

SUMMARY
The aim of this study was to identify sulfotransferase (SULT) isoform(s) responsible for the formation of indoxyl sulfate from indoxyl (3-hydroxyindole). Indoxyl was incubated together with the co-substrate 3'-phosphoadenosine 5'-phosphosulfate (PAPS) and either human or rat liver cytosol or recombinant sulfotransferase enzymes. Formation of indoxyl sulfate from indoxyl was measured by HPLC and used for determination of sulfonation rates. Both cytosols sulfonated indoxyl with apparent Km values of 6.8 ± 0.9 μM for human and 3.2 ± 0.6 μM for rat cytosol. To help identify the isoform(s) of SULT responsible for indoxyl sulfate formation, indoxyl was incubated with human and rat liver cytosols and PAPS in the presence of isoform-specific SULT inhibitors. No inhibition was observed by DHEA, a specific hydroxysteroid sulfotransferase inhibitor, nor by oestrone, an inhibitor of oestrogen sulfotransferase. However, an aryl (phenol) sulfotransferase inhibitor, 2,6-dichloro-4-nitrophenol (DCNP), inhibited the formation of indoxyl sulfate with a IC50 values of 3.2 μM for human and 1.0 μM for rat cytosol indicating that human and rat aryl (phenol) sulfotransferases are responsible for the formation of indoxyl sulfate. When indoxyl was incubated with SULT1A1*2, a human recombinant aryl SULT, an apparent Km value of 5.6 ± 1.8 μM was obtained. Kinetic studies with human and rat cytosols and human recombinant SULT1A1*2 gave similar kinetic values indicating that human and rat aryl sulfotransferases efficiently catalyze the formation of indoxyl sulfate, an important uremic toxin metabolite.
Keywords: Indoxyl, indoxyl sulfate, sulfotransferase, conjugation, metabolism
INTRODUCTION
Sulfo-conjugation is an important reaction in the metabolism of many drugs, xenobiotics and endogenous substances such as neurotransmitters and hormones (1). These metabolic reactions are catalyzed by sulfotransferases that involve the transfer of a sulfuryl group from 3'-phosphoadenosine 5'-phosphosulfate (PAPS) to acceptor molecules, thereby producing more water soluble and often less toxic metabolites. Although it is generally true for the sulfation of molecules to aid excretion from the body, sulfonation of certain allylic alcohols and polycyclic aromatic hydrocarbons bearing benzylic alcohol groups and sulfonation of N-hydroxy arylamines may produce metabolites with increased toxicity that cause mutagenic and carcinogenic responses (2-4). Therefore, sulfate conjugation may act as an important metabolic activation reaction for certain molecules, as well as a detoxication reaction. Indoxyl sulfate, a sulfate conjugate of indoxyl (3-hydroxyindole) is a known circulating uremic toxin promoting the progression of renal failure (5,6), and serum concentration of this metabolite is markedly increased in uremic patients (7). Indoxyl sulfate is mainly produced after bacterial decomposition of dietary tryptophan in intestines (8). The initial product of tryptophan metabolism in intestines is indole, which is hydroxylated to 3-hydroxyindole (indoxyl), and subsequently sulfonated to form indoxyl sulfate (5,9).
Primary observations from different laboratories indicated that sulfotransferase enzymes may be important for the formation of indoxyl sulfate from indoxyl (10, 11). The current study provides evidence that sulfotransferases catalyze the formation of indoxyl sulfate and demonstrates the sulfotransferase isoform(s) responsible for the production of this tryptophan metabolite.
MATERIALS AND METHODS
Reagents and chemicals
Indoxyl acetate, indoxyl sulfate, 3'-phosphoadenosine 5'-phosphosulfate (PAPS), adenosine 3',5'-diphosphate (PAP), dithiothreitol (DTT), 17β-oestradiol, oestrone, p-nitrophenol, p-nitrophenol sulfate, and porcine liver esterase were obtained from Sigma Chemical Co. (St Louis, MO, USA). Dehydroepiandrosterone (DHEA) and 2,6-dichloro-4-nitrophenol (DCNP) were from Fluka Chemika (Buchs, AG, Switzerland). Other chemicals were obtained at the highest available grade from commercial sources.
Liver cytosol and recombinant sulfotransferases
Human liver tissue was obtained from International Institute for the Advancement of Medicine (Exton, PA, USA) under IRB approval. Sprague-Dawley rats (male) were from Charles River (Wilmington, MA, USA) and handled with IACUC approval. Human and rat liver cytosolic fractions (100000g supernatant) were prepared by differential centrifugation in our laboratory as described previously (4). Cytosolic fractions were stored in 1 ml aliquots at −80°C and, after thawing, were kept at 4°C. Protein concentrations were determined by Biuret assay (Total Protein Reagent; Sigma Diagnostics, St Louis, MO, USA).
Recombinant human sulfotransferases SULT1A1*2 (thermostable phenol sulfotransferase allozyme), SULT2A1 (hydroxysteroid sulfotransferase), SULT1E1 (oestrogen sulfotransferase) and Sf-9 control cytosolic extract were obtained from Panvera (Madison, WI, USA) as cytosolic extracts from Sf-9 insect cells transfected with a baculovirus containing a corresponding individual human sulfotransferase cDNA. Recombinant rat hydroxysteroid sulfotransferase (STa) was a generous gift of Prof. Michael W. Duffel (University of Iowa, Iowa City, IA, USA).
Indoxyl (3-hydroxyindole) sulfonation assay
Because of the high spontaneous oxidation rate of indoxyl (10,12), this compound was generated in situ from indoxyl acetate by incubation with 3.3 units of porcine liver esterase at pH 7.0 for 2 min immediately prior to use in the sulfotransferase assay. All assays were run under argon atmosphere. The reaction mixture of 60 μl total volume contained 23.3 mM potassium phosphate buffer at pH 7.0, 10.6 mM dithiothreitol, 0.2 mM PAPS, 3.3 units porcine liver esterase, various concentrations of indoxyl acetate in acetone (final concentration of acetone in the assay was no more than 5% v/v). Reactions were pre-incubated for 2 min with porcine liver esterase to form indoxyl from indoxyl acetate. The sulfonation reaction was initiated by the addition of either human liver cytosol (0.1 mg protein), rat liver cytosol (0.1 mg protein), human SULT1A1*2 (2.2 μg protein), human SULT2A1 (4.5 μg protein), human SULT1E1 (3.8 μg protein), rat recombinant STa (1 μg protein) or Sf-9 control cytosolic extract (6.4 μg protein). After incubation for 2-10 min at 37°C, the reaction was terminated by addition of 60 μl ice-cold methanol. After centrifugation for 2 min at 12000g, 50 μl of the supernatant was injected to HPLC for determination the substrate-dependent formation of indoxyl sulfate.
Indoxyl sulfonation rate was determined by HPLC measuring the amount of indoxyl sulfate formed in the assay. HPLC analysis was carried out on an Alltech Econosphere C18 column (250 mm × 4.6 mm, 5 μm) using isocratic elution with water:methanol (88:12) containing 50 mM KH2PO4, 1.0 mM 1-octylamine, and 50 mM NH4Cl (pH adjusted to 5.45 before addition of methanol). All analysis of reaction mixtures was conducted at a flow rate of 2.0 ml/min, with detection by absorbance at 280 nm with a Hitachi photodiode array detector (Model L-7455). The concentration of the indoxyl sulfate formed in the reaction mixture was determined from a linear standard curve relating peak area to the concentration of indoxyl sulfate.
At least six different concentrations of indoxyl (from indoxyl acetate) were assayed, and these included concentrations both greater and less than the apparent Km values. Apparent Km and Vmax values are presented as ± the standard error obtained by non-linear least squares curve fitting (13) of the velocity data to the Michaelis-Menten equation.
Determination of SULT activities in cytosolic fractions and with recombinant SULTs
Presence of SULT activities in both human and rat liver cytosolic fractions were determined by using p-nitrophenol, DHEA and oestradiol as substrates for aryl, hydroxysteroid, and oestrogen sulfotransferase activities, respectively, by using either a PAPS/PAP conversion assay (14) or a SULT colorimetric method (15). The presence of isoform-specific SULT activities in human and rat recombinant forms were verified using a specific substrate for each recombinant form: p-nitrophenol (human SULT1A1*2), DHEA (human SULT2A1 and rat STa), oestradiol (human SULT1E1) and 1-naphthol (rat AST IV).
Inhibition of isoform-specific SULT activities in human and rat cytosolic fractions
Assays of inhibition of indoxyl sulfonation activity in cytosolic fractions were carried out by incubating for 10 min either human or rat liver cytosol with increasing amounts of known isoform-specific SULT inhibitors such as 2,6-dichloro-4-nitrophenol (DCNP) (an aryl SULT inhibitor), DHEA (a hydroxysteroid SULT inhibitor) or oestrone (oestrogen SULT inhibitor) in the presence of indoxyl as a substrate. The formation of indoxyl sulfate after 10 min was determined by the HPLC procedure as described above. IC50 values for the inhibition of specific SULT activity in the sulfonation of indoxyl, if present, were calculated.
RESULTS
Sulfonation of indoxyl by rat and human liver cytosols
The effect of variation of substrate concentration on indoxyl sulfonating activity of human liver and rat liver cytosols is shown in Fig. 1. Since SULT enzymes generally demonstrate pronounced substrate inhibition (16,17), we used a wide range of substrate concentrations in our experiments before determining Km and Vmax values for indoxyl sulfonation activity in human and rat cytosols. With the human and rat liver cytosols, substantial substrate inhibition was observed at concentrations above 20 μM and 64 μM, respectively (Fig. 1). Therefore, we used substrate concentrations below these levels for calculation of apparent Km and Vmax values. As shown in Fig. 2, seven data points for human cytosol and six data points for rat cytosol were used to calculate an apparent Km of 6.8 ± 0.9 μM and 3.2 ± 0.6 μM, respectively, for indoxyl sulfonation activity.
Fig. 1.
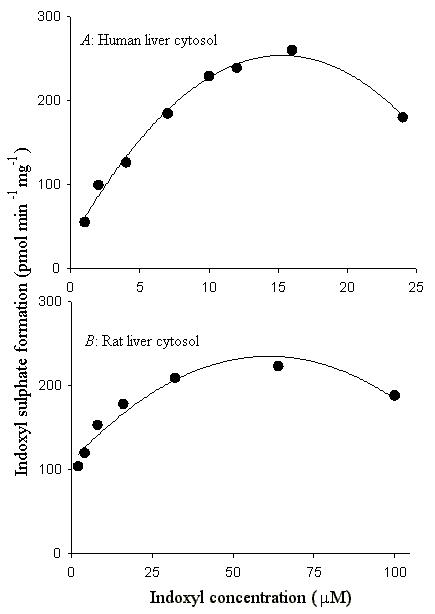
Effect of variation of indoxyl concentration on the sulfonation of indoxyl catalyzed by human (A) and rat (B) liver cytosol. Symbols represent observed values while the line represents a smooth curve indicating substrate inhibition.
Fig. 2.
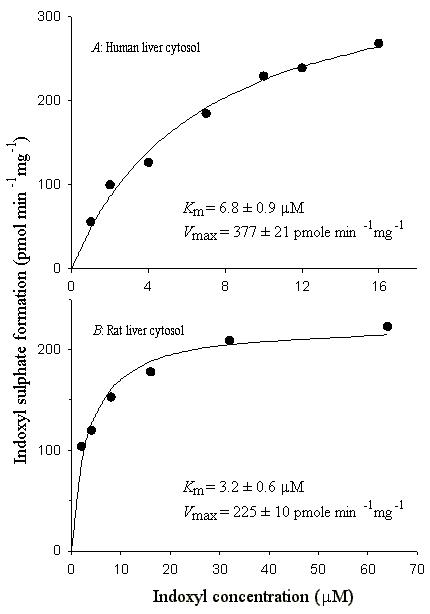
Initial velocity for the sulfonation of indoxyl to indoxyl sulfate by human (A) and rat (B) liver cytosol. Symbols represent observed values while the line represents non-linear least squares fitting of the observed data to the Michaelis–Menten equation.
Effect of isoform-specific SULT inhibitors on indoxyl sulfonating activity with rat and human cytosols
For evaluation of the contribution of various SULT isoforms to the sulfonation of indoxyl, presence of the SULT activity of each isoform was first measured at the optimal concentration for its respective prototype substrate in both human and rat cytosols, i.e., 5 μM DHEA for human DHEA SULT, 4 μM p-nitrophenol for human PST, 25 μM DHEA for rat hydroxysteroid SULT, 100 μM 1-naphthol for rat aryl SULT, and 25 μM oestrone for both human and rat oestrogen SULT. After determination of the presence of phenol and DHEA SULT activities in both human and rat cytosols, isoform-specific SULT inhibitors, DCNP, DHEA and oestrone, were used for measurement of aryl (phenol), hydroxysteroid (alcohol) SULT and oestrogen SULT activities on indoxyl sulfonating activity, respectively. Human thermostable phenol SULT (SULT1A1/2) is very sensitive to DCNP inhibition and is inhibited by this compound with an IC50 value of very low concentration, 2.3 μM (16,18,19). DCNP also inhibits thermolabile phenol SULT (SULT1A3) and DHEA SULT (SULT2A1) activity in human liver although with high IC50 values, such as 110 and 85 μM, respectively (19). Selective inhibition of rat aryl SULT activity by DCNP has also been reported (20). DHEA is a known competitive inhibitor of DHEA SULT activity in human liver cytosol (21) and hydroxysteroid SULT activity in rat liver cytosol (22), and is used to determine the contribution of this sulfotransferase family towards indoxyl sulfonation activity. Oestrone, a competitive substrate of oestrogen SULT, was used to determine the possible contribution of oestrogen sulfotransferase activity towards indoxyl sulfonation, although oestrogen SULT is expressed at only low levels in the human liver (23). DHEA (25 and 40 μM) and oestrone (25 and 40 μM) did not affect the sulfonation of indoxyl measured at the substrate concentration of 12 μM. In contrast, human and rat liver PST indoxyl sulfonation activity was inhibited by DCNP. The concentration of DCNP that inhibited indoxyl sulfonation in human liver cytosol by 50% (IC50) was 3.2 μM for the activity measured with 12 μM indoxyl, while the IC50 of DCNP inhibition of the same activity in rat liver cytosol was 1 μM.
Sulfonation of indoxyl by human and rat recombinant aryl (phenol) sulfotransferases
Recombinant human SULTs, SULT1A1*2 (TS-PST), SULT2A1 (DHEA-SULT) and SULT1E1 (oestrogen SULT) and a recombinant rat SULT, STa (hydroxysteroid SULT) were used to elucidate the responsible sulfotransferase isoforms for the sulfonation of indoxyl. With SULT2A1, SULT1E1 and STa, there was no indoxyl sulfate formed in the enzyme assay with various concentrations of indoxyl. However, SULT1A1*2 efficiently catalyzed the sulfonation of indoxyl under the assay conditions described in Materials and Methods. As anticipated, there was a pronounced substrate inhibition above 20 μM indoxyl with this recombinant enzyme. Apparent enzyme kinetic determinations, using substrate concentrations below 20 μM with recombinant SULT1A1*2, allowed saturable kinetics producing an apparent Km and Vmax values of 5.6 ± 1.8 μM and 1450 ± 190 pmol/min/mg, respectively (Fig. 3).
Fig. 3.
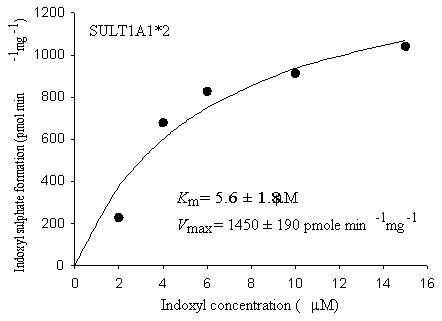
Initial velocity for the sulfonation of indoxyl to indoxyl sulfate by human recombinant SULT1A1*2. Symbols represent observed values while the line represents non-linear least squares fitting of the observed data to the Michaelis–Menten equation.
DISCUSSION
The present study shows that sulfonation of indoxyl to indoxyl sulfate is catalyzed by human and rat liver aryl (phenol) sulfotransferases with apparent Km's of 6.8 μM and 3.2 μM, respectively. The inhibition experiments suggest that the SULT1A1 isoform is responsible for the sulfonation activity in human and rat liver cytosol. This was further established by measurement of indoxyl sulfonation activity with human recombinant SULT1A1*2. The apparent Km measured for indoxyl sulfonation catalyzed by SULT1A1*2 (5.6 μM) was within experimental error of that measured for human liver cytosol. The other major allozyme, SULT1A1*1, was not available to us, but we expect that it would also catalyze sulfonation of indoxyl. There was no indoxyl sulfonation activity observed with the other sulfotransferase isoforms tested, and no inhibition of indoxyl sulfonation was observed in the presence of isoform-specific inhibitors for these sulfotransferases.
An early study with purified rat liver phenol sulfotransferases, PST I and II, showed that indoxyl served as a substrate for these rat liver phenol SULTs (10). Another study for the metabolism of 3-indolylacetic acid in human skin samples also demonstrated that indoxyl sulfate was one of the metabolites formed from parent molecule (11). These results are in good correlation with the results of our present study indicating that sulfonation of indoxyl is catalyzed by aryl (phenol) sulfotransferases. The apparent Km observed with both human and rat liver cytosols (6.8 μM for human and 3.2 μM for rat) and corresponding human recombinant form (5.6 μM for SULT1A1*2) were similar. Moreover, the binding affinity of indoxyl for SULT1A1*2 was comparable to the prototype substrate of this enzyme, p-nitrophenol (i.e., Km of 4 μM). This is a good indication that indoxyl serves as a very good substrate for both human and rat aryl (phenol) sulfotransferases.
The conjugation of phenolic type compounds in liver by aryl (phenol) sulfotransferases is generally a major detoxication reaction for the excretion of these compounds. Our observation, that indoxyl is an excellent substrate for sulfonation by both human and rat aryl sulfotransferases, is not surprising because indoxyl is similar chemically and electronically (i.e. planar molecular shape) to phenols. In this case, however, the metabolite indoxyl sulfate causes mor toxicity than its metabolic precursor tryptophan. Oral administration of indoxyl sulfate or its precursor, indole, stimulates the progression of chronic renal failure (5,6). Although the mechanism of the toxicity caused by indoxyl sulfate is unknown, it causes damage in proximal tubule cells and stimulates the progression of glomerular sclerosis and tubulointerstitial damage, as shown by histologic studies (24). Indoxyl sulfate also increases the expression of transforming growth factor (TGF)-β1 (25). TGF-β1 is a fibrogenic cytokine which causes increased production of extracellular matrix collagens and is a tissue inhibitor of the metalloproteinase TIMP-1. Increased expression of TGF-β1 leads to tubulointerstitial fibrosis and the consequent decline of renal function (25). Indoxyl sulfate may cause additional toxic effects due to inhibition of drug binding to albumin, causing drug toxicity in uremic patients; about 90% of indoxyl sulfate itself is bound to albumin (26).
Human liver contains at least two aryl sulfotransferase isoforms, thermostable or phenol preferring form (TS-PST, SULT1A1/2) and thermolabile or monoamine preferring form (TL-PST, SULT1A3). SULT1A1 preferentially catalyzes the sulfonation of micromolar concentrations of planar phenols (i.e., p-nitrophenol) and is very sensitive to inhibition by DCNP. SULT1A3 preferentially catalyzes the sulfonation of micromolar concentrations of phenolic monoamines and is relatively insensitive to inhibition by DCNP (18,27). Moreover, SULT1A1 is known to be genetically polymorphic which results in expression of SULT1A1 allozymes. Differential expression of these allozymes is thought to be responsible for the wide variation in SULT1A1 activity observed in human populations (14,27,28). Our experimental results based on DCNP inhibition studies and with recombinant SULT1A1*2 suggest that human polymorphic thermostable aryl SULTs are the only sulfotransferase isoforms involved in the formation of indoxyl sulfate. This determination is important for future elucidation of individual susceptibility of uremic patients to the toxic effects of this metabolite of tryptophan.
ACKNOWLEDGEMENTS
This study was supported by Gazi University and the University of Rhode Island Research Foundation.
REFERENCES
- 1.Duffel MW. Sulfotransferases. In: Guengerich FP, editor. Comprehensive Toxicology Vol.3, Biotransformation. Elsevier; Oxford: 1997. pp. 365–383. [Google Scholar]
- 2.Chou HC, Ozawa S, Fu PP, Lang NP, Kadlubar FF. Metabolic activation of methyl-hydroxylated derivatives of 7,12-dimethylbenz[a]anthracene by human liver dehydroepiandrosterone-steroid sulfotransferase. Carcinogenesis. 1998;19:1071–1076. doi: 10.1093/carcin/19.6.1071. [DOI] [PubMed] [Google Scholar]
- 3.Banoglu E. Current status of the cytosolic sulfotransferases in the metabolic activation of promutagens and procarcinogens. Curr. Drug Metab. 2000;1:1–30. doi: 10.2174/1389200003339234. [DOI] [PubMed] [Google Scholar]
- 4.King RS, Teitel CH, Kadlubar FF. In vitro bioactivation of N-hydroxy-2-amino-α-carboline. Carcinogenesis. 2000;21:1347–1354. [PubMed] [Google Scholar]
- 5.Niwa T, Ise M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J. Lab. Clin. Med. 1994;124:96–104. [PubMed] [Google Scholar]
- 6.Niwa T, Ise M, Miyazaki T. Progression of glomerular sclerosis in experimental uremic rats by administration of indole, a precursor of indoxyl sulfate. Am. J. Nephrol. 1994;14:207–212. doi: 10.1159/000168716. [DOI] [PubMed] [Google Scholar]
- 7.Niwa T, Takeda N, Tatematsu A, Maeda K. Accumulation of indoxyl sulfate, an inhibitor of drug binding, in uremic serum as demonstrated by internal-surface reversed-phase liquid chromatography. Clin. Chem. 1988;34:2264–2267. [PubMed] [Google Scholar]
- 8.Sims J, Renwick AG. The microbial metabolism of tryptophan in rats fed a diet containing 7.5% saccharin in a two-generation protocol. Food Chem. Toxicol. 1985;23:437–444. doi: 10.1016/0278-6915(85)90137-1. [DOI] [PubMed] [Google Scholar]
- 9.Banoglu E, Jha GG, King RS. Hepatic microsomal metabolism of indole to indoxyl, a precursor of indoxyl sulfate. Eur. J. Drug Metab. Pharmacokin. 2001;26(4):1–6. doi: 10.1007/BF03226377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sekura RD, Jakoby WB. Phenol Sulfotransferases. J. Biol. Chem. 1979;254:5658–5663. [PubMed] [Google Scholar]
- 11.Ademola JI, Wester RC, Maibach HI. Metabolism of 3-indolylacetic acid during percutaneous absorption in human skin. J. Pharm. Sci. 1993;82:150–154. doi: 10.1002/jps.2600820207. [DOI] [PubMed] [Google Scholar]
- 12.Russel GA, Kaupp G. Oxidation of carbanions. IV. Oxidation of indoxyl to indigo in basic solution. J. Am. Chem. Soc. 1969;91:3851–3859. [Google Scholar]
- 13.Perella FW. A practical curve-fitting microcomputer program for the analysis of enzyme kinetic data on IBM-PC compatible computers. Anal. Biochem. 1988;174:437–447. doi: 10.1016/0003-2697(88)90042-5. [DOI] [PubMed] [Google Scholar]
- 14.Duffel MW, Binder TP, Rao SI. Assay of purified aryl sulfotransferase suitable for reactions yielding unstable sulfuric acid esters. Anal. Biochem. 1989;183:320–324. doi: 10.1016/0003-2697(89)90486-7. [DOI] [PubMed] [Google Scholar]
- 15.Frame LT, Ozawa S, Nowell SA, Chou HC, DeLongchamp RR, Doerge DR, Lang NP, Kadlubar FF. A simple colorimetric assay for phenotyping the major human thermostable phenol sulfotransferase (SULT1A1) using platelet cytosols. Drug Metab. Dispos. 2000;28:1063–1068. [PubMed] [Google Scholar]
- 16.Campbell NRC, Van Loon JA, Weinshilboum RM. Human liver phenol sulfotrasferase: Assay conditions, biochemical properties and partial purification of isozymes of the thermostable form. Biochem. Pharmacol. 1987;36:1435–1446. doi: 10.1016/0006-2952(87)90108-0. [DOI] [PubMed] [Google Scholar]
- 17.Banoglu E, Duffel MW. Studies on the interactions of chiral secondary alcohols with rat hydroxysteroid sulfotransferase STa. Drug Metab. Dispos. 1997;25:1304–1310. [PubMed] [Google Scholar]
- 18.Rein G, Glover V, Sandler M. Multiple forms of phenolsulphotransferase in human tissues. Biochem. Pharmacol. 1982;31:1893–1897. doi: 10.1016/0006-2952(82)90493-2. [DOI] [PubMed] [Google Scholar]
- 19.Hernandez JS, Watson WG, Wood TC, Weinshilboum RM. Sulphation of estrone and 17β-estradiol in human liver: Catalysis by thermostable phenol sulfotransferase and by dehydroepiandrosterone sulfotransferase. Drug Metab. Dispos. 1992;20:413–422. [PubMed] [Google Scholar]
- 20.Koster H, Halsema I, Scholtens E, Meerman JHN, Pang S, Mulder GJ. Selective inhibition of sulfate conjugation in the rat. Biochem. Pharmacol. 1982;31:1919–1924. doi: 10.1016/0006-2952(82)90498-1. [DOI] [PubMed] [Google Scholar]
- 21.Watabe T, Hiratsuka A, Ogura K. Sulphotransferase-mediated covalent binding of the carcinogen 7,12-dihydroxymethyl[a]anthracene to calf thymus DNA and its inhibition by glutathione transferase. Carcinogenesis. 1987;8:445–453. doi: 10.1093/carcin/8.3.445. [DOI] [PubMed] [Google Scholar]
- 22.Chen G, Banoglu E, Duffel MW. Influence of substrate structure on the catalytic efficiency of hydroxysteroid sulfotransferase STa in the sulfation of alcohols. Chem. Res. Toxicol. 1996;9:67–74. doi: 10.1021/tx950065t. [DOI] [PubMed] [Google Scholar]
- 23.Falany CN, Krasnykh V, Falany JL. Bacterial expression and characterisation of a cDNA for human liver estrogen sulfotransferase. J. Steroid Biochem. Mol. Biol. 1995;52:529–539. doi: 10.1016/0960-0760(95)00015-r. [DOI] [PubMed] [Google Scholar]
- 24.Niwa T. Organic acids and the uremic syndrome: protein metabolite hypothesis in the progression of chronic renal failure. Sem. Nephrol. 1996;16:167–182. [PubMed] [Google Scholar]
- 25.Miyazaki T, Ise M, Seo H, Niwa T. Indoxyl sulfate increases the gene expressions of TGF-β1 and pro-α1(I) collagen in uremic rat kidneys. Kidney Int. 1997;52:S15–S22. [PubMed] [Google Scholar]
- 26.Sakai T, Takadate A, Otagiri M. Characterization of binding site of uremic toxins on human serum albumin. Biol. Pharmaceut. Bull. 1995;18:1755–1761. doi: 10.1248/bpb.18.1755. [DOI] [PubMed] [Google Scholar]
- 27.Raftogianis RB, Wood TC, Weinshilboum RM. Human phenol sulfotransferases SULT1A2 and SULT1A1. Biochem. Pharmacol. 1999;58:605–616. doi: 10.1016/s0006-2952(99)00145-8. [DOI] [PubMed] [Google Scholar]
- 28.Price RA, Spielman RS, Lucena AL, Van Loon JA, Maidak BL, Weinshilboum RM. Genetic polymorphism for human platelet thermostable phenol sulfotransferase (TS PST) activity. Genetics. 1989;122:905–914. doi: 10.1093/genetics/122.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]