

Abstract
In an attempt to identify leads that would enable the design of inhibitors with enhanced affinity for glycogen phosphorylase (GP), that might control hyperglycaemia in type 2 diabetes, three new analogs of β-D-glucopyranose, 2-(β-D-glucopyranosyl)-5-methyl-1, 3, 4-oxadiazole, -benzothiazole, and -benzimidazole were assessed for their potency to inhibit GPb activity. The compounds showed competitive inhibition (with respect to substrate Glc-1-P) with Ki values of 145.2 (±11.6), 76 (±4.8), and 8.6 (±0.7) μM, respectively. In order to establish the mechanism of this inhibition, crystallographic studies were carried out and the structures of GPb in complex with the three analogs were determined at high resolution (GPb-methyl-oxadiazole complex, 1.92 Å; GPb-benzothiazole, 2.10 Å; GPb-benzimidazole, 1.93 Å). The complex structures revealed that the inhibitors can be accommodated in the catalytic site of T-state GPb with very little change of the tertiary structure, and provide a rationalization for understanding variations in potency of the inhibitors. In addition, benzimidazole bound at the new allosteric inhibitor or indole binding site, located at the subunit interface, in the region of the central cavity, and also at a novel binding site, located at the protein surface, far removed (~ 32 Å) from the other binding sites, that is mostly dominated by the nonpolar groups of Phe202, Tyr203, Val221, and Phe252.
Keywords: type 2 diabetes, glycogen phosphorylase, β-D-glucopyranosyl analogs, inhibition, X-ray crystallography
Glycogen phosphorylase (GP), because of its central role in glycogen metabolism, has been exploited as a potential target for structure-based design of potent inhibitors, that may be relevant to the control of blood glucose concentrations in type 2 diabetes (Aiston et al. 2001, 2003; Latsis et al. 2002). Several regulatory binding sites, identified in GP, have been used as molecular targets (for review, see Oikonomakos 2002). These are the allosteric site that binds the activator AMP and the inhibitor glucose-6-P, the catalytic site that binds substrates glucose-1-P and glycogen, and the inhibitor glucose, the caffeine binding site, which binds caffeine and related compounds, and the new allosteric inhibitor or indole binding site that binds indole-2 carboxamides and β-D-glucopyranosyl ureas. More specifically, the catalytic site has been probed with glucose analog inhibitors, designed on the basis of information derived from the crystal structure of T-state GPb–α-D-glucose complex (Martin et al. 1991; Watson et al. 1994, 1995; Bichard et al. 1995; Oikonomakos et al. 1995, 2002a, b; Gregoriou et al. 1998; Somsák et al. 2001, 2003; Chrysina et al. 2003, 2004; Györgydeák et al. 2004). A common feature of these compounds is that upon binding at the catalytic site, they promote the (less active) T-state conformation of the enzyme through stabilization of the closed position of 280s loop (residues 282–287), which blocks access of the substrate to the catalytic site.
We report here on the kinetic and crystallographic study of three new lead compounds, derivatives of β-D-glucopy-ranose, 2-(β-D-glucopyranosyl)-5-methyl-1,3,4-oxadiazole, 2-(β-D-glucopyranosyl)-benzothiazole, and 2-(β-D-gluco-pyranosyl)-benzimidazole with GPb. The kinetic experiments reveal that the three compounds are potent competitive inhibitors with respect to the substrate Glc-1-P, whereas the crystallographic results show that each of the compounds binds at the catalytic site and stabilizes the closed position of the 280s loop. Additionally, benzimidazole can occupy the indole binding site and also a novel site that has not been observed previously. Benzimidazole is the first compound to show binding at this novel site.
Results and Discussion
The inhibitory efficiencies of methyl-oxadiazole (2), benzothiazole (3), and benzimidazole (4) were tested with GPb, assayed into the direction of glycogen synthesis (Table 1). The three compounds exhibited competition with respect to Glc-1-P, at constant concentrations of glycogen (1% w/v) and AMP (1 mM). Benzimidazole was found to be a better inhibitor (Ki = 8.6 ± 0.7 μM) than benzothiazole (Ki = 76 ± 4.8 μ M) or methyl-oxadiazole (Ki = 145.2 ± 11.6 μM).
Table 1.
Chemical structures of the C-(β-D-glucopyranosyl) azoles 2-4, showing the numbering system used and inhibition constants

For the chemical names used consult Hadady et al. (2004).
Taken from Martin et al. (1991).
Calculated from double-reciprocal plots with varied Glc-1-P concentrations (3–20 mM), constant concentrations of glycogen (1% w/v) and AMP (1 mM) and 0.2–1.0 mM inhibitor as described in Materials and Methods.
Calculated from double-reciprocal plots with 0.1–0.4 mM of inhibitor.
Calculated from double-reciprocal plots with 20–50 μM of inhibitor.
The overall architecture of the native T-state GPb with the location of the catalytic, the allosteric effector, the caffeine, and the indole inhibitor sites is shown in Figure 1 ▶. The data collection and refinement statistics are summarized in Table 2. Electron density maps (Fig. 2 ▶) clearly defined the position of each inhibitor within the catalytic site, consistent with the kinetic results. Apart from minor shifts in the sugar atoms, the mode of binding and the interactions that the glucopyranose moiety, in all three inhibitors, makes with GPb is almost identical with that previously reported for α-D-glucose, except from minor shifts observed (Martin et al. 1991). In fact, the hydrogen bonding network to the peripheral hydroxyls of the glucopyranose moiety is analogous to that observed for the α-D-glucose complex. Benzimidazole was also found to bind at the indole binding site of the enzyme (Fig. 2D ▶), identified recently (Oikonomakos et al. 2000; Rath et al. 2000a). Additional density (Fig. 2E ▶) at a novel site exposed to the solvent, close to the C terminus of the tower helix, was observed only in the GPb–benzimidazole complex structure. We describe the GPb/methyl-oxadiazole and GPb/benzothiazole interactions at the catalytic site and the GPb/ benzimidazole interactions at the three sites.
Figure 1.
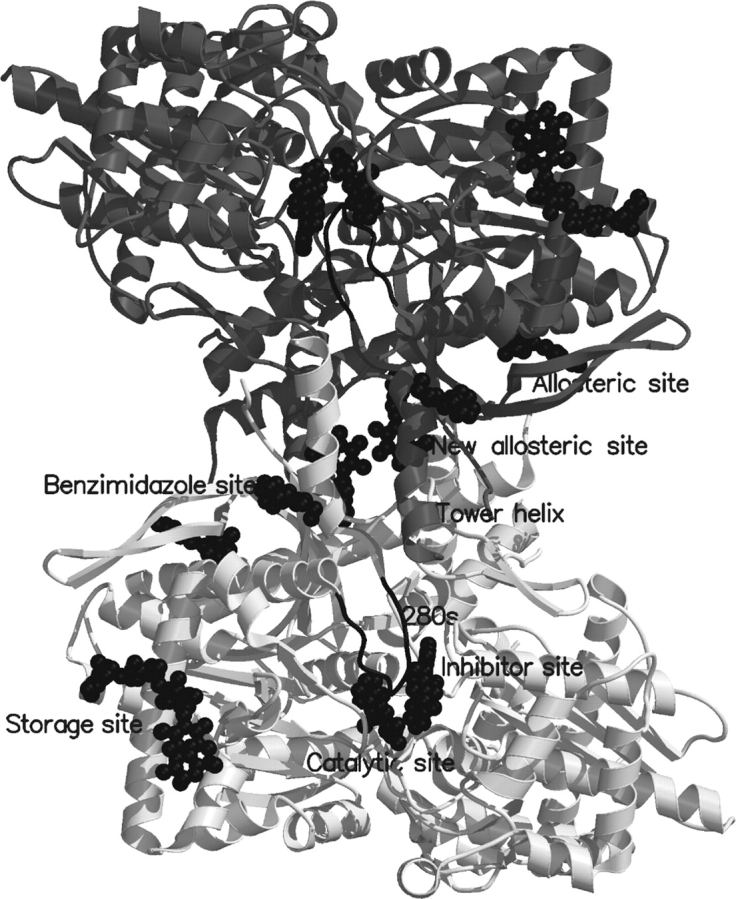
A schematic diagram of the muscle GPb dimeric molecule viewed down the molecular dyad. The positions are shown for the catalytic, allosteric, glycogen storage, the caffeine, the indole site, and a novel binding site for benzimidazole. The catalytic site, marked by benzimidazole, is buried at the center of the subunit and is accessible to the bulk solvent through a 15 Å-long channel. Binding of the competitive inhibitor benz-imidazole promotes the less active T state through stabilization of the closed position of the 280s loop (shown in black). The allosteric site, which binds the activator AMP (indicated in the figure), is situated at the subunit–subunit interface some 30 Å from the catalytic site. The inhibitor site or caffeine binding site, which binds purine compounds such as caffeine and flavopiridol (indicated) is located on the surface of the enzyme some 12 Å from the catalytic site and, in the T state, obstructs the entrance to the catalytic site tunnel. The glycogen storage site (with bound maltopentaose) is on the surface of the molecule some 30 Å from the catalytic site, 40 Å from the allosteric site and 50 Å from the new allosteric inhibitor site. The new allosteric or indole binding site, located inside the central cavity, formed on association of the two subunits, binds indole-2 carboxamide analogs, N-benzoyl-N′-β-D-glucopyranosyl urea, and benzimidazole (indicated). The novel binding site with bound benzimidazole, also located on the surface of the molecule, is some 31 Å from the catalytic site, 32 Å from the allosteric site, and 32 Å from the indole site.
Table 2.
X-ray crystallographic analysis of 2-(β-D-glucopyranosyl) azoles in complex with T-state GPb
| Methyl-oxadiazole | Benzothiazole | Benzimidazole | |
| Data collection and processing statistics | |||
| Experiment | T-state GPb soaked with 100 mM methyl-oxadiazole for 1 h and 40 min | T-state GPb soaked with 56 mM in 18% DMSO benzothiazole for 2 h | T-state GPb soaked with 100 mM benzimidazole for 2 h |
| No. of images (°) | 80 (64°) | 80 (64°) | 80 (64°) |
| Spacegroup | P43212 | P43212 | P43212 |
| Unit cell dimensions | a = b = 128.1, c = 115.4 | a = b = 128.1, c = 115.5 | a = b = 128.1, c = 115.4 |
| α = β = γ = 90° | α = β = γ = 90° | α = β = γ = 90° | |
| Resolution (Å) | 30.0–1.92 | 30.0–2.1 | 30.0–1.93 |
| No. of observations | 543,821 | 568,278 | 556,582 |
| No. of unique reflections | 72,818 (3552) | 56,472 (2754) | 72,470 (3551) |
| a | 0.053 (0526) | 0.067 (0.468) | 0.048 (0.512) |
| Rm (outermost shell) | |||
| Completeness (outermost shell) (%) | 98.8 (98.0) | 99.8 (99.4) | 100 (99.9) |
| Outermost shell (Å) | 1.95–1.92 | 2.14–2.10 | 1.96–1.93 |
| <I/σ(I)> (outermost shell)b | 21.1 (3.7) | 15.8 (3.8) | 20.9 (3.8) |
| Multiplicity (outermost shell) | 5.2 (5.0) | 4.1 (3.9) | 5.1 (5.1) |
| B-values (Å2) (Wilson plot) | 24.5 | 28.4 | 26.4 |
| Refinement statistics and model quality | |||
| Resolution range (Å) | 500–1.92 | 500–2.10 | 500–1.93 |
| No. of reflections used (free) | 72,730 (3717) | 56,369 (2854) | 72,379 (3682) |
| Residues included | (12–254) (261–314) (324–836) | (12–254) (261–314) (324–836) | (12–254) (261–314) (324–836) |
| No. of protein atoms | 6590 | 6590 | 6590 |
| No. of water molecules | 368 | 311 | 345 |
| No. of heteroatoms | 17 (methyl-oxadiazole) | 20 (Benzothiazole) | 20 (Benzimidazole) |
| 15 (PLP) | 15 (PLP) | 15 (PLP) | |
| Final R (c Rfree) | 0.183 (0.200) | 0.182 (0.204) | 0.186 (0.199) |
| R (Rfree) (outermost shell) | 0.252 (0.267) | 0.243 (0.259) | 0.260 (0.281) |
| r.m.s.d. in bond lengths (Å) | 0.005 | 0.006 | 0.005 |
| r.m.s.d. in bond angles (°) | 1.24 | 1.24 | 1.25 |
| r.m.s.d. in dihedral angles (°) | 21.9 | 21.9 | 21.9 |
| r.m.s.d. in improper angles (°) | 0.77 | 0.78 | 0.77 |
| Average B (Å2) for residues | (12–254) (261–314) (324–836) | (12–254) (261–314) (324–836) | (12–254) (261–314) (324–836) |
| Overall | 29.3 | 33.5 | 32.3 |
| Ca, C, N, O | 27.1 | 31.6 | 30.1 |
| Side chain | 31.4 | 35.4 | 34.4 |
| Average B (Å2) for heteroatoms | 18.1 (catalytic site) | 22.6 (catalytic site) | 20.2 (catalytic site) |
| 17.6 (PLP) | 20.9 (PLP) | 51.6 (indole site) | |
| 62.6 (novel site) | |||
| 19.7 (PLP) | |||
| Average B (Å2) for water molecules | 39 | 40.6 | 40.9 |
aRm = ∑i ∑h| <Ih> - Iih|/ ∑i ∑h Ih, where <Ih> and Iih are the mean and the ith measurement of intensity for reflection h, respectively.
b σ (I) is the standard deviation of I.
c Crystallographic R = ∑| |Fo| - |Fc| | / ∑ |Fo|, where |Fo| and |Fc| are the observed and calculated structure factor amplitudes respectively. Rfree is the corresponding R value for a randomly chosen 5% of the reflections that were not included in the refinement.
Figure 2.


Diagrams of the Fo - Fc electron density maps, contoured at 3σ, for the bound methyl-oxadiazole (A) and benzothiazole (B) at the catalytic site, and benzimidazole at the catalytic (C), indole (D), and the novel binding site (E). Electron density maps were calculated using the standard protocol as implemented in CNS v1.1 (Brünger et al. 1998) before incorporating ligand coordinates.
Methyl-oxadiazole (2)
There are no direct hydrogen bonding interactions between the atoms of the substituent introduced at C1 and the protein (Fig. 3A ▶). The two nitrogen atoms N1 and N2 form water-mediated interactions with Asn284 N (through Wat439, a water newly recruited and not present in the native GPb structure, and α-D-glucose, benzothiazole, or benzimidazole complexes), and Leu136 N and Asp283 OD1 (through Wat311), respectively. On binding to the enzyme, the ligand exploits an extended pattern of hydrogen bonds between Wat439 and residues Glu88 OE1, Asn133 ND2, and Asn282 through Wat437, and Phe285 N and O atoms through another water molecule (Wat405, which, compared to its position in the α-D-glucose complex, is shifted by ~ 1.2 Å). In addition, Wat311 (Wat233 in the α-D-glucose complex) is in contact with Wat72, which in turn, interacts with Gly135 N, Asp283 OD1, and with Glu88 OE2 and Gly137 N (through water Wat103) (Table 3). Two water molecules (Wat12 and Wat246 in the α-D-glucose complex) were displaced by the methyl group, in addition to those displaced by the glucopyranose moiety (Wat284, Wat297).
Figure 3.

(A) Interactions between methyl-oxadiazole and GPb at the catalytic site. (B) Comparison between GPb-methyl-oxadiazole complex (light gray) and GPb-α-D-glucose complex (dark gray) in the vicinity of the catalytic site. The side chain of Leu136 and water molecules are not shown for clarity reasons.
Table 3.
Hydrogen bond interactions of 2-β-D-glucopyranosyl) azoles at the catalytic site of GPb
| Methyl-oxadiazole | Benzothiazole | Benzimidazole | |||||
| Inhibitor atom | Protein atom | Distance (Å) | Angle (°) | Distance (Å) | Angle (°) | Distance (Å) | Angle (°) |
| O2 | Tyr573 OH | 3.0 | 147.9 | 3.0 | 148.0 | 3.0 | 147.1 |
| Glu672 OE1 | 3.2 | 176.2 | 3.2 | 172.0 | 3.2 | 176.2 | |
| Asn284 ND2 | 2.7 | 150.9 | 2.7 | 154.5 | 2.7 | 150.8 | |
| Wat316 | 2.9 | — | 2.9 | — | 2.9 | — | |
| Wat124 | 2.8 | — | 2.9 | — | 2.9 | — | |
| O3 | Ser674 N | 3.1 | 173.5 | 3.3 | 173.1 | 3.1 | 174.2 |
| Glu672 OE1 | 2.7 | 121.2 | 2.5 | 129.5 | 2.6 | 120.9 | |
| Gly675 N | 3.2 | 125.2 | 3.2 | 127.7 | 3.2 | 124.7 | |
| O4 | Gly675 N | 2.7 | 143.7 | 2.7 | 143.1 | 2.7 | 145.9 |
| Wat159 | 2.9 | — | 2.9 | — | 2.9 | — | |
| O6 | His377 ND1 | 2.7 | 159.4 | 2.8 | 167.5 | 2.7 | 164.2 |
| Asn484 OD1 | 2.7 | 136.9 | 2.7 | 138.9 | 2.8 | 137.9 | |
| O7 | His377 O | 3.2 | 104.2 | — | — | — | — |
| N1 | Wat311 | 2.7 | — | 2.8 | — | 2.7 | — |
| N2 | His377 O | — | — | — | — | 2.9 | 123.5 |
| Wat439 | 2.6 | — | — | — | — | — | |
| Total | 15 | 13 | 14 | ||||
Methyl-oxadiazole: Wat316 is hydrogen bonded to Tyr573 OH, Lys574 NZ, Asp283 OD2, and Wat72. Wat72, in turn, is hydrogen bonded to Gly135 N, Asp283 OD1, Wat78, and Wat311. Wat78, in turn, is hydrogen bonded to PLP999 O2P and Arg569 N. Wat124 is hydrogen bonded to Thr671 O, Ala673 N, and Wat130. Wat130, in turn, is hydrogen bonded to Val379 N, Thr671 O, and Wat126. Wat126, in turn, is hydrogen bonded to Gly670 O and Wat134. Wat311 is hydrogen bonded to Leu136 N, Asp283 OD1, Wat72, and Wat103. Wat103, in turn, is hydrogen bonded to Gly134 N, Gly137 N, and Glu88 OE2. Wat159 is hydrogen bonded to Thr676 OG1 and PLP999 O3P. Wat439 is hydrogen bonded to Asn284 N, Wat437, and Wat405. Wat437, in turn, is hydrogen bonded to Glu88 OE1, Asn133 ND2, and Asn282 O. Wat405, in turn, is hydrogen bonded to Phe285 N, O, and Wat201. Wat201, in turn, is hydrogen bonded to Ala383 O, Wat131, and Wat338.
Benzothiazole: Wat159 is hydrogen bonded to Thr676 OG1 and PLP999 O3P. Wat316 is hydrogen bonded to Tyr573 OH, Lys574 NZ, Asp 283 OD2, and Wat72. Wat72, in turn, is hydrogen bonded to Gly135 N, Asp283 OD1, and Wat78. Wat78, in turn, is hydrogen bonded to PLP999 O2P and Arg569 N. Wat124 is hydrogen bonded to Thr671 O, Ala673 N, and Wat130. Wat130, in turn, is hydrogen bonded to Val379 N, Thr671 O, and Wat126. Wat126, in turn, is hydrogen bonded to Gly670 O and Wat134. Wat311 is hydrogen bonded to Leu136 N, Asp283 OD1, Wat72, and Wat103. Wat103, in turn, is hydrogen bonded to Gly134 N, Gly137 N, Glu88 OE2, and Asn133 N.
Benzimidazole: Wat159 is hydrogen bonded to Thr676 OG1 and PLP999 O3P. Wat316 is hydrogen bonded to Tyr573 OH, Lys574 NZ, Asp283 OD2, and Wat72. Wat72, in turn, is hydrogen bonded to Gly135 N, Asp283 OD1, and Wat78. Wat78, in turn, is hydrogen bonded to PLP999 O2P and Arg569 N. Wat124 is hydrogen bonded to Thr671 O, Ala673 N, and Wat130. Wat130, in turn, is hydrogen bonded to Val379 N, Thr671 O, and Wat126. Wat126, in turn, is hydrogen bonded to Gly670 O and Wat134. Wat311 is hydrogen bonded to Leu136 N, Asp283 OD1. Wat72, and Wat103. Wat103, in turn, is hydrogen bonded to Gly134 N, Gly137 N, Glu88 OE2, and Asn133 N.
The structural results show that methyl-oxadiazole can be accommodated at the catalytic site with essentially no disturbance of the structure, except for small shifts of the side-chain atoms of Leu136 and Asp339 (Fig. 3B ▶). Thus, on ligand binding, the side chain of Leu136 rotated by ~ 90° (dihedral angle χ2 [CA-CB-CG-CD1]) to optimize contacts with the methyl-oxadiazole ring. The backbone atoms of residues Gly134, Gly135, and Leu136 of the glycine rich helix shifted by ~ 0.3–0.4 Å compared to their positions in the GPb–α-D-glucose complex. The plane of the carboxyl group of Asp339 (dihedral χ2 [CA-CB-CG-OD1]) has flipped ~ 104° about the CB-CG bond, which otherwise would place the —CH3 group close to Asp339 OD1. This change resulted in a shift of Wat338 (Wat319 in the α-D-glucose complex) by ~ 0.5 Å in order to make better interactions with Asp339 OD2, which contacts NE2 of His341 of the so-called β-pocket, a side channel from the catalytic site, lined by both polar and nonpolar groups, directed toward residue His341, but with no access to the bulk solvent (Martin et al. 1991). In addition, there were small changes in the side chains of Asn282, Thr378, and the water structure (Wat201, Wat437, Wat433), but negligible changes in the main-chain atoms of residues 283–287 of the 280s loop (not shown).
In general, methyl-oxadiazole, on binding to GPb, makes a total of 14 hydrogen bonds and 80 van der Waals interactions (6 nonpolar/nonpolar, 18 polar/polar, and 56 polar/ nonpolar) (Table 4). Methyl-oxadiazole is a good inhibitor (Ki = 145.2 ± 11.6 μM) and binds almost 12 times more tightly than α-D-glucose or 50 times more tightly than its parent compound β-D-glucose (Martin et al. 1991), possibly because of the additional interactions of the methyl-oxadiazole group with the protein.
Table 4.
Van der Waals interactions of methyl-oxadiazole, benzothiazole, and benzimidazole at the catalytic site of GPb
| Methyl-oxadiazole | Benzothiazole | Benzimidazole | ||||||
| Inhibitor atom | Protein atom | No. | Inhibitor atom | Protein atom | No. | Inhibitor atom | Protein atom | No. |
| C1 | Asn284 ND2 | 2 | C1 | Wat311 | 2 | C1 | Asn284 ND2 | 2 |
| Wat316 | Wat316 | Wat316 | ||||||
| C2 | Asn284 ND2 | 5 | C2 | Asn284 ND2 | 5 | C2 | Asn284 ND2 | 5 |
| His377 O | His377 O | His377 O | ||||||
| Glu672 OE1 | Glu672 OE1 | Glu672 OE1 | ||||||
| Wat124 | Wat124 | Wat316 | ||||||
| Wat316 | Wat316 | Wat124 | ||||||
| C3 | Glu672 OE1 | 3 | C3 | Glu672 OE1 | 3 | C3 | Glu672 OE1 | 3 |
| Gly675 N | Gly675 N | Gly675 N | ||||||
| Wat159 | Wat159 | Wat159 | ||||||
| C4 | Gly675 N | 2 | C4 | Gly675 N | 2 | C4 | Gly675 N | 2 |
| Wat159 | Wat159 | Wat159 | ||||||
| C5 | Leu136 N | 3 | C5 | Gly135 C | 3 | C5 | Gly135 C | 3 |
| Gly135 C | Leu136 N | Leu136 N | ||||||
| Wat159 | Wat159 | Wat159 | ||||||
| C6 | Gly135 C, O | 5 | C6 | Gly135 C, O | 5 | C6 | Gly135 C, O | 5 |
| Leu139 CD2 | Leu139 CD2 | Leu139 CD2 | ||||||
| His377 ND1 | His377 ND1 | His377 ND1 | ||||||
| Asn484 OD1 | Asn484 OD1 | Asn484 OD1 | ||||||
| C7 | Asn284 ND2 | 2 | C7 | Leu136 CB | 3 | C7 | Asn284 OD1 | 3 |
| Wat311 | Asn284 ND2 | Thr378 CB, CG2 | ||||||
| Wat311 | ||||||||
| C8 | Asn284 ND2, CG, OD1 | 5 | C8 | Leu136 CB, CD2 | 4 | C8 | Asn284 ND2, CG, OD1 | 5 |
| His377 CB | Asn284 N; | His377 CB, O | ||||||
| Wat439 | Wat311 | |||||||
| C9 | Asn284 OD1 | 6 | C9 | Leu136 CD2 | 4 | C9 | Leu136 CB, CD2 | 5 |
| Asp339 OD1 | Asp283 CA | |||||||
| Thr378 CB, CG2 | Asn284 N | Asn284 N, ND2 | ||||||
| Wat311 | ||||||||
| Wat338 | Wat311 | |||||||
| Wat439 | ||||||||
| O2 | Asn284 CG, OD1 | 2 | C10 | Asn282 O, N | 4 | C10 | Asn284 CA, N | 4 |
| His341 CE1 | Leu136 CD2 | |||||||
| Wat433 | Wat311 | |||||||
| O3 | Glu672 CG, CD, C | 10 | C11 | Asn284 N | 5 | C11 | Asn284 N, CA | 6 |
| Ala673 N, CA, CB | His341 CE1, NE2 | His341 CE1, NE2 | ||||||
| Wat201 | Wat201 | |||||||
| Ser674 C, CA | Wat338 | Wat405 | ||||||
| Gly675 CA | ||||||||
| Wat124 | ||||||||
| O4 | Asn484 OD1 | 9 | C12 | Wat338 | C12 | His341 NE2 | 3 | |
| Ser674 CA, N, CB, OG, C | Wat201 | |||||||
| Wat338 | ||||||||
| Gly675 O, C, CA | ||||||||
| O5 | Leu136 N, CA, CB | 5 | C13 | Asn284 OD1 | 1 | C13 | Asn284 ND2 | 4 |
| Leu136 CB | ||||||||
| His377 CB, ND1 | His377 O | |||||||
| Wat311 | ||||||||
| O6 | Leu139 CD2 | 7 | O2 | Asn284 CG, OD1 | 2 | O2 | Asn284 CG, OD1 | 2 |
| His377 CG, CE1 | ||||||||
| Val455 CG1, CG2, CB | ||||||||
| Asn484 CG | ||||||||
| O7 | Asn284 ND2, OD1 | 4 | O3 | Glu672 CG, CD, C; Ala673 N, CA, CB; Gly675 CA; Wat124 | 8 | O3 | Glu672 CG, CD, C Ala673 N, CA, CB; Gly675 CA; Ser674 C; Wat124 | 9 |
| His377 CB, C | ||||||||
| N1 | Asn284 ND2, OD1, CG, N | 5 | O4 | Asn484 OD1; Ser674 CA, N, CB, C; Gly675 O, C, CA | 8 | O4 | Asn484 OD1 | 9 |
| Wat311 | Ser674 CA, N, CB, OG, C | |||||||
| Gly675 O, C, CA | ||||||||
| N2 | Leu136 CB | 4 | O5 | His377 CB; Leu136 N, CA, CB | 4 | O5 | His377 CB, ND1 | 5 |
| Asn284 CG, | Leu136 N, CA, CB | |||||||
| ND2 | O6 | His377 CG, CE1; Leu139 CD2; Val455 CG1, CG2; Asn484 CG | 6 | O6 | His377 CG, CE1 | 6 | ||
| Wat439 | Leu139 CD2 | |||||||
| Val455 CG1, CG2 | ||||||||
| Asn484 CG | ||||||||
| N1 | Leu136 CB; Asn284 ND2 | 2 | N1 | Leu136 CB | 3 | |||
| Asn284 ND2 | ||||||||
| Wat311 | ||||||||
| S1 | Asn284 ND2, OD1 His377 CB, C, O; Thr378 CB, CG2 | 7 | N2 | Asn284 ND2, OD1, CG | 5 | |||
| His377 CB, C | ||||||||
| Total | 79 | Total | 79 | Total | 89 | |||
Benzothiazole (3)
As in the case of methyl-oxadiazole, there are no direct hydrogen bonding interactions between the atoms of the benzothiazole ring and the protein. N1 of the thiazole moiety forms water-mediated interactions with Leu136 N and Asp283 OD1 through Wat311, which shifts by ~ 1.3 Å to avoid clashes with the ligand and makes favorable contacts with Leu136 N and Asp283 OD1. On complex formation, three waters were displaced, Wat12, Wat143, and Wat302 (numbering from the α-D-glucose complex). Some rearrangement of the water structure in the vicinity involved Wat103 (Wat123 in the α-D-glucose complex) that shifted by ~ 0.8 Å to contact Gly134 N, Gly137 N, Glu88 OE2, and Asn133 N (Table 3). The hydrogen bonding interactions formed between the ligand and the protein are illustrated in Figure 4A ▶.
Figure 4.

(A) Interactions between benzothiazole and GPb at the catalytic site. (B) Comparison between the GPb–benzothiazole complex (light gray) and the GPb-α-D-glucose complex (dark gray) in the vicinity of the catalytic site.
Binding of benzothiazole causes a shift in the side chain of Asp339 as observed in the case of methyl-oxadiazole (dihedral angle χ2 [CA-CB-CG-OD1] rotates by ~ 79°), to create sufficient space for the ligand. Asn282 seems to adopt a different conformation by rotation of its dihedral angle χ2 (CA-CB-CG-ND2) by ~ 19°, which results in a small shift of Wat259 (Wat312 in α-D-glucose complex) by ~ 0.8 Å to retain the hydrogen bond contact with atom ND2 (not shown). The side chains of Asp283 (dihedral angle χ2 [CA-CB-CG-OD1] rotates by ~ 8°) and Asn284 (dihedral angle χ2 [CA-CB-CG-OD1] rotates by ~ 13°) are slightly perturbed on ligand binding leading to a change in the conformation of His571 (dihedral angle χ1 [N-CA-CB-CG] rotates by ~ 63° and χ2 [CA-CB-CG-ND1] by ~ 13°) and displacement of Wat171 (numbering from the α-D-glucose complex). Almost all atoms of His341 shift ~ 0.3–0.4 Å toward the ligand (Fig. 4B ▶). In fact, the phenyl of benzothiazole and His341 rings are inclined approximately 60°, and there are two nonpolar/nonpolar contacts between them (Table 4). There are also small shifts (~ 0.3–0.4 Å) in the side chain atoms of Thr378 toward the ligand in order to optimize the contacts to the sulphur atom. In addition, small changes were also observed for the backbone atoms of residues Gly134 and Gly135 (by ~0.4–0.5 Å) and the side chain of Leu136 (dihedral angle χ2 [CA-CB-CG-CD2] rotates by ~ 92°). However, the hydrogen bonding network of the glycine rich helix is maintained mainly through shifts of Wat72 (Wat85 in α-D-glucose complex) by ~ 0.7 Å that is hydrogen bonded to Gly135 N.
Benzothiazole was a better inhibitor (Ki = 76 ± 4.8 μM) than methyl-oxadiazole (Ki = 145.2 ± 11.6 μM). Benzothiazole, on binding to GPb, makes a total of 13 hydrogen bonds and 79 van der Waals interactions (10 nonpolar/non-polar, 7 polar/polar, and 62 polar/nonpolar), the most important of which are those to the side chains of Leu136 and His341. In the GPb–benzothiazole complex, S1 of thiazole makes seven van der Waals contacts to Asn284, His377, and Thr378. The structural results show that sulphur can be accommodated in this position with essentially no disturbance of the structure.
Benzimidazole (4)
Substitution of S1 in benzothiazole by nitrogen (N2 in benzimidazole) resulted in a better inhibitor with a Ki = 8.6 ± 0.7 μM. N2 of the imidazole moiety makes a direct hydrogen bond with O of His377, which shifts by ~ 0.8 Å toward the ligand (dihedral angle ψ [CB-CA-C-O] changes by ~ 21°) (Fig. 5A ▶). This interaction has been observed for the amide nitrogen of β-1-N-acetamido-D-glucopyranose (Ki = 32 μM) (Oikonomakos et al. 1995; Watson et al. 1995), spirohydantoin of glucopyranose (Ki = 3.1 μM) (Bichard et al. 1995; Gregoriou et al. 1998; Oikonomakos et al. 2002a) and D-glucopyranosyl formamide analogs (Ki:310–1800 μM) (Chrysina et al. 2003). N1 makes water-mediated interactions with Leu136 N and Asp283 OD1 through Wat311 (Wat233 in the α-D-glucose structure), which shifts by ~ 1.0 Å; Wat311, in turn, interacts with Gly135 N and Asp283 OD1 through Wat72, which shifts by ~0.7 Å, and Gly134 N, Gly137 N, Glu88 OE2, and Asn133 N through Wat103 (Wat123 in the α-D-glucose structure), which shifts by ~ 0.7 Å (Table 3).
Figure 5.

(A) Interactions between benzimidazole and GPb at the catalytic site. (B) Comparison between the GPb–benzimidazole complex (light gray) and the GPb–α-D-glucose complex (dark gray) in the vicinity of the catalytic site.
On ligand binding, two water molecules, Wat12 and Wat302 (numbering from α-D-glucose structure) are displaced, whereas Wat405 (Wat143 in the α-D-glucose structure) shifts by ~ 1.5 Å to avoid being in close contact with atom C11. In the α-D-glucose complex, Wat143 is hydrogen bonded to Asp339 OD1 (through Wat12), to Glu88 OE1 and Tyr280 O (through Wat302), and to His341 NE2, Ala383 O, and Glu385 O (through Wat299, Wat319, and Wat154, respectively). This hydrogen bonding pattern is partially retained in the benzimidazole complex (compared to the α-D-glucose structure) because of the displacement of Wat12 and Wat302 (numbering from the α-D-glucose complex). Thus, Wat504 is hydrogen bonded to Glu385 (directly); toAla383 O, N (through Wat201); to Arg292 NE, NH2 (through Wat131); and to His341 NE2 and Asp339 OD2 (through Wat201 and Wat338, respectively). In addition, ligand binding induces a conformational change in the side chain of Asp339 (dihedral angle χ2 [CA-CB-CG-OD1] changes by ~ 75°) that results in displacement of Wat246 (numbering from the α-D-glucose structure) to avoid clashes. Concomitant shifts, accompanying the conformational change of the Asp339 side chain, are those of Wat338 (Wat319 in the α-D-glucose structure) by ~ 0.7 Å to facilitate its hydrogen bonding interaction with OD2 of Asp339 at its new conformation and Wat129 (Wat151 in the α-D-glucose structure) by ~0.4 Å (not shown). The side chain of Leu136 adopts a different conformation (dihedral χ2 [CA-CB-CG-CD1] changes by ~ 88°) so that atoms CD1 and CD2 shift by ~ 1.8–2.1 Å. There are also shifts in Thr378 atoms (~ 0.3–0.8 Å) toward the ligand to optimize contacts with the C7 atom, and also small shifts in the His341 atoms (by ~ 0.3–0.4 Å), to optimize contacts with the ligand phenyl ring (Fig. 5B ▶). In addition, the side chain atoms of Asn282 rotate by ~ 12° (dihedral angle χ2 [CA-CB-CG-ND2]) and as a result Wat259 (Wat312 in the α-D-glucose structure) shifts by ~ 0.7 Å to allow its hydrogen bonding interaction with ND2 of Asn282. The conformation of His571 also changes (dihedral angle χ1 [N-CA-CB-CG] rotates by ~ 65° and χ2 [CA-CB-CG-ND1] by ~ 13°), and this change leads to displacement of Wat171 (numbering from the α-D-glucose structure).
Superposition of the three compounds bound at the catalytic site shows that the ligands superimpose well (not shown), except that the replacement of N2 by S changed the geometry of the imidazole ring and imposed a different orientation of the phenyl ring (shifted by ~ 0.8 Å compared to that of benzimidazole). The positions of the glucosyl components of each of the structures are similar, indicating that the glucosyl recognition site does not change substantially.
Benzimidazole, on binding to GPb, also occupies the indole binding site, shown to bind a number of indole-2-carboxamide analogs (Rath et al. 2000a) and N-benzoyl-N′-β-D-glucopyranosyl-urea (Oikonomakos et al. 2002b). The site is almost buried in the central cavity of the enzyme. Access to the central cavity (Fig. 1 ▶) is partially blocked at one end of the twofold axis, which relates the two subunits, by residues Arg33, His34, Arg60, and Asp61 from the cap region (residues 36–47) and α2 helix (residues 47–78) and their symmetry-related equivalents and at the other end by the tower helix residues Asn270, Glu273, and Ser276 of the tower helix (residues 262–276) and their symmetry-related equivalents.
The conformation of the bound benzimidazole at the indole binding site is similar to that described above for the catalytic site, except that the torsion angles O5-C1-C13-N1 and O5-C5-C6-O6 are -116° and -75°, respectively (different from those in the catalytic site -116° and 67°). Benzimidazole on binding at the indole binding site of GPb makes a total of nine hydrogen bonds (Table 5) and exploits 78 van der Waals interactions, 25 of which are interactions between nonpolar groups, and there are in total 20 contacts to the symmetry related subunit (Table 6; Fig. 6A ▶). The benzimidazole group exploits 55 van der Waals contacts, which are dominated by the contacts (26) to Arg60. Arg60 NH1 and NH2 are hydrogen bonded to the hydroxyl O2 group, which in turn, is hydrogen bonded to hydroxyl O3′ from the symmetry-related molecule of benzimidazole. There is also a hydrogen bond from N1 to Glu190 O, and Asn187 O (through Wat418) and from N2 to Thr38′ O (where the prime refers to residues from the symmetry-related subunit). The glucopyranose moiety makes 24 van der Waals contacts with the protein, of which 16 are to residues from the symmetry-related subunit. The glucopyranose and His57′ rings are inclined approximately 80°, and there are extensive contacts between them. There is a hydrogen bond from Lys191 NZ to the hydroxyl O6. In addition, hydroxyl groups O2 and O3 are hydrogen bonded to O3′ and O2′, respectively, of the glucopyranose from the symmetry-related bound benzimidazole molecule. However, there are no aromatic–aromatic, amino-aromatic, and CH-π interactions that are characteristic of the tight binding of the 4-fluorobenzyl moiety of CP320626 (IC50 = 334 ± 10 nM) (Oikonomakos et al. 2000).
Table 5.
Hydrogen bond interactions of benzimidazole at the indole and the novel binding site of GPb
| Indole binding site | Novel binding site | |||||
| Inhibitor atom | Protein atom | Distance (Å) | Angle (°) | Protein atom | Distance (Å) | Angle (°) |
| O2 | Arg60 NH1 | 3.3 | 142.7 | Gln219 NE2 | 3.1 | 154.2 |
| Arg50 NH2 | 3.2 | 147.0 | ||||
| Imi920′ O3 | 2.5 | — | ||||
| O3 | Imi920′ O2 | 2.5 | — | |||
| Imi920′ O3 | 2.7 | — | ||||
| O5 | — | Lys294 NZ | 3.1 | — | ||
| O6 | Lys191 NZ | 2.8 | — | Glu290 OE2 | 3.2 | 124.9 |
| — | Lys294 NZ | 2.8 | — | |||
| N1 | Glu190 O | 3.1 | 131.3 | Gln219 OE1 | 3.0 | 129.5 |
| Wat418 | 3.0 | — | ||||
| N2 | Thr38′ O | 2.9 | 142.3 | Lys294 NZ | 2.8 | — |
Indole binding site: Wat418 is hydrogen bonded to Glu190 O and Asn187 O.
Table 6.
Van der Waals interactions of benzimidazole at the indole and the novel binding site of GPb
| Indole binding site | New binding site | |||
| Inhibitor atom | Protein atom | No. of contacts | Protein atom | No. of contacts |
| C1 | Wat212; Thr38′ O | 2 | Lys294 NZ | 1 |
| C2 | Arg60 NH2; Imi920′ C3 | 2 | Tyr203 OH, CE2, CZ | 3 |
| C3 | Imi920′ O2, O3 | 2 | ||
| C4 | Thr38′ CG2; His57′ CE1, NE2 | 3 | ||
| C6 | Lys191 NZ; Leu39′ CD1 | 2 | Lys294 NZ | 1 |
| C7 | Arg60 CD, NE, CZ, NH2; Wat303; Phe37′ O; Val40′ CG2 | 7 | Phe202 CD1; Lys294 NZ | 2 |
| C8 | Arg60 NE, CZ, NH1, NH2; Lys191 CD; Thr38′ O | 6 | Lys294 NZ; Tyr203 CE2 | 2 |
| C9 | Arg60 CZ, NH1; Pro188 O; Glu190 O; Lys191 CB, CD; Wat418 | 7 | Gln219 OE1 | 1 |
| C10 | Arg60 CD, NH1; Pro188 O; Trp189 C, O; Glu190 C, O; Lys191 N, CA, CB | 10 | Gln219 CB, OE1; Phe252 CE1 | 3 |
| C11 | Arg60 CG, CD, NE; Pro188 O; Trp67 CZ3 | 5 | Val221 CG2; Ala272 CB; Gln219 CB | 3 |
| C12 | Val64 CG1; Arg60 CG, CD, NE; Val40′ CG2 | 5 | Val221 CG2; Phe202 CD1 | 2 |
| C13 | Arg60 NH1, NH2; Lys191 CD; Wat418; Thr38′ O | 5 | Gln219 OE1; Lys294 NZ | 2 |
| O2 | Arg60 CZ; Imi920′ C3 | 2 | Gln219 OE1, CD | 2 |
| O3 | Imi920′ C2, C3; His57′ NE2 | 3 | ||
| O4 | His57′ CE1, NE1 | 2 | ||
| O5 | Lys191 CE, CD, NZ; Wat212; Thr38′ O | 5 | ||
| O6 | Lys191 CE | 1 | Lys294 CE | 1 |
| N1 | Arg60 NH1; Lys191 CA, CD; | 4 | Gln219 CD | 1 |
| Wat212 | ||||
| N2 | Arg60 CZ, NH1, NH2; Lys191 CD; Thr38′ C | 5 | Lys294 CE; Tyr203 OH, CE2 | 3 |
| Total | 78 | 27 | ||
Figure 6.
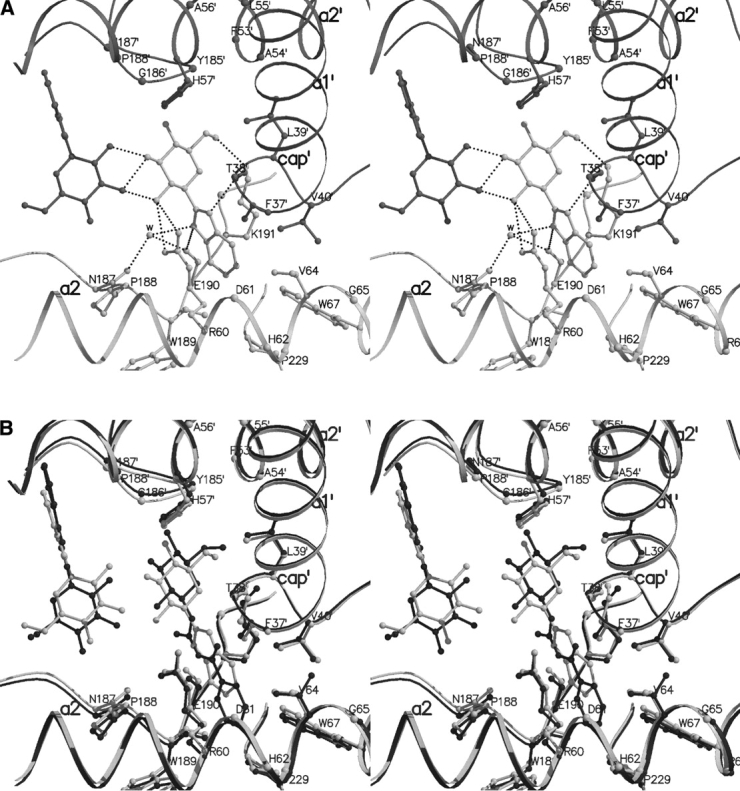
(A) Interactions between benzimidazole and GPb at the indole binding site. Residues from subunit 1 are shown in light gray and their symmetry-related equivalents (from subunit 2) are shown in dark gray. (B) Comparison between the GPb–benzimidazole complex (light gray) and the GPb–N-benzoyl-N′-β-D-glucopyranosyl-urea complex (dark gray) in the vicinity of the indole binding site.
The binding of benzimidazole at the indole binding site of GPb does not promote extensive conformational changes except for small shifts of residues Arg60 (dihedral angles χ2 and χ3 rotate by ~ 88° and ~ 64°, respectively), and Lys191 (dihedral angles χ3 and χ4 rotate by ~ 39° and ~ 161°), that undergo conformational changes to accommodate the ligand.
A structural comparison between the GPb–N-benzoyl-N′-β-D-glucopyranosyl urea and GPb–benzimidazole complex shows that the two complexes have very similar overall structures and do not differ significantly at the interfaces between monomers. The two structures closely resemble each other; the positions of the Cα, main-chain and side-chain atoms for residues 18–249, 262–312, and 326–829 deviate from their mean positions by 0.297 Å, indicating no significant changes between the complex structures. Figure 6B ▶ shows details of the contacts made at the indole binding site for GPb–N-benzoyl-N′-β-D-glucopyranosyl urea complex and the GPb–benzimidazole complex. The two ligands superimpose quite well except that the glucopyranose and the phenyl moieties of N-benzoyl-N′-β-D-glucopyranosyl urea are shifted ~ 1.0 Å and ~ 1.5 Å toward the symmetry-related residues Phe53′, His57′, and Pro188′, and the hydrophobic pocket lined by Val64, Thr67, and Pro229, respectively (compared to the position of benzimidazole).
There were also indications in both 2Fo–Fc and Fo–Fc electron density difference maps for binding of a benzimidazole molecule at a novel binding site located at the surface of the molecule ~ 31 Å from the catalytic site, ~ 32 Å from the indole binding site, and ~ 32 Å from the allosteric site (Fig. 1 ▶). The electron density was sufficiently well resolved, for the substituent group of benzimidazole and most of the atoms of glucopyranose, to indicate the conformation of the ligand (Fig. 2E ▶). The conformation of the bound benzimidazole at the new site is similar to that described above for the catalytic and the indole binding site, except that the torsion angles O5-C1-C13-N1 and O5-C5-C6-O6 are -154° and −32°, respectively.
The interactions of benzimidazole with GPb involve six hydrogen bonds and 27 van der Waals contacts, 11 of which are interactions between nonpolar groups (Fig. 7A ▶; Tables 5,6). The benzimidazole substituent pocket is formed by residue 202 (of the β8 strand-residues 198–209), residues 219–221 (of the β9 strand-residues 212–223, antiparallel to β8), residues 249 and 252 (of the loop of residues 248–260 between β11, residues 237–247, and tower helix-residues 261–274), residues 271 and 272 (of the tower helix), and residues 290 and 294 (of the α8 helix-residues 289–314). In the native 100 K GPb structure (Gregoriou et al. 1998), the site contains five located water molecules (Wat497, Wat534, Wat747, Wat823, and Wat828), and these are apparently displaced upon binding of benzimidazole. There are hydrogen bonds from N1 and N2 of the imidazole ring to the side-chain atoms Gln219 OE1 and Lys294 NZ, respectively. The glucopyranose moiety makes relatively few van der Waals interactions with the protein, and there are four potential hydrogen bonds from the sugar ring O5 and hydroxyl O2 and O6 groups to Gln219 NE2, Glu290 OE2, and Lys294 NZ.
Figure 7.
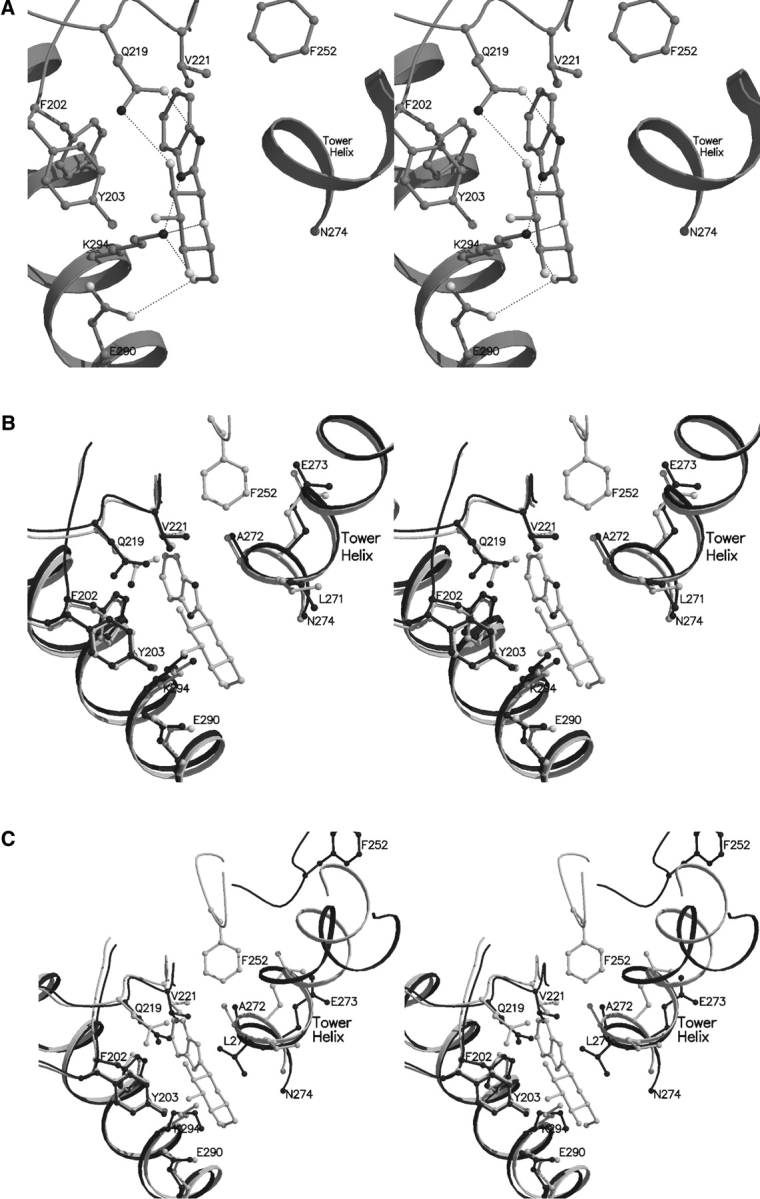
(A) Interactions between benzimidazole and GPb at the new ligand binding site. (B) Comparison between the GPb–benzimidazole complex (light gray) and native GPb (dark gray) in the vicinity of the new site. (C) Comparison between the GPb–benzimidazole complex (light gray) and human liver GPa (dark gray) in the vicinity of the new binding site.
Superposition of the GPb–benzimidazole complex structure on the native GPb structure over well-defined residues 18–249, 262–312, and 326–829 (r.m.s. deviation for Cα atoms: 0.313 Å) shows that the side chain of Gln219 shifts (dihedral angle χ2 rotates by ~ 18° and atoms CD, NE2, and OE1 shift by ~ 0.5–0.9 Å), to create sufficient space and facilitate binding through a hydrogen bond between hydroxyl O2 group and Gln219 OE1 (Fig. 7B ▶). Small changes were observed in residues Val221 (backbone atoms shifted by ~ 0.4 Å), Leu271 (dihedral angle χ1 rotates by 27° and χ2 by 127° to avoid clashes with the inhibitor), and residues Ala272 (all atoms shifted by ~ 0.5–0.7 Å), Glu273 (all atoms shifted by ~ 0.3–0.7 Å), and Glu290 (dihedral χ3 changed by ~ 65°) to form more favorable interactions with the ligand. It is also interesting, that on ligand binding, Phe252 becomes ordered, unlike the previous complex structures of GPb where its position was ambiguous, and as a result it was excluded from the model structure.
Comparison of the GPb–benzimidazole complex with the R state human liver GPa (Rath et al. 2000b) suggests that the ligand at the new binding site is likely to have lower affinity for the R state GPa (Fig. 7C ▶). Superposition of the structure of the liver GPa (subunit A) with the structure of the GPb–benzimidazole complex gave an r.m.s. deviation of 1.146 Å for Cα atoms, for ordered residues 24–205, 214–249, 270–281, 289–313, 327–553, 564–711, and 787–835. In the R state relative to the T state, one subunit is rotated so as to bring the two subunits closer at the α2′ (residues 48′–78′)-cap (42–45) interface and causing them to move further apart at the tower/tower′ interface of the dimer. In the inactive human liver GPa, the catalytic site is blocked by the 280s loop (residues 280–286) that follows the tower helix (residues 267–274). In the active human liver GPa, the tower helices are shorter by two turns and the 280s loop folds up in an ordered conformation and allows access of the substrate glycogen to the catalytic site and also creates the recognition site for inorganic phosphate substrate (Johnson and Lewis 2001). Thus, changes at the subunit interfaces are closely coupled to changes at the catalytic site. Modeling studies suggest that if GPb–benzimidazole were to be incorporated into the corresponding site of the R-state liver GPa structure, Gln219, Val221, and Leu271 of the tower helix would be required to move to open up the site in order to optimize contacts with benzimidazole. Hence, it would be anticipated that the affinity of benzimidazole for the R state would be less than that of the T state, but there is no experimental evidence for this. On the basis of our observations, we suggest that the binding of benzimidazole at the novel binding site stabilizes the T-state conformation of the enzyme. The physiological significance of this site has yet to be established.
In a crystallographic experiment with 5 mM benzimidazole (2 h soak) (data not shown), electron density maps indicated strong binding of benzimidazole at the catalytic site, but practically no binding at either the indole inhibitor site or the novel binding site. It seems, therefore, that the catalytic site is the primary binding site for benzimidazole binding.
In conclusion, benzimidazole was found to be the most potent inhibitor among the three compounds, with a Ki value of 8.6 μM. Replacement of N2 of benzimidazole by either oxygen (O7) or sulphur (S1) in methyl-oxadiazole or benzothiazole, respectively, disrupted the hydrogen bond formed between N2 and His377 O in the GPb–benzimidazole complex and resulted in less potent inhibitors. The van der Waals interactions with the residues of the catalytic site are reduced from 89 in benzimidazole to 79 in methyl-oxadiazole or benzothiazole. Benzothiazole (Ki = 76 μM) was a better inhibitor than methyl-oxadiazole (Ki = 145.2 μM). A possible explanation for the difference in Ki may lie in the number of the nonpolar/nonpolar contacts of the two ligands with the protein. In the GPb–benzothiazole structure there are 10 nonpolar/nonpolar interactions, while in the GPb–methyl-oxadiazole there are only six nonpolar/nonpolar interactions. In addition, the number of water molecules displaced in the complex with methyl-oxadiazole is reduced to two, compared with three observed in the complex with benzothiazole, possibly leading to an increase in entropy. The ability of the ligands to displace waters, to exploit existing or recruit new waters appears to be important for the variation observed in the potency of the inhibitors studied. The structural results also show that the three compounds interact with the side chains of Asn282, Asp283, and Asp284 holding the 280s loop in its closed T-state conformation, therefore inhibiting access of the substrate to the catalytic site. These observations will be of value in the design of further potent and specific inhibitors of the enzyme. Nevertheless, further investigation is required to explore whether the new (benzimidazole) binding site, located in a surface cavity formed by residues Phe202, Tyr203, Val221, and Phe252, could be exploited as a target for the design of new inhibitors.
Materials and methods
The synthesis of methyl-oxadiazole (2), benzothiazole (3), and benzimidazole (4) was described in Hadady et al. (2004). Rabbit muscle GPb was isolated, purified, recrystallized, and assayed as previously described (Oikonomakos et al. 1995). Kinetic studies were performed in the direction of glycogen synthesis in the presence of constant concentrations of glycogen (1% w/v), AMP (1 mM), and various concentrations of Glc-1-P (3–20 mM) and the inhibitors. The kinetic results are summarized in Table 1.
Native T-state GPb crystals, grown in the tetragonal lattice, spacegroup P43212 (Oikonomakos et al. 1985), were soaked with 100 mM methyl-oxadiazole (for 1 h and 40 min) or 56 mM benzothiazole in 18% DMSO (for 2 h) or 100 mM benzimidazole (for 2 h), prior to data collection. Diffraction data were collected from single crystals in room temperature using synchrotron radiation source at Daresbury Laboratory (beamline PX-9.6, λ = 0.87 Å) and EMBL-Hamburg outstation (beamline X13, λ = 0.8015 Å). Data for the 5 mM benzimidazole soak (2 h) (not shown) were collected at a resolution of 2.26 Å on an Image Plate RAXIS IV mounted on a Rigaku Ru-H3RHB generator with a belt drive rotating anode (λ = 1.5418 Å). Data reduction and integration followed by scaling and merging of the intensities obtained were performed with Denzo and Scalepack, respectively, as implemented in HKL suite (Otwinowski and Minor 1997).
The crystal structure of GPb-α-D-glucose was used as a starting model for crystallographic refinement against the experimental data by applying a standard protocol as implemented by CNS (Brünger et al. 1998), involving rigid body refinement followed by positional and individual B-factor refinement (with bulk solvent correction). 2Fo–Fc and Fo–Fc electron density maps calculated were visualized using the program for molecular graphics “O” (Jones et al. 1991). Ligand models, constructed and minimized using the program SYBYL (Tripos Associates Inc.), were fitted to the electron density maps after adjustment of the torsion angles of the computed models. Alternate cycles of manual rebuilding with “O” and refinement with CNS improved the quality of the models.
The stereochemistry of the protein residues was validated by PROCHECK (Laskowski et al. 1993; CCP4, 1994). Hydrogen bonds and van der Waals interactions were calculated with the program CONTACT as implemented in CCP4 (1994) applying a distance cutoff of 3.3 Å and 4.0 Å, respectively. The program calculates the angle O. . .H. . .N (where the hydrogen position is unambiguous) and the angle source . . . oxygen-bonded carbon atom. Suitable values are 120° and 90°. A Luzatti plot (Luzatti 1952) suggests an average positional error for all structures of approximately 0.22–0.25 Å. GPb complex structures were superimposed over well-defined residues using LSQKAB (CCP4 1994). Comparisons of the water molecules in the complex structures were made taking into consideration their equivalent positions. The schematic representation of the crystal structures presented in all figures were prepared with the programs MolScript (Kraulis 1991) and BobScript (Esnouf 1997) and rendered with Raster3D (Merritt and Bacon 1997). The coordinates of the new structures have been deposited with the RCSB Protein Data Bank (http://www.rcsb.org/pdb) with codes 1XL0 (methyl-oxadiazole complex), 1XL1 (benzothiazole complex), and 1XKX (benzimidazole complex).
Acknowledgments
This work was supported by the Greek General Secretariat for Research and Technology (GSRT) through the programs PENED-204/2001 (to M.N.K.), ENTER-EP6/2001 (to E.D.C.), the Joint Research and Technology project between Greece and Hungary (2004–2006) (to N.G.O and L.S.), the Hungarian Scientific Research Fund (OTKA T37210 to P.G., T46081 to L.S.), the Hungarian Ministry of Health (ETT 030/2003 to P.G., T.D., and L.S.), the SRS Daresbury Laboratory through IHPP HPRI-CT-1999-00012, and the EMBL-Hamburg outstation under FP6 “Structuring the European Research Area Programme” contract no. RII3/CT/2004/5060008. Zs.H. thanks CMS Chemicals (Oxford, UK) for a stipend supporting her Ph.D. studies with L.S. in Debrecen. We also acknowledge the assistance of the staff at SRS and EMBL-Hamburg outstation for providing excellent facilities for X-ray data collection.
Abbreviations
GPb, muscle glycogen phosphorylase b
PLP, pyridoxal 5′-phosphate
glucose, α-D-glucose
Glc-1-P, α-D-glucose 1-phosphate
methyl-oxadiazole, 2-(β-D-glucopyranosyl)-5-methyl-1,3,4-oxadiazole
benzothiazole, 2-(β-D-glucopyranosyl)-benzothiazole
benzimidazole, 2-(β-D-glucopyranosyl)-benzimidazole
r.m.s. deviation, root-mean-square deviation.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041216105.
References
- Aiston, S., Hampson, L., Gómez-Foix, A.M., Guinovart, J.J., and Agius, L. 2001. Hepatic glycogen synthesis is highly sensitive to phosphorylase activity. J. Biol. Chem. 276 23858–23866. [DOI] [PubMed] [Google Scholar]
- Aiston, S., Coghlan, M.P., and Agius, L. 2003. Inactivation of phosphorylase is a major component of the mechanism by which insulin stimulates hepatic glycogen synthesis. Eur. J. Biochem. 270 2773–2781. [DOI] [PubMed] [Google Scholar]
- Bichard, C.J.F., Mitchell, E.P., Wormald, M.R., Watson, K.A., Johnson, L.N., Zographos, S.E., Koutra, D.D., Oikonomakos, N.G., and Fleet, G.W.J. 1995. Potent inhibition of glycogen phosphorylase by a spirohydantoin of glucopyranose: First pyranose analogues of hydantocidin. Tetrahedron Lett. 36 2145–2148. [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography and NMR system; A new software suite for macro-molecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Collaborative Computing Project Number 4. 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50 760–763. [DOI] [PubMed] [Google Scholar]
- Chrysina, E.D., Oikonomakos, N.G., Zographos, S.E., Kosmopoulou, M.N., Bischler, N., Leonidas, D.D., Kovács, L., Docsa, T., Gergely, P., and Somsák, L. 2003. Crystallographic studies on α- and β-D-glucopyranosyl formamide analogues, inhibitors of glycogen phosphorylase. Biocatal. Biotrans-for. 21 233–242. [Google Scholar]
- Chrysina, E.D., Kosmopoulou, M.N., Kardakaris, R., Bischler, N., Leonidas, D.D., Kannan, T., Loganathan, D., and Oikonomakos, N.G. 2004. Binding of β-D-glucopyranosyl bismethoxyphosphoramidate to glycogen phosphorylase b: Kinetic and crystallographic studies. Bioorg. Med. Chem. 13 765–772. [DOI] [PubMed] [Google Scholar]
- Esnouf, R.M. 1997. An extensively modified version of Molscript that includes greatly enhanced coloring capabilities. J. Mol. Graph. Model. 15 132–134. [DOI] [PubMed] [Google Scholar]
- Gregoriou, M., Noble, M.E.M., Watson, K.A., Garman, E.F., Krulle, T.M., Fuente, C., Fleet, G.W.J., Oikonomakos, N.G., and Johnson, L.N. 1998. The structure of a glycogen phosphorylase glucopyranose spirohydantoin complex at 1.8 Å resolution and 100 K: The role of the water structure and its contribution to binding. Protein Sci. 7 915–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györgydeák, Z., Hadady, Z., Felföldi, N., Krakomperger, A., Nagy, V., Tóth, M., Brunyánszki, A., Docsa, T., Gergely, P., and Somsák, L. 2004. Synthesis of N-(β-D-glucopyranosyl)-and N-(2-acetamido-2-deoxy-β-D-gluco-pyranosyl)amides as inhibitors of glycogen phosphorylase. Bioorg. Med. Chem. 12 4861–4870. [DOI] [PubMed] [Google Scholar]
- Hadady, Zs., Tóth, M., and Somsák, L. 2004. C-(β-D-Glucopyranosyl) heterocycles as potential glycogen phosphorylase inhibitors. Arkivoc vii 140–149. [Google Scholar]
- Johnson, L.N. and Lewis, R.J. 2001. Structural basis for control by phosphorylation. Chem. Rev. 101 2209–2242. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 110–119. [DOI] [PubMed] [Google Scholar]
- Kraulis, P. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24 946–950. [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 283–291. [Google Scholar]
- Latsis, T., Andersen, B., and Agius, L. 2002. Diverse effects of two allosteric inhibitors on the phosphorylation state of glycogen phosphorylase in hepatocytes. Biochem. J. 368 309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzatti, V. 1952. Traitement statistique des erreurs dans la determination des structure crystallines. Acta Crystallogr. 5 802–810. [Google Scholar]
- Martin, J.L., Veluraja, K., Johnson, L.N., Fleet, G.W.J., Ramsden, N.G., Bruce, I., Oikonomakos, N.G., Papageorgiou, A.C., Leonidas, D.D., and Tsitoura, H.S. 1991. Glucose analogue inhibitors of glycogen phosphorylase: The design of potential drugs for diabetes. Biochemistry 30 10101–10116. [DOI] [PubMed] [Google Scholar]
- Merritt, E.A. and Bacon, D.J. 1997. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 277 505–524. [DOI] [PubMed] [Google Scholar]
- Oikonomakos, N.G. 2002. Glycogen phosphorylase as a molecular target for type 2 diabetes therapy. Curr. Protein Pept. Sci. 3 561–586. [DOI] [PubMed] [Google Scholar]
- Oikonomakos, N.G., Melpidou, A.E., and Johnson, L.N. 1985. Crystallisation of pig skeletal phosphorylase b. Biochim. Biophys. Acta 832 248–256. [DOI] [PubMed] [Google Scholar]
- Oikonomakos, N.G., Kontou, M., Zographos, S.E., Watson, K.A., Johnson, L.N., Bichard, C.J.F., Fleet, G.W.J., and Acharya, K.R. 1995. N-acetyl-β-D-glucopyranosylamine: A potent T state inhibitor of glycogen phosphorylase. A comparison with α-D-glucose. Protein Sci. 4 2469–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikonomakos, N.G., Skamnaki, V.T., Tsitsanou, K.E., Gavalas, N.G., and Johnson, L.N. 2000. A new allosteric site in glycogen phosphorylase b as a target for drug interactions. Structure 8 575–584. [DOI] [PubMed] [Google Scholar]
- Oikonomakos, N.G., Skamnaki, V.T., Ösz, E., Szilágyi, L., Somsák, L., Docsa, T., Tóth, B., and Gergely, P. 2002a. Kinetic and crystallographic studies of glucopyranosylidene spirothiohydantoin binding to glycogen phosphorylase b. Bioorg. Med. Chem. 10 261–268. [DOI] [PubMed] [Google Scholar]
- Oikonomakos, N.G., Kosmopoulou, M., Zographos, S.E., Leonidas, D.D., Chrysina, E.D., Somsák, L., Nagy, V., Praly, J.-P., Docsa, T., Tóth, B., et al. 2002b. The binding of N′-acetyl- and Benzoyl-N′-β-D-glucopyranosyl ureas to glycogen phosphorylase b: Kinetic and crystallographic studies. Eur. J. Biochem. 269 1–13. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Rath, V.L., Ammirati, M., Danley, D.E., Ekstrom, J.L., Gibbs, E.M., Hynes, T.R., Mathiowetz, A.M., McPherson, R.K., Olson, T.V., Treadway, J.L., et al. 2000a. Human liver glycogen phosphorylase inhibitors bind at a new allosteric site. Chem. Biol. 7 677–682. [DOI] [PubMed] [Google Scholar]
- Rath, V.L., Ammirati, M., LeMotte, P.K., Fennell, K.F., Mansour, M.N., Danley, D.E., Hynes, T.R., Schulte, G.K., Wasilko, D.J., and Pandit, J. 2000b. Activation of human liver glycogen phosphorylase by alteration of the secondary structure and packing of the catalytic core. Mol Cell. 1 139–148. [PubMed] [Google Scholar]
- Somsák, L., Kovács, L., Tóth, M., Ösz, E., Szilágyi, L., Györgydeák, Z., Dinya, Z., Docsa, T., Tóth, B., and Gergely, P. 2001. Synthesis of and a comparative study on the inhibition of muscle and liver glycogen phosphorylases by epimeric pairs of D-gluco- and D-xylopyranosylidene-spiro-(thio)hydantoins and N-(D-glucopyranosyl) amides. J. Med. Chem. 44 2843–2848. [DOI] [PubMed] [Google Scholar]
- Somsák, L., Nagy, V., Hadady, Z., Docsa, T., and Gergely, P. 2003. Glucose analog inhibitors of glycogen phosphorylases as potential antidiabetic agents: Recent developments. Curr. Pharm. Des. 9 1177–1189. [DOI] [PubMed] [Google Scholar]
- Watson, K.A., Mitchell, E.P., Johnson, L.N., Son, J.C., Bichard, C.J.F., Orchard, M.G., Fleet, G.W.J., Oikonomakos, N.G., Leonidas, D.D., Kontou, M., et al. 1994. Design of inhibitors of glycogen phosphorylase: A study of α- and β-C-glucosides and 1-thio-β-D-glucose compounds. Biochemistry 33 5745–5758. [DOI] [PubMed] [Google Scholar]
- Watson, K.A., Mitchell, E.P., Johnson, L.N., Cruciani, G., Son, J.C., Bichard, C.J.F., Fleet, G.W.J., Oikonomakos, N.G., Kontou, M., and Zographos, S.E. 1995. Glucose analogue inhibitors of glycogen phosphorylase: From crystallographic analysis to drug prediction using GRID force-field and GOLPE variable selection. Acta Crystallogr. D Biol. Crystallogr. 51 458–472. [DOI] [PubMed] [Google Scholar]