

Abstract
A recurring obstacle for structural genomics is the expression of insoluble, aggregated proteins. In these cases, the use of alternative salvage strategies, like in vitro refolding, is hindered by the lack of a universal refolding method. To overcome this obstacle, fractional factorial screens have been introduced as a systematic and rapid method to identify refolding conditions. However, methodical analyses of the effectiveness of refolding reagents on large sets of proteins remain limited. In this study, we address this void by designing a fractional factorial screen to rapidly explore the effect of 14 different reagents on the refolding of 33 structurally and functionally diverse proteins. The refolding data was analyzed using statistical methods to determine the effect of each refolding additive. The screen has been miniaturized for automation resulting in reduced protein requirements and increased throughput. Our results show that the choice of pH and reducing agent had the largest impact on protein refolding. Bis-mercaptoacetamide cyclohexane (BMC) and tris (2-carboxyethylphosphine) (TCEP) were superior reductants when compared to others in the screen. BMC was particularly effective in refolding disulfide-containing proteins, while TCEP was better for nondisulfide-containing proteins. From the screen, we successfully identified a positive synergistic interaction between nondetergent sulfobetaine 201 (NDSB 201) and BMC on Cdc25A refolding. The soluble protein resulting from this interaction crystallized and yielded a 2.2 Å structure. Our method, which combines a fractional factorial screen with statistical analysis of the data, provides a powerful approach for the identification of optimal refolding reagents in a general refolding screen.
Keywords: protein folding, fractional factorial screen, crystal structure, inclusion bodies, high-throughput refolding, structural genomics
The identification of 20,000–25,000 genes from the human genome project has resulted in a wealth of potential targets for structural biology investigation and pharmaceutical design (International Human Genome Sequencing Consortium 2004). Since the completion of the project, expectations have been high that the number of protein crystal structures would dramatically increase but, in reality, there has only been a moderate rise in the number of crystal structures, due largely to a lack of sufficient quantities of protein suitable for structural studies (Service 2002). Although the technology responsible for expressing recombinant proteins is highly developed (Chambers et al. 2004), it is still difficult to produce enough soluble protein for these structural studies. The ultimate goal of determining crystal structures on a genome-wide scale requires methods designed to improve the yield of functional protein.
Historically, optimization of soluble protein expression has been the first strategy when trying to obtain protein for structural studies. In contrast, refolding insoluble protein has often been a strategy of last resort due to the unpredictable and time-consuming nature of the refolding process. However, the literature shows that numerous proteins can be refolded into their active forms, and that certain additives can assist in the refolding process. The combination of these additives dictates the efficiency of refolding as well as the utility of this method to gain soluble protein. Some of the more effective additives include reducing agents, thiol shuffling enzymes, polar and nonpolar reagents, various detergents, and chaperonins; numerous excellent reviews have previously discussed these and other refolding additives in more detail (Rudolph and Lilie 1996; De Bernardez Clark 1998; Lilie et al. 1998; Voziyan et al. 2000; Clark 2001; Middelberg 2002). Due to the unpredictable nature of the refolding process, the development of a systematic method for identifying useful refolding conditions is needed. Fractional factorial refolding screens have emerged as a way to compensate for this unpredictability. Fractional factorial screens contain a representative subset of reagent combinations contained in full factorial screens and are designed to maximize the number of refolding variables explored while minimizing the amount of data collection (Hofmann et al. 1995; Chen and Gouaux 1997; Armstrong et al. 1999; Tobbell et al. 2002). These screens have been used successfully to refold proteins, but the choice of refolding additives included in these screens is based on historical precedent and does not take into account novel reagents shown to improve protein renaturation. More recently, Vincentelli et al. (2004) designed an automated, 96-well refolding strategy that incorporated a fractional factorial buffer design utilizing both the traditional refolding additives used in previous refolding screens as well as a newer class of refolding agents known as NDSBs.
Although prior refolding screens identify useful conditions for protein refolding, they stop short of using statistical methods to determine the utility of each reagent when used in a general screen on a diverse protein data set. In this study, we investigate the effects of additives on the refolding of 33 proteins using a fractional factorial refolding screen. We include reagents such as the reductants BMC and TCEP, and the detergent-mimic NDSB 201 in our matrix as a way of assessing their utility in refolding a variety of proteins. These reagents have been shown to be beneficial to protein refolding, extraction, and stability (Vuillard et al. 1995a,b; Woycechowsky et al. 1999; Chong and Chen 2000; English et al. 2002). The screen has been miniaturized for automation, resulting in reduced protein requirements, increased throughput, and enhanced reproducibility. To assess the applicability of the screen to a wide spectrum of proteins, we refolded multiple members from five gene families, as well as single members from additional families. The data gathered from refolding 33 proteins were analyzed using statistical methods to identify individual reagents, and reagent interactions having a significant effect on protein refolding. Every buffer condition successfully refolded at least one protein, and of the 14 reagents tested, 12 reagents significantly improved protein refolding. Finally, this screen was used successfully to identify a positive synergistic interaction between reagents that resulted in the production of soluble, functional protein leading to diffraction quality crystals and the solution of a protein structure. The results obtained support the use of a fractional factorial screen in combination with statistical analysis to identify suitable reagents to be included in a general refolding screen and provide a systematic method for optimizing the refolding process.
Results
Refolding screen design and the use of automation
Additives such as the reducing agents BMC and TCEP, the detergent Tween 80, and the detergent-mimic NDSB 201, were identified from the literature as useful refolding agents and evaluated for their suitability in a refolding screen (Vuillard et al. 1995a; Goldberg et al. 1996; Woycechowsky et al. 1999; Arakawa and Kita 2000; English et al. 2002). A fractional factorial design was used to sample multiple components in 32 buffers that included seven factors assessed at two levels (salt, PEG, GdnHCl, divalent metal ions, sucrose, arginine, and ligand) and three factors assessed at four levels (pH, detergent, and reductant) (Table 1). Miniaturization of the refolding and assay reactions to a 96-well plate format reduced the protein requirements to <500 μg of unfolded protein per triplicate screen and allowed the introduction of automatic pipetting systems at multiple steps in the refolding process, resulting in improved data quality and throughput.
Table 1.
Thirty-two condition fractional factorial screen
| Buffera | Detergentb | Reductantc | Saltd | PEG 3350e | GdnHClf | Cationg | Sucroseh | Argininei | Ligandj | |
| 1 | MES 5.5 | 0 | BMC | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2 | MES 6.5 | DDM | BMC | 1 | 1 | 0 | 0 | 1 | 1 | 1 |
| 3 | BORATE 9.5 | DDM | GSH:GSSG | 1 | 0 | 0 | 1 | 1 | 0 | 0 |
| 4 | MES 6.5 | T80 | BMC | 1 | 0 | 1 | 1 | 0 | 1 | 1 |
| 5 | TRIS 8.2 | NDSB | DTT | 1 | 1 | 0 | 1 | 0 | 0 | 1 |
| 6 | TRIS 8.2 | T80 | BMC | 1 | 1 | 0 | 0 | 1 | 1 | 0 |
| 7 | MES 5.5 | DDM | DTT | 0 | 0 | 0 | 1 | 1 | 1 | 1 |
| 8 | MES 5.5 | T80 | DTT | 0 | 1 | 1 | 0 | 0 | 1 | 1 |
| 9 | MES 5.5 | NDSB | TCEP | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| 10 | TRIS 8.2 | T80 | TCEP | 0 | 0 | 1 | 1 | 0 | 0 | 0 |
| 11 | MES 6.5 | NDSB | GSH:GSSG | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| 12 | MES 5.5 | DDM | GSH:GSSG | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| 13 | TRIS 8.2 | DDM | TCEP | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| 14 | MES 6.5 | NDSB | DTT | 1 | 0 | 1 | 0 | 1 | 0 | 0 |
| 15 | BORATE 9.5 | NDSB | TCEP | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 16 | BORATE 9.5 | T80 | DTT | 0 | 0 | 0 | 1 | 1 | 1 | 0 |
| 17 | TRIS 8.2 | 0 | DTT | 1 | 0 | 1 | 0 | 1 | 0 | 1 |
| 18 | TRIS 8.2 | DDM | BMC | 1 | 0 | 1 | 1 | 0 | 1 | 0 |
| 19 | TRIS 8.2 | 0 | GSH:GSSG | 0 | 1 | 0 | 1 | 0 | 1 | 1 |
| 20 | BORATE 9.5 | 0 | BMC | 0 | 1 | 1 | 1 | 1 | 0 | 1 |
| 21 | TRIS 8.2 | NDSB | GSH:GSSG | 0 | 0 | 1 | 0 | 1 | 1 | 1 |
| 22 | MES 6.5 | 0 | DTT | 1 | 1 | 0 | 1 | 0 | 0 | 0 |
| 23 | MES 5.5 | T80 | GSH:GSSG | 1 | 0 | 0 | 1 | 1 | 0 | 1 |
| 24 | MES 6.5 | DDM | TCEP | 0 | 0 | 1 | 1 | 0 | 0 | 1 |
| 25 | MES 5.5 | NDSB | BMC | 0 | 1 | 1 | 1 | 1 | 0 | 0 |
| 26 | MES 6.5 | 0 | GSH:GSSG | 0 | 0 | 1 | 0 | 1 | 1 | 0 |
| 27 | BORATE 9.5 | 0 | TCEP | 1 | 0 | 0 | 0 | 0 | 1 | 1 |
| 28 | BORATE 9.5 | T80 | GSH:GSSG | 1 | 1 | 1 | 0 | 0 | 0 | 0 |
| 29 | BORATE 9.5 | NDSB | BMC | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 30 | MES 6.5 | T80 | TCEP | 0 | 1 | 0 | 0 | 1 | 0 | 1 |
| 31 | MES 5.5 | 0 | TCEP | 1 | 1 | 1 | 1 | 1 | 1 | 0 |
| 32 | BORATE 9.5 | DDM | DTT | 0 | 1 | 1 | 0 | 0 | 1 | 0 |
a 50 mM buffer.
bDDM, 0.3 mM; Tween 80, 0.5 mM; NDSB 201, 1 M.
c BMC, TCEP, and DTT, 5 mM; GSH:GSSG, 1 mM GSH:0.1 mM GSSG.
d 0=10.56 mM NaCl, 0.44 mM KCl; 1=264 mM NaCl, 11 mM KCl.
e 0=no PEG 3350; 1=0.06% PEG 3350 w/v.
f 0=no GdnHCl; 1=550 mM GdnHCl.
g 0=1.1 mM EDTA; 1=2 mM MgCl2, 2 mM CaCl2.
h 0=no sucrose; 1=440 mM sucrose.
i 0=no arginine; 1=550 mM arginine.
j 0=no ligand; 1=presence of ligand (target:ligand, kinases:100 μM AMP-PNP, phosphatases:100 μM o-phospho-L-tyrosine, proteases, RNase A, sRNase A, helicase:1–10 μM assay substrate, dehydrogenases:20 μM NADH or NADP, lysozyme:10 μg/mL Micrococcus lysodeikticus).
Protein target selection
To ensure this screen met the criteria of broad applicability, we investigated the refolding of 33 proteins from different families of varying molecular weights (14–80 kDa), pIs (5.3–9.4), and disulfide content. The protein set in this study was comprised of 11 kinases, 9 proteases, 5 dehydrogenases, 4 phosphatases, and 4 single proteins (hepatitis C virus NS3 helicase, lysozyme, RNase A, and sRNase A), representing three other gene families. In addition to simple monomeric proteins, our data set also includes examples with complex quaternary structures such as a homodimer (steroid dehydrogenase), homotetramers (inosine 5′-monophosphate dehydrogenase [IMPDH] and lactate dehydrogenase [LDH]), and an α2β2 heterodimer (interleukin-1β converting enzyme [ICE]). Our initial focus was on soluble, active proteins with well-characterized activities that were then unfolded in a high concentration of denaturant. To ensure that the screen’s utility extended to insoluble proteins, we also included three proteins purified from inclusion bodies (ICE, promemapsin 2, and Cdc25A).
In vitro folding of 33 proteins
The range of significant refolding across the 33 protein set varied dramatically from 0.1% to 65% of the total protein being refolded. The feasibility and practicality of scaling-up refolding reactions yielding <1% activity has not been tested. However, we have previously shown that ICE, which typically refolds with ~1% efficiency, can generate sufficient protein for solving high resolution crystal structures (Wilson et al. 1994; Wei et al. 2000). When all buffers in the primary screen are considered, the refolding data indicate that every buffer resulted in the successful refolding of at least one protein (Fig. 1A ▶). The four buffers refolding the largest number of proteins were Buffer 22>Buffer 6>Buffer 29>Buffer 1. These buffers cover the entire pH range; three of the four contain BMC, and all lack GdnHCl. Grouping the refolding data by protein family indicates that the best refolding conditions are specific for each family (Fig. 1B ▶). The unpredictability of the best refolding conditions (shown in black) suggests that there is no universal refolding buffer in this screen and provides one of the strongest reasons why a fractional factorial screen is so useful. Given that many protein refolding studies in the literature use an iterative approach to identify optimal refolding conditions, the data from this set of proteins supports the utility of using a broad screen to more efficiently explore conditions resulting in refolding.
Figure 1.
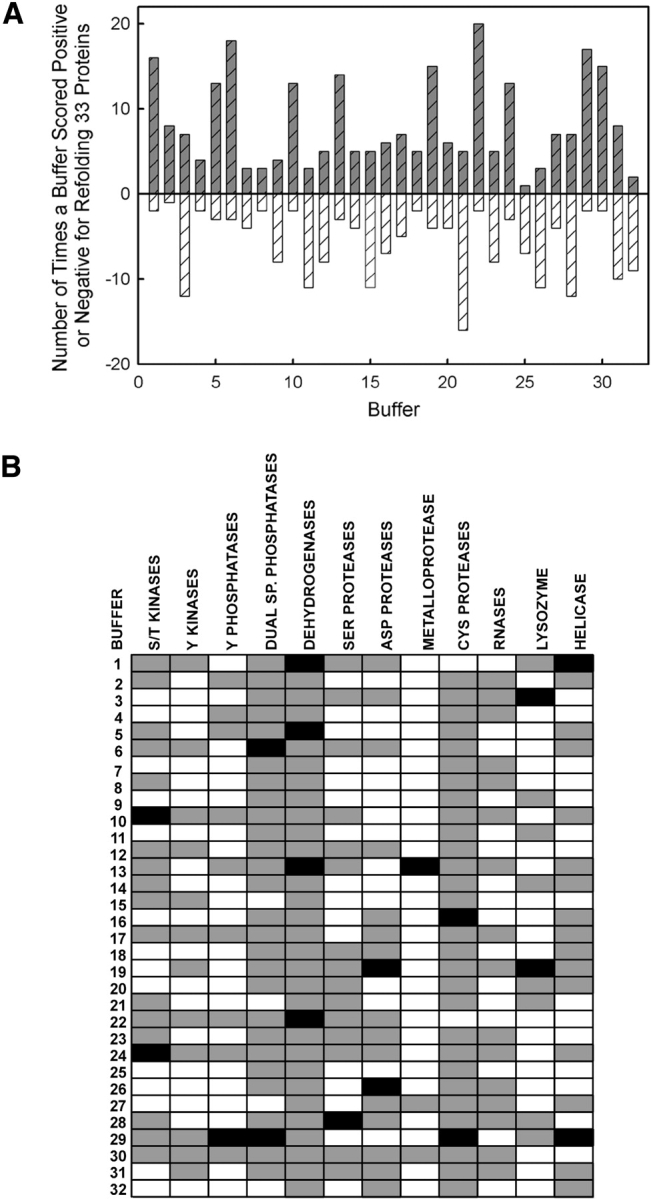
Protein refolding from the primary screen. (A) The effect of each buffer on the in vitro refolding of 33 proteins. The upper and lower activity quartiles for each protein were determined, and each buffer was scored positive for activity in the upper quartile and negative for activity in the lower quartile for all proteins. (B) In vitro refolding of protein families. The activity for each refolded protein was normalized, sorted into quartiles, scored, and a weighted average calculated for each buffer. Black represents high refolding (0.76–1.0); gray represents intermediate refolding (0.26–0.75); and white represents low refolding (0–0.25).
Identification of reagent effects on refolding
Triplicate refolding data sets were subjected to a rank transformation and the significance of each protein/reagent combination (p<0.05) was determined by analysis of variance followed by pair-wise comparisons for four level factors. Refolding factors shown to have a significant positive effect on refolding this set of proteins include dodecyl maltoside, NDSB 201, Tween 80, arginine, ligand, PEG 3350, salt, and sucrose. The presence of divalent metal ions and GdnHCl had a negative effect on refolding (Fig. 2 ▶). The design of the screen with four levels for both pH and reductant required that the baseline be assigned to the activity set with the lowest amount of refolding. This analysis indicates an optimal pH of 8.2 for refolding this set of proteins when compared to all other pH levels. In addition, expansion of the pH range resulted in the refolding of three proteins that did not refold at pH 6.5 or pH 8.2. Likewise, BMC and TCEP significantly enhanced the refolding of proteins when compared to the more commonly used dithiothreitol (DTT) and reduced: oxidized glutathione (GSH:GSSG). A comparison of both compounds was conducted on refolding proteins in the data set whose crystal structures have been published. The results of this comparison demonstrate that BMC significantly aids in refolding 64% of the proteins with disulfide bonds, while TCEP significantly aids in refolding 75% of the proteins lacking disulfide bonds.
Figure 2.

The effect of each reagent on protein refolding. The total number of significant effects (p<0.05) for each reagent from a pairwise comparison of the rank transformed refolding activity was determined.
Secondary screens for reagent optimization
Reagents shown to have a significant positive effect on protein refolding from the primary screen analysis were chosen for secondary screens. Five proteins (Cdc25A, IMPDH, DYRK3, MAPKAP-K5, and lysozyme) were selected to confirm the observed effects of BMC, Tween 80, dodecyl maltoside, pH, TCEP, and GSH:GSSG, respectively, on protein refolding. These experiments confirmed all six of the initial observations made from the primary screen in which a reagent had a positive effect, thus illustrating the power of this approach. The optimal reagent concentrations determined from these secondary screens were Tween 80, 0.5 mM; dodecyl maltoside, 0.3 mM; pH, 7.0; TCEP, 5–10 mM; and GSH:GSSG, 2.5–10 mM GSH: 0.25–1 mM GSSG.
Secondary screen of interacting reagents resulting in a high resolution crystal structure
The fractional factorial used in the primary screen was designed to identify main effects of the refolding additives. In addition, the resolution of the screen is sufficient to identify some interactions between reagents. The entire data set from the primary screen was examined for interactions between two reagents resulting in enhanced refolding. A positive synergistic interaction between NDSB 201 and BMC on the refolding of Cdc25A was identified, and a secondary screen designed to vary both reagents simultaneously (Fig. 3A ▶). When either NDSB 201 or BMC were present individually, the maximum refolding was less than twofold over the baseline condition that lacked both reagents. However, the combination of both reagents resulted in up to a 36-fold increase in refolded protein confirming the positive interaction observed in the primary screen. The best condition for refolding Cdc25A resulted from 0.6 M NDSB 201 and 5 mM BMC, and this was used to refold the protein on a larger scale. The final yield of soluble, active protein suitable for crystallization after refolding and purification was 1.5%. The protein crystallized under conditions similar to those reported previously and the resultant 2.2 Å crystal structure of refolded Cdc25A is identical to that published for soluble Cdc25A (Fig. 3B,C ▶) (Fauman et al. 1998).
Figure 3.
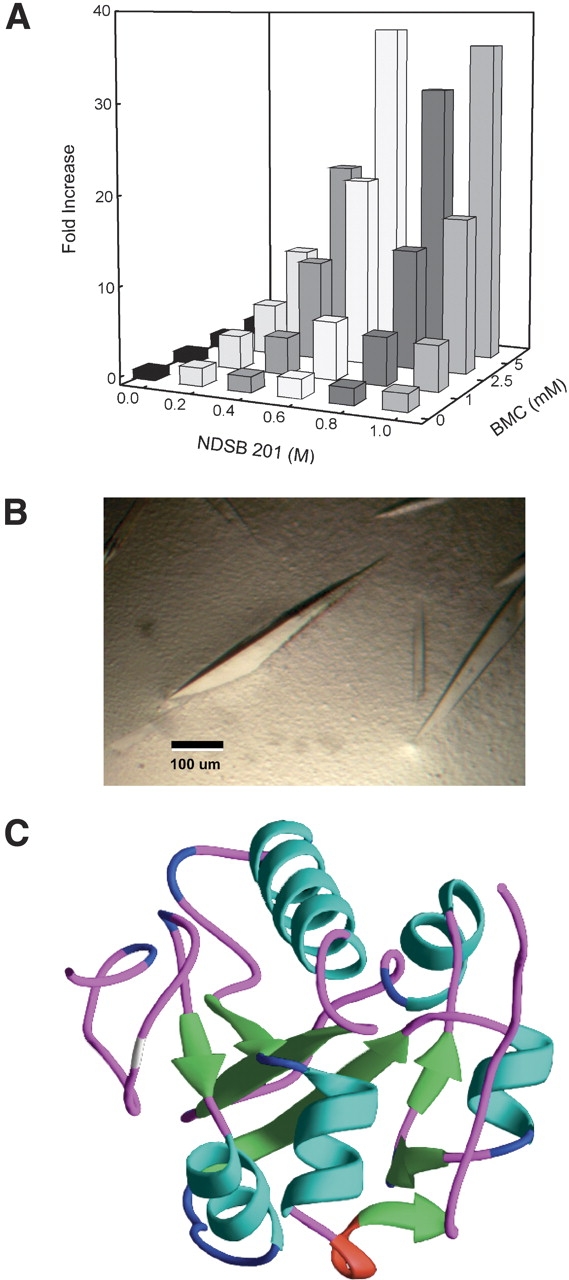
Reagent interaction effects on Cdc25A refolding and crystallization. (A) The interaction between NDSB 201 and BMC on Cdc25A refolding identified from the primary screen was tested by varying NDSB 201 (0–1 M) and BMC (0–5 mM) concentrations to identify the optimal reagent combination. Activity was measured as described and a 36-fold increase over baseline lacking both reagents was observed. (B) Cdc25A crystals. (C) Ribbon diagram for the 2.2 Å crystal structure of refolded Cdc25A.
Discussion
A significant barrier facing structural genomic projects is the generation of soluble, functional eukaryotic protein for structural studies. Meeting this demand has proven to be a challenge, given the low success rate for expressing soluble eukaryotic proteins compared to prokaryotic proteins (Yee et al. 2002; Chambers et al. 2004). An alternative approach for generating sufficient quantities of soluble protein is refolding the insoluble protein expressed in the inclusion bodies of Escherichia coli. In theory, refolding these proteins should be a straightforward process given that the refolding literature is replete with the effects of individual reagents on the refolding of single proteins. In practice, however, there is no universal method or buffer for reliably refolding a given protein of interest and identification of initial refolding conditions remains a major hurdle.
One way to overcome this obstacle is by the introduction of refolding screens to rapidly identify initial conditions that result in folded protein (Hofmann et al. 1995; Chen and Gouaux 1997; Armstrong et al. 1999; Tobbell et al. 2002; Maxwell et al. 2003; Scheich et al. 2004; Tresaugues et al. 2004; Vincentelli et al. 2004). These screens were designed to test a variety of refolding additives in a minimal number of experiments. Although these screens have been successful in refolding multiple proteins, a comprehensive statistical analysis of the importance of the reagents for generalized protein refolding is minimal. Our method uses a fractional factorial design combined with statistical analysis to directly compare the effects of both well-known, and lesser-known, refolding reagents on a large and diverse set of proteins. The data gathered from this study was used to determine the general utility of each reagent for the better design of future refolding screens.
Based on our analysis, pH and reductants had the largest impact on refolding our set of 33 proteins. The effect of pH on protein refolding has been well documented on a protein-specific basis, but previous analysis regarding the optimal pH for protein refolding has been limited. Our data demonstrates a direct comparison of four pH levels and provides examples where pH extremes are crucial for protein refolding. Likewise, the data from a refolding screen designed by Vincentelli et al. (2004) showed that a broad pH range was important for protein solubility, underscoring the importance of exploring pH when designing a generalized refolding screen. Reducing agents also play an important role in refolding proteins; however, the use of compounds for protein refolding beyond the more traditional reductants (DTT, GSH:GSSG, and βME) remains protein-specific. BMC is a dithiol that improves protein refolding both in vitro and in vivo, and is thought to mimic protein disulfide isomerase (PDI) by catalyzing native disulfide bond formation (Woycechowsky and Raines 2000). TCEP is a nonthiol-containing molecule and is a stronger reductant than DTT at pH values below 8 (Getz et al. 1999). The results from this protein data set strongly support the inclusion of BMC and TCEP in a refolding screen. Proteins containing disulfide bonds were more effectively refolded using BMC than its well-studied counterpart, GSH:GSSG. In contrast, proteins lacking disulfide bonds were more effectively refolded using TCEP than DTT. The utility of alternative reductants, such as 4-mercaptobenzeneacetate (4-MPA) shown in the literature to aid protein folding (Gough et al. 2002), suggests that other compounds may also be useful, and could be explored in future refolding screens.
Although important, pH and reductants are not the only variables to consider when designing a refolding screen. Studies have shown that a single protein can refold under markedly different conditions (Hofmann et al. 1995; Armstrong et al. 1999). Our data set contained two phosphatases with 65% sequence identity and nearly identical structural folds. Even with such a high level of identity, one of the proteins refolded productively in twice as many buffer conditions as the other. One way to overcome the unpredictable nature of protein refolding is to include an array of reagents known to improve refolding as a way to maximize the opportunity to recover functional protein. As such, our screen also includes all the reagents originally described in a fractional factorial screen by Chen et al. (Chen and Gouaux 1997) as well as the detergent Tween 80 and the detergent-mimic NDSB 201. The latter two were added because they inhibit aggregation during the refolding process resulting in increased yields of soluble protein (Goldberg et al. 1996; Arakawa and Kita 2000; Chong and Chen 2000). NDSBs lack the hydrophobic tail of detergents, thereby preventing micelle formation and have been shown to be especially helpful in refolding at higher protein concentrations (Expert-Bezancon et al. 2003). Vincentelli et al. (2004) included NDSBs 195, 201, and 256 in their refolding screen and found them to be useful refolding additives. The remaining reagents in our screen improved the refolding of at least one protein with the exception of GdnHCl and divalent metal ions. The results from our analysis suggest that inclusion of all the reagents discussed, aside from GdnHCl and divalent metal ions, will increase the chance of successfully applying a broad refolding screen. The inclusion of alternative refolding agents like cyclodextrins, which have been used successfully in prior refolding studies (Machida et al. 2000; Scheich et al. 2004), could be explored in future fractional factorial screens.
While the effects of reagent interactions on refolding have been touched upon previously (Tobbell et al. 2002), the optimization of a positive reagent interaction for generating crystallization quality protein is unique. Reagent interactions can be identified depending on the resolution of the fractional factorial screen. The importance of using appropriate experimental designs and statistical methods to analyze the refolding data is particularly relevant when looking beyond the main effects for these interactions. SAmBA, a software program used previously to design a refolding matrix (Vincentelli et al. 2004), is good for setting up the experimental design but lacks the complementary statistical methods needed to analyze the data. The reagent interactions in our screen were not immediately discernable, and could only be identified using statistical analysis. Using this method, we were able to identify potential interactions, and interestingly, a third of these interactions were between pH and the various reductants. The interaction between NDSB 201 and BMC on the refolding of Cdc25A was selected for follow-up due to the novelty of the reagents. In addition, the low refolding efficiency of the protein made it a more challenging example to pursue. The resultant crystal structure of Cdc25A supports the literature in promoting the utility of refolding for generating soluble protein for structural genomics programs (Maxwell et al. 2003).
The matrix described here allowed the rapid exploration of 14 different reagents on the refolding of 33 proteins representing significant diversity in structure and function. Moreover, this screen incorporated recently described reagents shown to improve the refolding process while decreasing the total number of conditions from >8000 data points in a full factorial to a mere 32 data points. While other refolding screens have used light scattering as a measurement of refolding (Tresaugues et al. 2004; Vincentelli et al. 2004), protein activity provides a useful alternative method to measure refolding, and has low protein requirements of <500 μg of unfolded protein per triplicate primary screen. In addition, the small reaction volumes allow future screening designs to include more difficult to obtain refolding reagents such as chaperonins.
The identification of important new reagent effects and interactions that enhance refolding highlights the need to identify optimal buffer conditions for refolding proteins in a methodical, fast, and economical way. In this regard, the combination of automation, fractional factorial screens, and a thorough analysis of the data using statistical software provide a powerful tool to expand on existing refolding methodology. The data presented here demonstrates the strength of this strategy as a way to overcome the bottleneck of obtaining soluble, functional protein for structural genomics programs.
Materials and methods
Protein expression and purification
The proteins expressed and purified in this study have been published elsewhere: kinases (Takahashi et al. 1989; Lindberg and Hunter 1990; McTigue et al. 1999; Chambers et al. 2004), phosphatases (Cool et al. 1989; Fauman et al. 1998; Andersen et al. 2000; Austen et al. 2004), proteases (Thompson et al. 1995; Hong et al. 2000; Wei et al. 2000; Austen et al. 2004), IMPDH (Fleming et al. 1996), and helicase (Kim et al. 1998). Recombinant proteins were expressed in E. coli or insect cells using a multisystem expression vector (Chambers 2002). Proteins were flanked with a cleavable (His)6 tag, allowing purification by metal affinity chromatography, followed by size-exclusion and ion-exchange chromatography when necessary. Commercial enzymes were purified by size-exclusion chromatography. Protein concentrations were determined from the A280 using calculated extinction coefficients (Gill and Von Hippel 1989).
Refolding matrix design
A fractional factorial design was constructed using the Design of Experiments (DOE) function within the JMP statistical analysis software package (JMP v. 4, SAS Institute). Three sets of reagents (buffer pH, detergent, and reductant) were grouped and considered as single factors at four levels by the previously reported strategy of combining a pair of two level factors (Montgomery 1991). The four levels for each reagent are pH (5.5, 6.5, 8.2, and 9.5), reducing agents (GSH:GSSG, TCEP, BMC, and DTT), and detergent (dodecyl maltoside, Tween 80, NDSB 201, and no detergent). The remaining reagents (ligand, divalent metal ions, arginine, GdnHCl, NaCl/KCl, PEG 3350, and sucrose) were either present or absent (two levels), giving the fractional factorial design shown in Table 1. All refolding buffers were made and stored in deep, 96-well blocks and frozen at −80°C.
Refolding protocol
Proteins were unfolded overnight in 6 M GdnHCl and 5 mM βME at 25°C and then concentrated to 1 mg/mL. Prior to use, deep, 96-well blocks housing enough refolding buffer to perform each primary screen in triplicate were thawed and ligands added to the appropriate wells. Daughter plates (round-bottom polypropylene) of refolding buffers were made using an Apricot Designs pipetting station (Perkin-Elmer). The plates were cooled to 4°C, unfolded protein added to a final concentration of 50 μg/mL, and incubated overnight with rocking at 4°C.
Activity measurements
Refolded proteins (5–40 μL) were assayed and data collected on a Spectramax (absorbance) or an Fmax (fluorescence) plate reader using the Spectramax Pro software for data analysis (Molecular Devices Corp.). Negative control plates containing everything but refolded protein were subtracted from experimental data. A coupled assay using the appropriate peptide phosphoacceptor substrates and measuring NADH conversion at 340 nm was used to detect kinase activity (Fox et al. 1998). Phosphatase activity was measured by monitoring pNPP hydrolysis at 405 nm (Dunphy and Kumagai 1991). Protease activity was measured by monitoring cleavage of the appropriate peptide substrates (Nakajima et al. 1979). Dehydrogenase activity was measured by monitoring 340 nm as described (Fleming et al. 1996; Prabhakar et al. 1998). The activity of lysozyme, RNase A, and helicase were measured as described (Goldberg et al. 1991; Kim et al. 1998; Schultz et al. 1998).
Analysis of primary screen data
The raw refolding data was subjected to a rank transformation (Conover and Iman 1981) and significance (p<0.05) for each reagent/protein combination was determined by analysis of variance using SAS Institute statistical software. Dunnett’s test was applied to the four level factors for pairwise comparisons of the individual levels (Dunnett 1955). For reductant and pH, the reagent level with the poorest refolding was set as the baseline. Interactions were obtained from a stepwise regression model using the rank transformed data sets.
Secondary screen of main effects
Reagents that were shown to have a significant positive effect on refolding were subsequently investigated individually. Significance was determined after a rank transformation of the raw data and analysis of variance of the protein/reagent combinations. The effects of BMC, Tween 80, TCEP, and GSH:GSSG were examined by measuring the activity of refolded Cdc25A (Dunphy and Kumagai 1991), IMPDH (Fleming et al. 1996), MAPKAP-K5 (New et al. 1998), and lysozyme (Goldberg et al. 1991), respectively. The effects of dodecyl maltoside and pH were examined using DYRK3 (Himpel et al. 2000). The core buffers used in the secondary screens were chosen by determining the best buffer from the 32 conditions that refolded the protein and contained the reagent to be investigated. A minimal buffer was used in cases where the best buffer contained reagents that had a potential negative effect on refolding. To examine the effects of BMC, Cdc25A was refolded in Buffer 6 containing 0–5 mM BMC. Likewise, IMPDH was refolded in Buffer 10 containing 0–2 mM Tween 80; DYRK3 was refolded in Buffer 24, containing 0–1.2 mMdodecyl maltoside, and Buffer 10, with a pH range of 5.5–10; MAPKAP-K5 was refolded in minimal buffer containing 50 mM Tris [pH 8.2], 10.56 mM NaCl, 0.44 mM KCl, 1.1 mM EDTA, and 0–100 mM TCEP; and lysozyme was refolded in the same buffer containing a 10:1 ratio of reduced and oxidized glutathione (GSH:GSSG; 0–100:0–10 mM).
Secondary screen of interacting reagents
The primary screen suggested an interaction between NDSB 201 and BMC on Cdc25A refolding and a secondary screen was designed to vary both reagents simultaneously. Cdc25A was refolded in Buffer 29 containing 0–1 M NDSB and 0–5 mM BMC and assayed as described.
Cdc25A refolding
Inclusion bodies containing Cdc25A were isolated from E. coli after extensive washing, and were solubilized in 8.0 M GdnHCl. The soluble material was purified by size-exclusion chromatography and the peak fractions were pooled for refolding. Denatured protein was added to a final concentration of 50 μg/mL in Buffer 29 containing 0.6MNDSB 201 and 5 mM BMC while being stirred at 4°C. The solution was incubated for 24 h at 4°C and NDSB 201 was removed by dialysis. The refolded protein was purified by size-exclusion and cation-exchange chromatography, and the activity measured as described. The final sample was dialyzed against crystallography buffer and concentrated to 10 mg/mL.
Cdc25A crystallization and structural determination
Crystallization of refolded Cdc25A was carried out using the hanging-drop vapor diffusion technique at room temperature. The protein was added in a 1:1-μL ratio to crystallization buffer containing 18% (w/v) PEG 6000, 100 mM Na-Citrate [pH 6.2], and 200 mM KCl. The structure was solved using molecular replacement with the published Cdc25A coordinates (PDB 1C25; Fauman et al. 1998).
Acknowledgments
We thank Anne Behrens, Debby Brennan, Joyce Coll, and Max Dawson for their help in refining the screen and refolding numerous proteins; Lora Swenson for crystallizing Cdc25A; Dr. Stephan Ogenstad for helpful discussions regarding statistical analysis; and Drs. Stephen Chambers, Scott Raybuck, and John Thomson for critical review of the manuscript.
Abbreviations
AMP-PNP, 5′-adenylylimidodiphosphate
βME, β-mercaptoethanol
BMC, bis-mercaptoacetamide cyclohexane
Cdc25A, cell division cycle 25A phosphatase
DDM, n-dodecyl-β-D-maltopyranoside
DTT, dithiothreitol
DYRK3, dual specificity Yak1-related kinase 3
GdnHCl, guanidine hydrochloride
GSH, reduced glutathione
GSSG, oxidized glutathione
ICE, interleukin-1β converting enzyme
IMPDH, inosine 5′-monophosphate dehydrogenase
LDH, lactate dehydrogenase
MAPKAP-K5, mitogen-activated protein kinase-activated protein kinase 5
MES, 2-[N-morpholino] ethanesulfonic acid
NADH, β-nicotinamide-adenine- dinucleotide, reduced
NADP+, β-nicotinamide-adenine-dinucleotide phosphate
NDSB, nondetergent sulfobetaine
PEG 3350, polyethylene glycol 3350 Da
pNPP, p-nitrophenylphosphate
RNase A, ribonuclease A
TCEP, tris(2-carboxyethylphosphine)
Tris-HCl, tris (hydroxymethyl)aminomethane hydrochloride
Tween 80, polyoxyethylene (80) sorbitan monolaurate.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051433205.
References
- Andersen, H.S., Iversen, L.F., Jeppesen, C.B., Branner, S., Norris, K., Rasmussen, H.B., Moller, K.B., and Moller, N.P. 2000. 2-(Oxalylamino)- benzoic acid is a general, competitive inhibitor of protein-tyrosine phosphatases. J. Biol. Chem. 275 7101–7108. [DOI] [PubMed] [Google Scholar]
- Arakawa, T. and Kita, Y. 2000. Protection of bovine serum albumin from aggregation by Tween 80. J. Pharm. Sci. 89 646–651. [DOI] [PubMed] [Google Scholar]
- Armstrong, N., de Lencastre, A., and Gouaux, E. 1999. A new protein folding screen: Application to the ligand binding domains of a glutamate and kainate receptor and to lysozyme and carbonic anhydrase. Protein Sci. 8 1475–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austen, D.A., Fulghum, J.R., Lu, F., Petrillo, R.A., and Chambers, S.P. 2004. Development of a high-throughput protein expression strategy. BioProcess. J. 3 41–47. [Google Scholar]
- Chambers, S.P. 2002. High-throughput protein expression for the post-genomic era. Drug Discov. Today 7 759–765. [DOI] [PubMed] [Google Scholar]
- Chambers, S.P., Austen, D.A., Fulghum, J.R., and Kim, W.M. 2004. High-throughput screening for soluble recombinant expressed kinases in Escherichia coli and insect cells. Protein Expr. Purif. 36 40–47. [DOI] [PubMed] [Google Scholar]
- Chen, G.Q. and Gouaux, E. 1997. Overexpression of a glutamate receptor (GluR2) ligand binding domain in Escherichia coli: Application of a novel protein folding screen. Proc. Natl. Acad. Sci. 94 13431–13436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, Y. and Chen, H. 2000. Preparation of functional recombinant protein from E. coli using a nondetergent sulfobetaine. Biotechniques 29 1166–1167. [DOI] [PubMed] [Google Scholar]
- Clark, E.D. 2001. Protein refolding for industrial processes. Curr. Opin. Biotechnol. 12 202–207. [DOI] [PubMed] [Google Scholar]
- Conover, W.J. and Iman, R.L. 1981. Rank transformations as a bridge between parametric and nonparametric statistics. Am. Stat. 35 124–129. [Google Scholar]
- Cool, D.E., Tonks, N.K., Charbonneau, H., Walsh, K.A., Fischer, E.H., and Krebs, E.G. 1989. cDNA isolated from a human T-cell library encodes a member of the protein–tyrosine–phosphatase family. Proc. Natl. Acad. Sci. 86 5257–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bernardez Clark, E. 1998. Refolding of recombinant proteins. Curr. Opin. Biotechnol. 9 157–163. [DOI] [PubMed] [Google Scholar]
- Dunnett, C.W. 1955. A multiple comparison procedure for comparing several treatments with a control. J. Am. Stat. Assoc. 50 1096–1121. [Google Scholar]
- Dunphy, W.G. and Kumagai, A. 1991. The cdc25 protein contains an intrinsic phosphatase activity. Cell 67 189–196. [DOI] [PubMed] [Google Scholar]
- English, B.P., Welker, E., Narayan, M., and Scheraga, H.A. 2002. Development of a novel method to populate native disulfide-bonded intermediates for structural characterization of proteins: Implications for the mechanism of oxidative folding of RNase A. J. Am. Chem. Soc. 124 4995–4999. [DOI] [PubMed] [Google Scholar]
- Expert-Bezancon, N., Rabilloud, T., Vuillard, L., and Goldberg, M.E. 2003. Physical-chemical features of non-detergent sulfobetaines active as protein-folding helpers. Biophys. Chem. 100 469–479. [DOI] [PubMed] [Google Scholar]
- Fauman, E.B., Cogswell, J.P., Lovejoy, B., Rocque, W.J., Holmes, W., Montana, V.G., Piwnica-Worms, H., Rink, M.J., and Saper, M.A. 1998. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell 93 617–625. [DOI] [PubMed] [Google Scholar]
- Fleming, M.A., Chambers, S.P., Connelly, P.R., Nimmesgern, E., Fox, T., Bruzzese, F.J., Hoe, S.T., Fulghum, J.R., Livingston, D.J., Stuver, C.M., et al. 1996. Inhibition of IMPDH by mycophenolic acid: Dissection of forward and reverse pathways using capillary electrophoresis. Biochemistry 35 6990–6997. [DOI] [PubMed] [Google Scholar]
- Fox, T., Coll, J.T., Xie, X., Ford, P.J., Germann, U.A., Porter, M.D., Pazhanisamy, S., Fleming, M.A., Galullo, V., Su, M.S., et al. 1998. A single amino acid substitution makes ERK2 susceptible to pyridinyl imidazole inhibitors of p38 MAP kinase. Protein Sci. 7 2249–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getz, E.B., Xiao, M., Chakrabarty, T., Cooke, R., and Selvin, P.R. 1999. A comparison between the sulfhydryl reductants tris (2-carboxyethyl)- phosphine and dithiothreitol for use in protein biochemistry. Anal. Biochem. 273 73–80. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and Von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Goldberg, M.E., Rudolph, R., and Jaenicke, R. 1991. A kinetic study of the competition between renaturation and aggregation during the refolding of denatured-reduced egg white lysozyme. Biochemistry 30 2790–2797. [DOI] [PubMed] [Google Scholar]
- Goldberg, M.E., Expert-Bezancon, N., Vuillard, L., and Rabilloud, T. 1996. Non-detergent sulphobetaines: A new class of molecules that facilitate in vitro protein renaturation. Fold. Des. 1 21–27. [DOI] [PubMed] [Google Scholar]
- Gough, J.D., Williams Jr., R.H., Donofrio, A.E., and Lees, W.J. 2002. Folding disulfide-containing proteins faster with an aromatic thiol. J. Am. Chem. Soc. 124 3885–3892. [DOI] [PubMed] [Google Scholar]
- Himpel, S., Tegge, W., Frank, R., Leder, S., Joost, H.G., and Becker, W. 2000. Specificity determinants of substrate recognition by the protein kinase DYRK1A. J. Biol. Chem. 275 2431–2438. [DOI] [PubMed] [Google Scholar]
- Hofmann, A., Tai, M., Wong, W., and Glabe, C.G. 1995. A sparse matrix screen to establish initial conditions for protein renaturation. Anal. Biochem. 230 8–15. [DOI] [PubMed] [Google Scholar]
- Hong, L., Koelsch, G., Lin, X., Wu, S., Terzyan, S., Ghosh, A.K., Zhang, X.C., and Tang, J. 2000. Structure of the protease domain of memapsin 2 (β-secretase) complexed with inhibitor. Science 290 150–153. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature 431 931–945. [DOI] [PubMed] [Google Scholar]
- Kim, J.L., Morgenstern, K.A., Griffith, J.P., Dwyer, M.D., Thomson, J.A., Murcko, M.A., Lin, C., and Caron, P.R. 1998. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: The crystal structure provides insights into the mode of unwinding. Structure 6 89–100. [DOI] [PubMed] [Google Scholar]
- Lilie, H., Schwarz, E., and Rudolph, R. 1998. Advances in refolding of proteins produced in E. coli. Curr. Opin. Biotechnol. 9 497–501. [DOI] [PubMed] [Google Scholar]
- Lindberg, R.A. and Hunter, T. 1990. cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Mol. Cell. Biol. 10 6316–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida, S., Ogawa, S., Xiaohua, S., Takaha, T., Fujii, K., and Hayashi, K. 2000. Cycloamylose as an efficient artificial chaperone for protein refolding. FEBS Lett. 486 131–135. [DOI] [PubMed] [Google Scholar]
- Maxwell, K.L., Bona, D., Liu, C., Arrowsmith, C.H., and Edwards, A.M. 2003. Refolding out of guanidine hydrochloride is an effective approach for high-throughput structural studies of small proteins. Protein Sci. 12 2073–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue, M.A., Wickersham, J.A., Pinko, C., Showalter, R.E., Parast, C.V., Tempczyk-Russell, A., Gehring, M.R., Mroczkowski, B., Kan, C.C., Villafranca, J.E., et al. 1999. Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: A key enzyme in angiogenesis. Struct. Fold. Des. 7 319–330. [DOI] [PubMed] [Google Scholar]
- Middelberg, A.P.J. 2002. Preparative protein refolding. Trends Biotechnol. 20 437–443. [DOI] [PubMed] [Google Scholar]
- Montgomery, D.C. 1991. Design and analysis of experiments, 3rd ed. John Wiley and Sons, New York.
- Nakajima, K., Powers, J.C., Ashe, B.M., and Zimmerman, M. 1979. Mapping the extended substrate binding site of cathepsin G and human leukocyte elastase. Studies with peptide substrates related to the α 1-protease inhibitor reactive site. J. Biol. Chem. 254 4027–4032. [PubMed] [Google Scholar]
- New, L., Jiang, Y., Zhao, M., Liu, K., Zhu, W., Flood, L.J., Kato, Y., Parry, G.C., and Han, J. 1998. PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 17 3372–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar, P., Laboy, J.I., Wang, J., Budker, T., Din, Z.Z., Chobanian, M., and Fahien, L.A. 1998. Effect of NADH-X on cytosolic glycerol-3- phosphate dehydrogenase. Arch. Biochem. Biophys. 360 195–205. [DOI] [PubMed] [Google Scholar]
- Rudolph, R. and Lilie, H. 1996. In vitro folding of inclusion body proteins. FASEB J. 10 49–56. [PubMed] [Google Scholar]
- Scheich, C., Niesen, F.H., Seckler, R., and Bussow, K. 2004. An automated in vitro protein folding screen applied to a human dynactin subunit. Protein Sci. 13 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, L.W., Quirk, D.J., and Raines, R.T. 1998. His...Asp catalytic dyad of ribonuclease A: Structure and function of the wild-type, D121N, and D121A enzymes. Biochemistry 37 8886–8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Service, R.F. 2002. Tapping DNA for structures produces a trickle. Science 298 948–950. [DOI] [PubMed] [Google Scholar]
- Takahashi, M., Buma, Y., and Hiai, H. 1989. Isolation of ret protooncogene cDNA with an amino-terminal signal sequence. Oncogene 4 805–806. [PubMed] [Google Scholar]
- Thompson, A., Huber, G., and Malherbe, P. 1995. Cloning and functional expression of a metalloendopeptidase from human brain with the ability to cleave a β-APP substrate peptide. Biochem. Biophys. Res. Commun. 213 66–73. [DOI] [PubMed] [Google Scholar]
- Tobbell, D.A., Middleton, B.J., Raines, S., Needham, M.R.C., Taylor, I.W.F., Beveridge, J.Y., and Abbott, W.M. 2002. Identification of in vitro folding conditions for procathepsin S and cathepsin S using fractional factorial screens. Protein Expr. Purif. 24 242–254. [DOI] [PubMed] [Google Scholar]
- Tresaugues, L., Collinet, B., Minard, P., Henckes, G., Aufrere, R., Blondeau, K., Liger, D., Zhou, C.Z., Janin, J., Van Tilbeurgh, H., et al. 2004. Refolding strategies from inclusion bodies in a structural genomics project. J. Struct. Funct. Genomics 5 195–204. [DOI] [PubMed] [Google Scholar]
- Vincentelli, R., Canaan, S., Campanacci, V., Valencia, C., Maurin, D., Frassinetti, F., Scappucini-Calvo, L., Bourne, Y., Cambillau, C., and Bignon, C. 2004. High-throughput automated refolding screening of inclusion bodies. Protein Sci. 13 2782–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voziyan, P.A., Jadhav, L., and Fisher, M.T. 2000. Refolding a glutamine synthetase truncation mutant in vitro: Identifying superior conditions using a combination of chaperonins and osmolytes. J. Pharm. Sci. 89 1036–1045. [DOI] [PubMed] [Google Scholar]
- Vuillard, L., Madern, D., Franzetti, B., and Rabilloud, T. 1995a. Halophilic protein stabilization by the mild solubilizing agents nondetergent sulfobetaines. Anal. Biochem. 230 290–294. [DOI] [PubMed] [Google Scholar]
- Vuillard, L., Marret, N., and Rabilloud, T. 1995b. Enhancing protein solubilization with non-detergent sulfobetaines. Electrophoresis 16 295–297. [DOI] [PubMed] [Google Scholar]
- Wei, Y., Fox, T., Chambers, S.P., Sintchak, J., Coll, J.T., Golec, J.M., Swenson, L., Wilson, K.P., and Charifson, P.S. 2000. The structures of caspases-1, -3, -7 and -8 reveal the basis for substrate and inhibitor selectivity. Chem. Biol. 7 423–432. [DOI] [PubMed] [Google Scholar]
- Wilson, K.P., Black, J.A., Thomson, J.A., Kim, E.E., Griffith, J.P., Navia, M.A., Murcko, M.A., Chambers, S.P., Aldape, R.A., Raybuck, S.A., et al. 1994. Structure and mechanism of interleukin-1 β converting enzyme. Nature 370 270–275. [DOI] [PubMed] [Google Scholar]
- Woycechowsky, K.J. and Raines, R.T. 2000. Native disulfide bond formation in proteins. Curr. Opin. Chem. Biol. 4 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woycechowsky, K.J., Wittrup, K.D., and Raines, R.T. 1999. A smallmolecule catalyst of protein folding in vitro and in vivo. Chem. Biol. 6 871–879. [DOI] [PubMed] [Google Scholar]
- Yee, A., Chang, X., Pineda-Lucena, A., Wu, B., Semesi, A., Le, B., Ramelot, T., Lee, G.M., Bhattacharyya, S., Gutierrez, P., et al. 2002. An NMR approach to structural proteomics. Proc. Natl. Acad. Sci. 99 1825–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]