, b and c from [α-32P]-m7Gppp-RNA as shown in Figure 4A was indeed catalysed by the intrinsic enzymatic activity of GST-AtDcp2-PD-His10 and not by the activity of any contaminating enzyme in the protein preparation.
In addition to TLC analysis, the mixture generated by prolonged incubation of [α-32P]-m7Gppp-RNA with GST-AtDcp2-PD-His10 was subject to analysis by SDS/urea polyacrylamide gel electrophoresis following alkaline phosphatase treatment. It is noteworthy that free inorganic phosphate migrates at the dye front, while the migration of small nucleotides such as 7-methyl GMP and 7methy GDP is retarded in the SDS/urea gel electrophoresis (34). Being a non-specific phosphatase, alkaline phosphatase can fully dephosphorylate all possible 32P-labelled nucleotides arising from decapping of [α-32P]-m7Gppp-RNA, including [32P]-7-methyl GMP, [α-32P]-7-methyl GDP and [α-32P]-7-methyl GTP. As shown in Figure 4C, treatment of reaction mixture with alkaline phosphatase resulted in the formation of the fast-migrating [32P]-inorganic phosphate ([32P]-Pi), while the mixture without alkaline phosphatase treatment contained a negligible amount of [32P]-Pi.
Taken together, the results shown in Figure 4A–C confirm that 7-methyl GDP and by inference 5′-monophosphate mRNA, are the major products of the AtDcp2-catalysed decapping reaction. In addition to the major radioactive spot (spot b), corresponding to 7-methyl-GDP, two minor radioactive spots (spots a and c) were also detected in the TLC plate. Since they correspond to the minor components of the products of the AtDcp2-catalysed decapping reaction, we did not conduct further experiments to confirm their identities. Comparison of their mobilities in the TLC (Figure 4A) and those reported by other researchers, spots a and c likely correspond to 7-methyl GTP and 7-methyl GMP, respectively. It is noteworthy that in all analyses, only the radioactivity associated with spot b was used to measure the decapping activity of AtDcp2 and its mutants.
Both the human and the yeast decapping enzymes require the presence of either Mg2+ or Mn2+ ions for activity (11,17). For the human enzyme, Mn2+ is the preferred cofactor in vitro (17). For the truncated yeast decapping enzyme Dcp2ΔCp, both by itself and in the presence of the yeast Dcp1p protein, Mn2+ was the preferred bivalent cation for decapping (11). The decapping activity of the GST-AtDcp2-PD-His10 protein was absolutely dependent on either Mn2+ or Mg2+ cofactors, with activity four fold higher with Mn2+ compared to Mg2+ (Figure 4D). Reaction rates show an initial rapid rate of decapping followed by a second more gradual rate of decapping (Figure 4D). Similar biphasic kinetics was also observed for the yeast decapping enzyme Dcp2ΔCp in the presence of Dcp1p (11).
Fluoride ions have little effect on the catalytic activity of AtDcp2
Many Nudix hydrolases are highly susceptible to inhibition by fluoride ions (35,36). Nudix enzymes active on Ap4A, coenzyme A and ADP-ribose have been demonstrated to be strongly inhibited by fluoride ions (35,37,38). The IC50 values for fluoride inhibition for the Ap4A hydrolase from Lupinus angustifolius, the coenzyme A pyrophosphatase from Caenorhabditis elegans and ADP-ribose pyrophosphatase from human were 3, 3 and 20 μM respectively, demonstrating that even at low micromolar concentrations of fluoride ions Nudix hydrolases are strongly inhibited (35,37,38). In contrast to these, other Nudix hydrolases, the GST-AtDcp2-PD-His10 protein was not inhibited by fluoride ions up to a concentration 1 mM NaF (Figure 5A), and showed only a small inhibition at 25 mM NaF (Figure 5B). To determine if the lack of inhibition was due to the presence of Mn2+ as cofactor, fluoride sensitivity of AtDcp2 was also tested in the presence of Mg2+ ions. In the presence of Mg2+ ions, there was no inhibition by fluoride of concentrations up to 1 mM and there was only a minor inhibitory effect in the presence of 25 mM NaF (Figure 5C). It was concluded that the mRNA decapping enzyme from A. thaliana did not show the exquisite sensitivity to fluoride ions that is characteristic of many Nudix hydrolases.
Figure 5.

The effect of fluoride ions on mRNA decapping. (A) Decapping activity of GST-AtDcp2-PD-His10 protein determined in the presence and absence of 50–1000 μM of NaF with 5 mM Mn2+ as the cofactor. Five micrograms of protein was used for each assay (B) Time course of decapping catalysed by GST-AtDcp2-PD-His10 in the presence and absence of 25 mM NaF with 5 mM Mn2+ as the cofactor. A total of 5 μg of the protein was used for each assay. (C) Decapping activity of GST-AtDcp2-PD-His10 protein in the presence of 5 mM Mg2+ and 50 μM to 25 mM of NaF. SEM, standard error of mean.
Mutation of the essential glutamates within the Nudix box abolishes decapping activity of the AtDcp2 protein
In spite of its insensitivity to fluoride ions, AtDcp2 retains the catalytic mechanism characteristic of Nudix enzymes. The key residues for catalysis within the catalytic site have been previously investigated in many Nudix hydrolases. Two glutamates, three residues apart in the conserved sequence REXXEE have been shown to be significant in the hydrolysis of Nudix substrates. The first glutamate in the REXXEE sequence acts as a general base that deprotonates a water molecule, which in turn attacks the β-phosphorus of the substrate (39). The mutation of this residue results in substantial loss of enzymatic activity in Nudix hydrolases (40). The third glutamate in the REXXEE sequence contributes a ligand to the enzyme-bound bivalent cation cofactor (39,41). Disruption of the third glutamate in the E. coli MutT enzyme induced a dramatic loss in enzymatic activity (41).
The analogous glutamates (Glu-154 and Glu-158) within the Nudix box of the AtDcp2 enzyme were mutated to alanine, as single substitution mutations. As shown in Figure 6, E154A and E158A mutations resulted in 350-fold and 160-fold reductions in decapping activity, respectively. The results confirm that the conserved Nudix box in the AtDcp2 decapping enzyme is catalytically critical and that the mechanism of catalysis common to the Nudix superfamily is conserved in hydrolysis of capped mRNA carried out by this enzyme.
Figure 6.

Effects of mutation of two conserved glutamates in the Nudix box on decapping activity of AtDcp2. (A) The mutated residues (underlined) within the Nudix box. (B) Decapping activities of the GST-AtDcp2-PD-His10 protein and the mutants. The decapping assays were carried out with 8 μg of purified recombinant enzyme from three separate expressions. SEM, standard error of mean.
Dependency of mRNA decapping on binding to the mRNA body
An mRNA body of 23 nucleotides has been shown to be essential for decapping by the human hDcp2 protein (7). The hDcp2 decapping enzyme is unable to hydrolyse cap analog, m7G(5′)ppp(5′)G and decapping of capped mRNA by the hDcp2 enzyme is not inhibited by the addition of cap analog as a competitor (7,12,17). Taken together, these studies suggest the requirement of a minimal number of nucleotides in the capped mRNA for its recognition by hDcp2 as substrates. In addition, uncapped transcripts are efficient competitors of capped mRNA in the hDcp2-catalysed decapping reaction, suggesting that mRNA binding by hDcp2 can be independent of the 7-methyl diguanosine triphosphate cap (17). To determine whether the Arabidopsis AtDcp2 enzyme bound to the mRNA moiety of the capped mRNA or to the cap, the GST-AtDcp2-PD-His10 protein was assayed for decapping activity with a fixed amount (0.14 nM) of [α-32P] capped mRNA in the presence or absence of 164 μM of uncapped Omp mRNA or cap analog as competitors. As shown in Table 1, production of [α-32P] 7-methyl GDP from [α-32P] capped mRNA was reduced by uncapped Omp mRNA even though it does not have the 7-methyl diguanosine triphosphate cap. In contrast, the cap analog, 7-methyl diguanosine triphosphate, did not reduce mRNA decapping. From these results, we conclude that the RNA strand rather than the cap mediates binding of the capped mRNA to AtDcp2. Such property is consistent with the mechanism proposed by Piccirillo et al. (17) for the human decapping enzyme—the decapping enzyme first binds the RNA moiety of the substrate and then scans the RNA body to locate the cap.
Table 1.
The dependency of decapping on mRNA/cap/product binding
| Competitor | Decapped (%) | SEM (+/−) |
|---|---|---|
| Panel A | ||
| None | 68.4 | 0.7 |
| Uncapped Omp mRNA(164 μM) | 37.8 | 2.1 |
| 7-methyl diguanosine triphosphate (164 μM) | 69 | 1 |
| Panel B | ||
| None | 44.3 | 0.3 |
| 7-methyl GDP (2 mM) | 25.3 | 1.4 |
| 7-methyl GDP (0.2 mM) | 29.4 | 0.1 |
(A) The extent of decapping of the substrate was measured after 60 min in assays containing GST-AtDcp2-PD-His10 protein, 5 mM Mn2− and a fixed amount (0.14 nM) of radiolabelled capped Omp mRNA, supplemented with 164 μM each of the competitors: uncapped Omp and cap analog dinucleotide. The SEM of three separate assays for each competitor are also indicated. All other conditions are as described in the ‘Materials and methods’ section. (B) The extent of decapping of the substrate by the recombinant enzyme in the presence and absence of the reaction product 7-methyl GDP after 60 min.
AtDcp2 activity is inhibited by the reaction product 7-methyl GDP
In contrast to the inability of the 7-methyl diguanosine triphosphate to inhibit AtDcp2 activity, the reaction product 7-methyl GDP at 200 μM effectively suppressed the AtDcp2-catalysed decapping activity (Table 1). Our results suggest that the active site of the decapping enzyme adopts a configuration that binds poorly to the cap structure of capped mRNA but relatively efficiently to the reaction product 7-methyl GDP.
Deletion of a key loop region in the putative RNA-binding domain interferes with the AtDcp2 catalysed decapping activity
Multiple sequence alignment of Dcp2 enzymes from a range of species was used to identify the RNA binding domain in the AtDcp2 protein (Figure 7A). In the human decapping enzyme, hDcp2, a region consisting of Box B and the preceding loop region was shown to be important for RNA binding (17). The Box B region is conserved in the Dcp2-type decapping enzymes (Figure 7A). From the structure of the Schizosaccharomyces pombe Dcp2p enzyme, it is clear that Box B forms the C-terminal helix in the α/β/α sandwich of the Nudix fold (18). To define the structural features in the loop region preceding the Box B region responsible for RNA binding, we conducted bioinformatics analysis of this region in the AtDcp2 sequence. The analysis revealed a motif (VITHGVSGLKL) in this loop region of AtDcp2 that closely resembles the VIGXXGXXUK segment (Figure 7B) commonly found in the KH domain of several RNA-binding proteins including ERA, Fragile X protein, hnRNP K and Poly(C) binding proteins (42–45). We therefore termed this motif as KH-like motif. Homology modelling of the Nudix fold of the AtDcp2 protein (residues 98–250) using the known structure of the S. pombe Dcp2p decapping enzyme, revealed that this KH-like motif likely resides in the loop region that extends to the start of the Box B helix (Figure 7C). We postulate that the KH-like motif in AtDcp2 (Figure 7B) contains determinants that facilitate the decapping reaction by binding to the RNA moiety of the capped mRNA substrate.
Figure 7.

Effects of mutation of residues within the putative RNA-binding KH-like domain and Box B domain on catalytic activity of AtDcp2. (A) Sequence alignment of Arabidopsis thaliana (AtDcp2) to reveal the putative KH-like domain. Oryza sativa (osDcp2), human (hDcp2), Xenopus laevis (xlDcp2) and Caenorhabditis elegans (ceDcp2) Dcp2 decapping enzymes were used for the alignments. The position of the KH-like motif and Box B within the AtDcp2 protein is shown in the sequence alignment. (B) The consensus sequence of the conserved 10 residue region found in KH domains (42,43), the sequence corresponding to the consensus sequence found in the KH domain in the RNA-binding site of ERA protein from E. coli (42) and the sequence of a KH-like motif in the AtDcp2 decapping enzyme. (C) Comparison of the structure of the Ap4A hydrolase from L. angustifolius (PDB ID–1JKN) with the homology model of the Nudix fold of the AtDcp2 protein (residues 98–250) modelled after the structure of the S.pombe Dcp2p enzyme (PDB ID – 2A6T). Box B helix in the AtDcp2 homology model and the analogous helix 4 in the lupin enzyme structure are marked in red. The 11-residue region forming the near-consensus of a KH motif, found within the loop region preceding Box B, is coloured in yellow. This region was deleted in mutant AtDcp2Δ222–232. The side chains of three lysines and a single glycine, which were mutated individually to alanines, are also shown. (D) Decapping activities of the GST-AtDcp2-PD-His10 protein and the mutant proteins carrying mutations in the putative RNA-binding site. All decapping efficiencies were obtained from decapping assays carried out using 8 μg of recombinant enzyme from three separate expressions. SEM, standard error of mean.
To ascertain the functional significance of the 11-residue KH-like motif, it was deleted or mutated in AtDcp2. As shown in Figure 7D, deletion of the KH-like motif led to a 92-fold reduction in the decapping activity. Replacement of Lys-231 and Gly-226 in this segment (VITHGVSGLK) by alanine reduced the decapping activity of AtDcp2 by ∼68% and 34%, respectively. In summary, our results suggest that the KH-like motif contains determinants essential for decapping activity. Presumably, these determinants facilitate decapping by mediating association of AtDcp2 with the RNA backbone of the capped mRNA substrate. This notion is supported by comparison of the homology model of the AtDcp2 protein with the structure of the related Nudix enzyme Ap4A hydrolase from Lupinus angustifolius (46,47). It was evident from the comparison that the Box B helix of AtDcp2 is equivalent to helix 4 in the lupin enzyme structure (Figure 7C). The loop region preceding helix 4 in the lupin Ap4A hydrolase was demonstrated to contribute critical substrate binding residues that lined the hydrophobic pocket, where the ATP-MgFX substrate bound (47). It is likely that the KH-like motif in this loop region is suitably located in the AtDcp2 structure to mediate or facilitate binding of the decapping enzyme to the RNA substrate.
Two lysine residues in Box B region contribute to efficient hydrolysis of capped mRNA by AtDcp2
Since the Box B region in human decapping enzyme was found to participate in RNA binding, we conducted a mutagenesis study to ascertain if the Box B region of AtDcp2 contains determinants that are important for activity. Both Lys-243 and Lys-248 in the Box B region of AtDcp2 were individually modified to alanine to eliminate their polar side chains. As shown in Figure 7D, the mutations resulted in 56–68% reductions in decapping activity (Figure 7D), supporting the notion that both lysine residues in the Box B region contribute to the ability of AtDcp2 to efficiently hydrolyse capped mRNA. Presumably, the positively charged side chains of both lysines mediate binding of AtDcp2 to the RNA moiety of the capped mRNA substrate.
DISCUSSION
The roles of Glu-154 and Glu-158 in the decapping reaction
This study describes the functional characterization of a Dcp2 type decapping enzyme from plants. The AtDcp2 enzyme hydrolyses the mRNA cap by substitution at β-phosphorus, the conserved mechanism of catalysis for the Nudix superfamily, yielding 7-methyl GDP and 5′-monophosphate mRNA as the products. The individual conversion of both essential catalytic site glutamates, Glu-154 and Glu-158, to alanine resulted in more than 150-fold reductions in catalytic activity of the AtDcp2 enzyme, consistent with their functions as the effector of catalysis (Glu-154) and a ligand donor (Glu-158) that coordinates a divalent cation, respectively (39). Relevant to our findings presented in Figure 6, mutations of homologous glutamates in the conserved Nudix box motif of both the human and yeast decapping enzymes abolished their decapping activities (7,11).
Implications of product inhibition of AtDcp2 by 7-methyl GDP
Decapping was inhibited by the product 7-methyl GDP, by uncapped mRNA but not by the cap analog, 7-methyl diguanosine triphosphate. It appears that presentation of the cap to the cleavage site requires the cooperation of the mRNA body and that the catalytic site binds to the product 7-methyl GDP more strongly than to the cap in capped mRNA. The 7-methyl GDP similarly inhibits the inherent mRNA decapping activity in the L-A virus (48) and the activity of the nuclear Nudix small nucleolar RNA decapping enzyme X29 (49). Such product inhibition by 7-methyl GDP constitutes a potential mechanism to prevent decapping of intact translatable mRNAs and accordingly 7-methyl GDP has a short cellular lifetime. The 7-methyl GDP formed by Dcp2 enzymes is converted rapidly to 7-methyl GMP by the scavenger mRNA decapping activity in vivo (50).
The lack of sensitivity to fluoride ions is a distinguishing feature of AtDcp2 within the Nudix hydrolase superfamily of enzymes
Extreme sensitivity to fluoride has been shown previously to be a widespread characteristic of Nudix hydrolases. All enzymes so far tested have shown this (35,36,38,51). Contrary to other Nudix enzymes, the AtDcp2 protein was not inhibited by fluoride ions at concentrations of up to 1 mM. Only at 25 mM, did the fluoride ions impose a minor inhibitory effect. Fluoride compounds mimic phosphate groups. It has been proposed that Nudix enzymes and ATPases are inhibited by complexes formed with the fluoride compounds and the reaction products (36,52). Since our results demonstrate that AtDcp2 is refractory to inhibition by fluoride ions, we propose that AtDcp2 active site adopts a configuration with limited accessibility to the complexes formed by fluoride ions with 7-methyl GDP. It is likely all other Dcp2-type decapping enzymes also share such a property. In this respect, the Dcp2-type decapping enzyme appears to be distinct from many other Nudix hydrolases. Future investigations to solve the 3D structure of AtDcp2 will provide the structural basis for this distinct property.
The structural basis of RNA binding by AtDcp2
RNA-binding proteins interact with RNA molecules in two structurally defined modes: (i) groove binding and (ii) β-sheet binding (53,54). For the proteins adopting the groove-binding mode, a secondary structural element such as a helix or a loop is inserted into the groove of the RNA substrate (53,54). It is proposed here that AtDcp2 adopts the groove-binding mode to bind RNA—the loop region preceding Box B and/or Box B extending into grooves of the RNA molecule. Katsamba and co-workers (55) proposed a two-step ‘lure and lock’ model of protein–RNA association. In this model, electrostatic interactions are the key for the initial association (the lure step). This is then followed by rapid induced fit binding (the lock step), resulting in a stable protein–RNA complex (55). Since mutation of Lys-231 in the loop region and Lys-243 and Lys-248 in the Box B region reduces the decapping activity of AtDcp2 (Figure 7), we propose that they are responsible for the initial electrostatic interactions in the lure step to ‘lure’ the RNA molecule to AtDcp2, the bound RNA molecule then induces conformational changes to establish additional interactions between the RNA moiety with determinants in other parts of AtDcp2 to form a stable protein–RNA complex.
Piccirillo et al. (17) reported that the carboxyl-terminal portion including Box B in the human decapping enzyme hDcp2 can mediate RNA binding. They also demonstrated that hDcp2 and single-stranded DNA could form stable complexes, and the ability of hDcp2 to form the complexes was significantly diminished upon deletion of the C-terminal segment containing Box B. We have attempted to use the three different methods described by Piccirillo et al. (17) and Moller et al. (56) and to ascertain if AtDcp2 can form stable complexes with [α-32P]-m7Gppp-RNA. These methods include: (i) the Northwestern RNA-binding assay, (ii) cross-linking of AtDcp2 with [α-32P]-m7Gppp-RNA by ultraviolet light irradiation and (iii) cross-linking of AtDcp2 with [α-32P]-m7Gppp-RNA by formaldehyde treatment. In contrast to the findings by Piccirillo and co-workers, we failed to detect stable AtDcp2-[α-32P]-m7Gppp–RNA complexes. A possible explanation for our failure to detect the stable complexes is that the binding of AtDcp2 to capped mRNA is transient in nature. An appropriate approach for future studies to measure the affinity of AtDcp2 for capped mDNA and to define the structural motifs critical for RNA binding is the use of surface plasmon resonance to determine the affinities of AtDcp2 and its mutants for capped mRNA immobilized on sensor chips.
Possible functional significance of the putative PDZ domain-binding motif at the C-terminus of AtDcp2
Bioinformatics analysis identified the ‘GNSA’ motif at the C-terminus of AtDcp2 as a putative PDZ domain-binding motif. In addition, we conducted further analysis by homology searches in the NCBI database and found that similar putative PDZ domain-binding motifs also exist in almost 80% of vertebrate and plant Dcp2 decapping enzymes (data not shown). One approach to ascertain if the GNSA can indeed mediate AtDcp2 binding to PDZ domain-containing proteins is to use the recombinant GST-AtDcp2 proteins as the baits to search for binding proteins in Arabidopsis tissue lysates. Proteins that can bind the GST-AtDcp2 but not GST-AtDcp2-PD (Figure 1) are likely those that target the GNSA-motif. Identification of these proteins will shed light on regulatory properties of AtDcp2.
CONCLUSIONS
Based upon the findings described in this article, we propose a model of the catalytic mechanism and regulation of AtDcp2 (Figure 8). We have demonstrated that AtDcp2 catalyses the following reaction.
In the reaction, AtDcp2 selectively catalyses cleavage of the diphosphate linkage between the β- and γ-phosphates in the cap of the capped mRNA substrate (m7Gppp-RNA) to generate 7-methyl GDP and 5′-monophosphate mRNA (p-RNA) as the reaction products. Similar to other known Nudix hydrolase superfamily members, the AtDcp2-catalysed decapping reaction involves nucleophilic attack of the pyrophosphate bond by a water molecule (Equation 1). One of the two conserved glutamates in the Nudix box is involved in polarizing and appropriately positioning the water molecule, while the other conserved glutamate coordinates a divalent cation (Figure 8). It is proposed that the recognition of the substrates by AtDcp2 involves binding of the RNA moiety of the capped mRNA to the Box B region and a KH-like motif in the loop preceding the Box B region. One lysine in the KH-like motif and two lysines in the Box B region likely mediate in part the binding of AtDcp2 to the RNA moiety. Furthermore, the activity of AtDcp2 is subject to feedback inhibition by its reaction product 7-methyl GDP and uncapped RNA.
Figure 8.

A model of the catalytic mechanism, regulatory domains and regulatory properties of AtDcp2. Based upon the structure of the yeast Dcp2 and the results shown in Figure 6, we postulate that the two conserved glutamates in the Nudix box in AtDcp2 are catalytically critical residues. Hydrolysis of capped mRNA (m7-Gppp-RNA) involves nucleophilic attack of the γ-phosphate of m7-Gppp-RNA by a water molecule. The enhanced nucleophilicity and proper alignment of this water molecule are governed by electrostatic interactions and hydrogen bonding with a divalent cation (Mn2+) and one of the glutamates in the Nudix box. The second conserved glutamate in the Nudix box is involved in chelating the divalent cation. In addition, a second divalent cation is involved in binding the α- and β-phosphates of the substrate. The RNA moiety of m7-Gppp-RNA interacts with Box B and the preceding loop structure (KH-like motif). Lys-231 of the loop structure, and Lys-243 and Lys-248 of Box B are the likely determinants involved in binding RNA.
In spite of these findings, several important outstanding questions remain: (i) What are the exact roles of Glu-154 and Glu-158 of the Nudix box in catalysis? (ii) How does AtDcp2 selectively target the pyrophosphate bond linking the β- and γ-phosphates but not that linking the α- and β-phosphates and the phosphoester linkage between γ-phosphate and the RNA moiety of the capped RNA substrate? (iii) What is the structural basis of RNA binding to the loop and Box B regions? (iv) How does the active site of AtDcp2 target the cap at the 5′-end of the capped RNA substrates? (v) What are the upstream regulators of AtDcp2? Xu et al. (8) recently reported that AtDcp2 interacts with Dcp1 and VARICOSE (VCS), the plant homolog of an important component called Hedls/Ge-1 of the mRNA processing bodies (P bodies) in the cytoplasm. Furthermore, they demonstrated that AtDcp2 activity is stimulated upon binding to these proteins. Exactly how these regulatory proteins modulate AtDcp2 activity is unclear. The availability of the intact recombinant AtDcp2 permits future investigation to answer these questions by biochemical and structural approaches.
ACKNOWLEDGEMENTS
We are indebted to Dr Lena Burri, University of Melbourne for the pSP73-Omp clone and the Sir John and Lady Higgins Foundation for sponsoring the PhD studies of the candidate Dilantha Gunawardana. Funding to pay the Open Access publication charges for this article was provided by The University of Melbourne.
Conflict of interest statement. None declared.
REFERENCES
- 1.Merrick WC. Cap-dependent and cap-independent translation in eukaryotic systems. Gene. 2004;332:1–11. doi: 10.1016/j.gene.2004.02.051. [DOI] [PubMed] [Google Scholar]
- 2.Moteki S, Price D. Functional coupling of capping and transcription of mRNA. Mol. Cell. 2002;10:599–609. doi: 10.1016/s1097-2765(02)00660-3. [DOI] [PubMed] [Google Scholar]
- 3.Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fitzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005;169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cougot N, Babajko S, Seraphin B. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 2004;165:31–40. doi: 10.1083/jcb.200309008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muhlrad D, Decker CJ, Parker R. Deadenylation of the unstable mRNA encoded by the yeast MFA2 gene leads to decapping followed by 5′–>3′ digestion of the transcript. Genes Dev. 1994;8:855–866. doi: 10.1101/gad.8.7.855. [DOI] [PubMed] [Google Scholar]
- 6.Coller J, Parker R. Eukaryotic mRNA decapping. Annu. Rev. Biochem. 2004;73:861–890. doi: 10.1146/annurev.biochem.73.011303.074032. [DOI] [PubMed] [Google Scholar]
- 7.van Dijk E, Cougot N, Meyer S, Babajko S, Wahle E, Seraphin B. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. Embo. J. 2002;21:6915–6924. doi: 10.1093/emboj/cdf678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu J, Yang JY, Niu QW, Chua NH. Arabidopsis DCP2, DCP1, and VARICOSE form a decapping complex required for postembryonic development. Plant Cell. 2006;18:3386–3398. doi: 10.1105/tpc.106.047605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Decker CJ, Parker R. A turnover pathway for both stable and unstable mRNAs in yeast: evidence for a requirement for deadenylation. Genes Dev. 1993;7:1632–1643. doi: 10.1101/gad.7.8.1632. [DOI] [PubMed] [Google Scholar]
- 10.Hsu CL, Stevens A. Yeast cells lacking 5′–>3′ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5′ cap structure. Mol. Cell Biol. 1993;13:4826–4835. doi: 10.1128/mcb.13.8.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steiger M, Carr-Schmid A, Schwartz DC, Kiledjian M, Parker R. Analysis of recombinant yeast decapping enzyme. RNA. 2003;9:231–238. doi: 10.1261/rna.2151403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc. Natl Acad. Sci. USA. 2002;99:12663–12668. doi: 10.1073/pnas.192445599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunckley T, Parker R. The DCP2 protein is required for mRNA decapping in Saccharomyces cerevisiae and contains a functional MutT motif. Embo. J. 1999;18:5411–5422. doi: 10.1093/emboj/18.19.5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu W, Dunn CA, Jones CR, D'Souza G, Bessman MJ. The 26 Nudix hydrolases of Bacillus cereus, a close relative of Bacillus anthracis. J. Biol. Chem. 2004;279:24861–24865. doi: 10.1074/jbc.M403272200. [DOI] [PubMed] [Google Scholar]
- 15.Sheikh S, O’Handley SF, Dunn CA, Bessman MJ. Identification and characterization of the Nudix hydrolase from the Archaeon, Methanococcus jannaschii, as a highly specific ADP-ribose pyrophosphatase. J. Biol. Chem. 1998;273:20924–20928. doi: 10.1074/jbc.273.33.20924. [DOI] [PubMed] [Google Scholar]
- 16.Bessman MJ, Frick DN, O’Handley SF. The MutT proteins or “Nudix” hydrolases, a family of versatile, widely distributed, “housecleaning” enzymes. J. Biol. Chem. 1996;271:25059–25062. doi: 10.1074/jbc.271.41.25059. [DOI] [PubMed] [Google Scholar]
- 17.Piccirillo C, Khanna R, Kiledjian M. Functional characterization of the mammalian mRNA decapping enzyme hDcp2. RNA. 2003;9:1138–1147. doi: 10.1261/rna.5690503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.She M, Decker CJ, Chen N, Tumati S, Parker R, Song H. Crystal structure and functional analysis of Dcp2p from Schizosaccharomyces pombe. Nat. Struct. Mol. Biol. 2006;13:63–70. doi: 10.1038/nsmb1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwasaki S, Takeda A, Motose H, Watanabe Y. Characterization of Arabidopsis decapping proteins AtDCP1 and AtDCP2, which are essential for post-embryonic development. FEBS Lett. 2007;581:2455–2459. doi: 10.1016/j.febslet.2007.04.051. [DOI] [PubMed] [Google Scholar]
- 20.Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–2139. doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- 21.Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11:1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Gopalan V, Tan TW, Lee BT, Ranganathan S. Xpro: database of eukaryotic protein-encoding genes. Nucleic Acids Res. 2004;32:D59–D63. doi: 10.1093/nar/gkh051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tompa P, Csermely P. The role of structural disorder in the function of RNA and protein chaperones. Faseb. J. 2004;18:1169–1175. doi: 10.1096/fj.04-1584rev. [DOI] [PubMed] [Google Scholar]
- 25.Gottesman S. Proteases and their targets in Escherichia coli. Annu. Rev. Genet. 1996;30:465–506. doi: 10.1146/annurev.genet.30.1.465. [DOI] [PubMed] [Google Scholar]
- 26.Levchenko I, Smith CK, Walsh NP, Sauer RT, Baker TA. PDZ-like domains mediate binding specificity in the Clp/Hsp100 family of chaperones and protease regulatory subunits. Cell. 1997;91:939–947. doi: 10.1016/s0092-8674(00)80485-7. [DOI] [PubMed] [Google Scholar]
- 27.Longhi S, Receveur-Brechot V, Karlin D, Johansson K, Darbon H, Bhella D, Yeo R, Finet S, Canard B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003;278:18638–18648. doi: 10.1074/jbc.M300518200. [DOI] [PubMed] [Google Scholar]
- 28.Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J. Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- 29.Beebe KD, Shin J, Peng J, Chaudhury C, Khera J, Pei D. Substrate recognition through a PDZ domain in tail-specific protease. Biochemistry. 2000;39:3149–3155. doi: 10.1021/bi992709s. [DOI] [PubMed] [Google Scholar]
- 30.Walsh NP, Alba BM, Bose B, Gross CA, Sauer RT. OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell. 2003;113:61–71. doi: 10.1016/s0092-8674(03)00203-4. [DOI] [PubMed] [Google Scholar]
- 31.Wilken C, Kitzing K, Kurzbauer R, Ehrmann M, Clausen T. Crystal structure of the DegS stress sensor: how a PDZ domain recognizes misfolded protein and activates a protease. Cell. 2004;117:483–494. doi: 10.1016/s0092-8674(04)00454-4. [DOI] [PubMed] [Google Scholar]
- 32.Keiler KC, Silber KR, Downard KM, Papayannopoulos IA, Biemann K, Sauer RT. C-terminal specific protein degradation: activity and substrate specificity of the Tsp protease. Protein Sci. 1995;4:1507–1515. doi: 10.1002/pro.5560040808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang S, Williams CJ, Wormington M, Stevens A, Peltz SW. Monitoring mRNA decapping activity. Methods. 1999;17:46–51. doi: 10.1006/meth.1998.0706. [DOI] [PubMed] [Google Scholar]
- 34.Bergman N, Opyrchal M, Bates EJ, Wilusz J. Analysis of the products of mRNA decapping and 3′-to-5′ decay by denaturing gel electrophoresis. RNA. 2002;8:959–965. doi: 10.1017/s1355838202025049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maksel D, Gooley PR, Swarbrick JD, Guranowski A, Gange C, Blackburn GM, Gayler KR. Characterization of active-site residues in diadenosine tetraphosphate hydrolase from Lupinus angustifolius. Biochem. J. 2001;357:399–405. doi: 10.1042/0264-6021:3570399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guranowski A. Fluoride is a strong and specific inhibitor of (asymmetrical) Ap4A hydrolases. FEBS Lett. 1990;262:205–208. doi: 10.1016/0014-5793(90)80190-t. [DOI] [PubMed] [Google Scholar]
- 37.Gasmi L, Cartwright JL, McLennan AG. Cloning, expression and characterization of YSA1H, a human adenosine 5′-diphosphosugar pyrophosphatase possessing a MutT motif. Biochem. J. 1999;344 (Pt 2):331–337. [PMC free article] [PubMed] [Google Scholar]
- 38.AbdelRaheim SR, McLennan AG. The Caenorhabditis elegans Y87G2A.14 Nudix hydrolase is a peroxisomal coenzyme A diphosphatase. BMC Biochem. 2002;3:5. doi: 10.1186/1471-2091-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mildvan AS, Xia Z, Azurmendi HF, Saraswat V, Legler PM, Massiah MA, Gabelli SB, Bianchet MA, Kang LW, et al. Structures and mechanisms of Nudix hydrolases. Arch. Biochem. Biophys. 2005;433:129–143. doi: 10.1016/j.abb.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 40.Harris TK, Wu G, Massiah MA, Mildvan AS. Mutational, kinetic, and NMR studies of the roles of conserved glutamate residues and of lysine-39 in the mechanism of the MutT pyrophosphohydrolase. Biochemistry. 2000;39:1655–1674. doi: 10.1021/bi9918745. [DOI] [PubMed] [Google Scholar]
- 41.Lin J, Abeygunawardana C, Frick DN, Bessman MJ, Mildvan AS. The role of Glu 57 in the mechanism of the Escherichia coli MutT enzyme by mutagenesis and heteronuclear NMR. Biochemistry. 1996;35:6715–6726. doi: 10.1021/bi953071x. [DOI] [PubMed] [Google Scholar]
- 42.Chen X, Court DL, Ji X. Crystal structure of ERA: a GTPase-dependent cell cycle regulator containing an RNA binding motif. Proc. Natl Acad. Sci. USA. 1999;96:8396–8401. doi: 10.1073/pnas.96.15.8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siomi H, Matunis MJ, Michael WM, Dreyfuss G. The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic Acids Res. 1993;21:1193–1198. doi: 10.1093/nar/21.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993;74:291–298. doi: 10.1016/0092-8674(93)90420-u. [DOI] [PubMed] [Google Scholar]
- 45.Sidiqi M, Wilce JA, Vivian JP, Porter CJ, Barker A, Leedman PJ, Wilce MC. Structure and RNA binding of the third KH domain of poly(C)-binding protein 1. Nucleic Acids Res. 2005;33:1213–1221. doi: 10.1093/nar/gki265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fletcher JI, Swarbrick JD, Maksel D, Gayler KR, Gooley PR. The structure of Ap4A hydrolase complexed with ATP-MgF(x) reveals the basis of substrate binding. Structure. 2002;10:205–213. doi: 10.1016/s0969-2126(02)00696-2. [DOI] [PubMed] [Google Scholar]
- 47.Swarbrick JD, Bashtannyk T, Maksel D, Zhang XR, Blackburn GM, Gayler KR, Gooley PR. The three-dimensional structure of the Nudix enzyme diadenosine tetraphosphate hydrolase from Lupinus angustifolius L. J. Mol. Biol. 2000;302:1165–1177. doi: 10.1006/jmbi.2000.4085. [DOI] [PubMed] [Google Scholar]
- 48.Naitow H, Tang J, Canady M, Wickner RB, Johnson JE. L-A virus at 3.4 Å resolution reveals particle architecture and mRNA decapping mechanism. Nat. Struct. Biol. 2002;9:725–728. doi: 10.1038/nsb844. [DOI] [PubMed] [Google Scholar]
- 49.Peculis BA, Reynolds K, Cleland M. Metal determines efficiency and substrate specificity of the nuclear NUDIX decapping proteins X29 and H29K (Nudt16) J. Biol. Chem. 2007;282:24792–24805. doi: 10.1074/jbc.M704179200. [DOI] [PubMed] [Google Scholar]
- 50.van Dijk E, Le Hir H, Seraphin B. DcpS can act in the 5′–3′ mRNA decay pathway in addition to the 3′–5′ pathway. Proc. Natl Acad. Sci. USA. 2003;100:12081–12086. doi: 10.1073/pnas.1635192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.AbdelRaheim SR, Cartwright JL, Gasmi L, McLennan AG. The NADH diphosphatase encoded by the Saccharomyces cerevisiae NPY1 Nudix hydrolase gene is located in peroxisomes. Arch. Biochem Biophys. 2001;388:18–24. doi: 10.1006/abbi.2000.2268. [DOI] [PubMed] [Google Scholar]
- 52.Coll RJ, Murphy AJ. Fluoride-inhibited calcium ATPase of sarcoplasmic reticulum. Magnesium and fluoride stoichiometry. J. Biol. Chem. 1992;267:21584–21587. [PubMed] [Google Scholar]
- 53.Jones S, Daley DT, Luscombe NM, Berman HM, Thornton JM. Protein-RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29:943–954. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Draper DE. Themes in RNA-protein recognition. J. Mol. Biol. 1999;293:255–270. doi: 10.1006/jmbi.1999.2991. [DOI] [PubMed] [Google Scholar]
- 55.Katsamba PS, Myszka DG, Laird-Offringa IA. Two functionally distinct steps mediate high affinity binding of U1A protein to U1 hairpin II RNA. J. Biol. Chem. 2001;276:21476–21481. doi: 10.1074/jbc.M101624200. [DOI] [PubMed] [Google Scholar]
- 56.Moller K, Rinke J, Ross A, Buddle G, Brimacombe R. The use of formaldehyde in RNA-protein cross-linking studies with ribosomal subunits from Escherichia coli. Eur. J. Biochem. 1977;76:175–187. doi: 10.1111/j.1432-1033.1977.tb11583.x. [DOI] [PubMed] [Google Scholar]