

Abstract
The depolymerization of polysaccharides, particularly those containing acid-sensitive components, into intact constituent repeating units can be very difficult. We describe a method using ozonolysis for depolymerizing polysaccharides containing β-d-aldosidic linkages into short-chain polysaccharides and oligosaccharides. This method is carried out on polysaccharides that have been fully acetylated whereby β-d-aldosidic linkages are selectively oxidized by ozone to form esters, from which the polysaccharides are subsequently cleaved with a nucleophile. Ozone oxidation of aldosidic linkages proceeds under strong stereoelectronic control, and reaction rates depend on the conformations of glycosidic linkages. Thus, β-d-aldosidic linkages with different conformations can have very different reaction rates even in the absence of substantial chemical differences. These rate differences allowed for very high selectivity in cleaving β-d-linkages of polysaccharides. Several polysaccharides from group B Streptococcus and other bacterial species were selectively depolymerized with this method. The repeating units of the group B Streptococcus polysaccharides all contain an acid-sensitive sialic acid residue in a terminal position on a side chain and several β-d-residues including galactose, glucose, and N-acetylglucosamine; however, with each polysaccharide, one type of linkage was more reactive than others. Selective cleavage of the most sensitive linkage occurs randomly throughout the polymer chain, yielding fragments of controllable and narrowly distributed sizes and the same repeating-unit structure. The average size of the molecules decreases exponentially, and desired sizes can be obtained by stopping the reaction at appropriate time points. With this method the labile sialic acid residue was not affected.
Polysaccharides play a key role in biological recognition and have been used as vaccines against bacterial infections. Bacterial polysaccharides, which make up the surface capsule of many pathogenic microorganisms, tend to have very long chains (with a molecular weight of up to millions of daltons), but short-chain polymer or oligomer fragments have frequently proven preferable for some applications such as vaccines. For instance, short-chain polysaccharides, because of their lower viscosity and better solubility, may be preferable to native polymers for use in highly cross-linked glycoconjugate vaccines. The only commercially available glycoconjugate vaccine, Haemophilus influenzae type b, uses oligosaccharides rather than intact polysaccharides (1, 2). We are currently developing glycoconjugate vaccines against group B Streptococcus (GBS), the foremost cause of neonatal infections in the United States (3, 4). In previous studies, with oligosaccharides derived from the type III GBS polysaccharide by endoglycosidase digestion, it was determined that the antigenicity of the polysaccharides was size-dependent (5–7); however, this research was hindered by the lack of availability for testing of different-sized saccharides from other GBS capsular polysaccharides. The GBS capsular polysaccharides types I–VIII (8–10) that we are studying contain acid-labile sialic acids that are critical to protective epitopes; therefore, chemical methods based on acid hydrolysis (11) or chromium trioxide oxidation in acetic acid (12) cannot be used to prepare oligosaccharides. Other methods based on periodate oxidation, β-elimination, deamination, and enzymatic hydrolysis are of very limited use (13–15). Because of these problems, we sought to develop a general chemical method for the selective depolymerization of these and other polysaccharides. Herein we report such a method based on ozonolytic cleavage of β-d-aldosidic linkages.
In the 1970s, ozone was reported to react with acetal functions in a
specific manner to give esters in which one of the alkoxy residues in
the acetal was retained in the acyloxy group (16–19). Methyl
β-d-glycopyranosides, representing a special kind of
acetal, were also reported to react with ozone to yield the
corresponding aldonic esters, but conformationally rigid
α-d-glycosides were inert toward ozone. It was proposed
in these earlier studies that the ozonolysis reaction proceeds under
strong stereoelectronic control and only if the acetal function
(Structure 1) can
assume a conformation in which both oxygens of the acetal have one of
their lone pair orbitals oriented trans-antiperiplanar to the
alkylidene C—H bond. This orientation is only possible for
β-d-glycosides in the 4C1
conformation (Structure
2), and ozonolysis
can selectively convert them to aldonic acid esters.
![]()
Given the dependence of the oxidation of acetals such as β-d-glycosides on the conformation around the glycosidic bond, it should be expected that oxidation will be more rapid at linkages where the conformation is close to the preferred one and much slower at linkages where it is not. This fact suggested the potential use of ozone to selectively oxidize β-d-linkages in polysaccharides. Differences in rates of oxidation may have chemical or stereoelectronic origins, and these differences might be reflected in the rate of cleavage for different kinds of glycosyl linkages, which in turn might differ with the type of glycosyl residues because of chemical differences. Hence, uronic acids might not be expected to be oxidized at the same rates as 2-amino-2-deoxy sugars, and galactoses might not have the same reaction rate as fucoses. Even when the same kind of residue appears more than once in a repeating sequence and chemoselectivity is thereby undermined, appreciable differences in the conformations around the glycosidic bond might lead to different rates of cleavage. For example, a β-d-galactopyranosyl residue involved in a 1→4 linkage might have a different conformation around its glycosidic linkage than a β-d-galactopyranosyl residue involved in a 1→3 linkage. If one kind of linkage in a complex heteropolysaccharide cleaved at 10 times the combined rate of the others because of a combination of these chemical and stereoelectronic factors, we might expect 90% selectivity. Thus the ozonolytic method theoretically offers promise as a means for selectively oxidizing one kind of linkage in complex polysaccharides.
By using the above rationale, we have developed a method of selectively
depolymerizing polysaccharides based on ozonolysis. This method uses
three steps (Scheme): (i) All hydroxyl groups on the
polysaccharide, e.g., type III GBS (Structure
3), are protected as
ester groups. (ii) The protected polysaccharide is
“ozonolyzed” to selectively oxidize the most reactive glycosidic
linkages into ester linkages. (iii) The ester linkages are
cleaved with a nucleophile, thus cleaving the polymer while
simultaneously removing the ester-protecting groups on the sugar rings.

MATERIALS AND METHODS
Materials.
Capsular polysaccharides from GBS were isolated and purified as described (8–10). Ozone was generated from compressed air through an ozone generator (More-Zon10, Taiwan).
Peracetylation.
A 20-mg sample of polysaccharide was dissolved in 10 ml of formamide, and 2 ml of pyridine and 1 ml of Ac2O were added. The solution was stirred magnetically at room temperature for 16 h. Upon completion of the reaction, the mixture was dialyzed extensively against distilled water and freeze-dried.
Ozonolysis.
The peracetylated polysaccharide was dissolved in 10 ml of ethyl acetate. A stream of ozone/air gas (21% O3) was passed through the solution at room temperature at a flow rate of 3.17 ml/sec. At various time points, 0.1 ml of the reaction solution was dried, and the molecular size distribution of the products was determined by gel-filtration column chromatography. The reaction was terminated when the products reached the desired molecular size by stopping the addition of ozone, e.g., starting with a 61-kDa type III GBS polysaccharide, it took 1 h of ozonolysis to decrease the average molecular size to 20 kDa and 10 h to 10 kDa. After ozonolysis, ethyl acetate was removed on a rotary evaporator.
Saponification of the Ester Intermediate.
The oxidized material was hydrolyzed with 5 ml of 0.1 M NaOH at room temperature for 2 h and then neutralized with CH3COOH. Resulting oligosaccharides and short-chain polysaccharides with carboxylate termini were liberated, and the acetyl protecting groups were also removed. The products were separated on gel-filtration columns.
Gel Filtration Chromatography.
The 0.1-ml samples taken at various times were hydrolyzed with 1 ml of 0.1 M NaOH at room temperature for 0.5 h and then neutralized with 0.1 M HCl. They were then loaded on a Superose 12 column (Pharmacia) with a fractionation range of 1–300 kDa on an FPLC system to determine the average molecular size and the size distribution of the products. The saccharides were eluted with PBS (pH 7.2; 0.01 M sodium phosphate/0.15 M NaCl/0.05% azide) at 0.5 ml/min. The fractions were monitored by a differential refractometer. The Superose 12 column was calibrated with dextran standards of molecular mass 150, 110, 40, 20, 10, and 6 kDa. The molecular sizes of the aliquots were obtained from the column calibration curve. The final products of small oligosaccharides (<10 kDa) were separated on a Superdex 75 column (Pharmacia) with a fractionation range of 0.5–30 kDa and eluted with PBS containing 0.3 mM phosphate, instead of 0.01 M phosphate, with 0.05% azide (pH 7.2) at a flow rate of 1.0 ml/min. The Superdex 75 column was calibrated with oligosaccharide standards (20), which were one to five repeating units of type III GBS oligosaccharides obtained from endo-β-galactosidase digestion of the polysaccharides as described (9). Fractions of different-sized saccharides were pooled and analyzed by NMR spectroscopy and electrospray mass spectrometry.
Compositional Analysis of the Oligosaccharide Products.
Samples (0.2 mg) of the oligosaccharide fractions from the type II and type III GBS polysaccharide were hydrolyzed with 1 ml of 2 M trifluoroacetic acid at 110°C for 4 h, converted to alditol acetate derivatives as described (21), and subjected to GC/MS analysis on an HP 6890/5973 GC/MSD instrument with a DB17 capillary column. The analysis was carried out with an initial temperature of 180°C, held for 1 min, followed by a 2°C/min increase to 260°C, and held for 10 min.
Instrumental Analysis.
Proton NMR experiments were performed on a Bruker AMX500 spectrometer with a proton resonance frequency of 500.13 MHz. All 1H spectra were recorded at 70°C in 2H2O and chemical shifts are referenced relative to the H2HO resonance at 4.26 ppm. Electrospray mass spectra were acquired on a VG Micromass Quattro instrument in the negative ion mode scanned from 150 to 2,000 atomic mass units in 9.9 sec.
RESULTS AND DISCUSSION
We have applied this method to the depolymerization of 11 types of bacterial capsular polysaccharides, including GBS type I–VIII, Staphylococcus aureus type 5 (22), and Bacteroides fragilis polysaccharide A (23). Each of these polysaccharides contains at least one β-d-aldosidic linkage in its backbone. The polysaccharides from GBS and B. fragilis contain highly acid-labile sialic acid or pyruvyl residues, which are critical to their biological or immunological functions and which, in our hands, were lost by other chemical methods for breaking the polysaccharides down into smaller fragments. However, with the present method, they were not affected. Because all results were similar, we will present only the results of our studies with polysaccharides of GBS types II and III in detail.
Depolymerization of the Type II GBS Polysaccharide.
The type II GBS polysaccharide has a heptasaccharide repeating unit with the following structure (8):
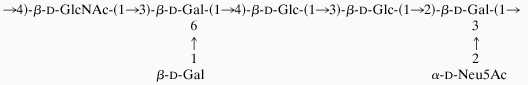 |
All galactose, Glc, and GlcNAc residues are linked through β-d-aldosidic linkages, whereas the sialic acid residue is linked through an α-ketosidic linkage. After ozonolysis followed by base hydrolysis, gel-filtration chromatography on a Superdex 75 column gave several peaks (Fig. 1). As judged by the column calibration (20), fractions 1–4 have average molecular masses of 1.3, 2.7, 4.3, and 5.7 kDa, respectively, which correspond to saccharides of one to four repeating units. The molecular weight of the one-repeating-unit of peak 1 was also confirmed by negative ion electrospray mass spectrometry, where an ion at m/z 1,359.5 corresponding to [M + Na − 2H]− (expected 1,359.7) was observed. The elution of the oligosaccharide products in distinct peaks of different repeating units strongly indicated that the cleavage of polysaccharide linkages was highly selective. Random cleavage of all linkages, however, should lead to a mixture of sizes and compositions and, therefore, result in a single, broad, and continuous peak.
Figure 1.

Elution profile on a Superdex 75 column of 20-mg oligosaccharides generated from type II GBS polysaccharide after 22 h of ozonolysis. Fractions 1–4 correspond to one to four repeating units, with average molecular sizes of 1.3, 2.7, 4.3, and 5.7 kDa, respectively. The molecular sizes were determined from oligosaccharide standards obtained by glycosidase digestion of type III GBS polysaccharide.
Structural analysis of these oligosaccharides by NMR spectroscopy (Fig. 2) showed a spectrum of the native polysaccharide (Fig. 2, spectrum PS) that was expected for a heptasaccharide repeating unit, with six anomeric protons between 4.4 and 5.2 ppm from the hexose residues and a pair of characteristic resonances from the H-3 protons of sialic acid residue at 2.82 (dd) and 1.80 (t) ppm. The NMR spectrum of fraction 4 (data not shown) was identical to that of the polymer. Fraction 3, an oligosaccharide of three repeating units, gave a spectrum almost identical (Fig. 2, spectrum 3) to that of the polymer. Fraction 2 oligomer (Fig. 2, spectrum 2) gave 11 anomeric signals from 11 hexose residues. These signals, along with two sialic acids and a terminal aldonic acid, revealed the presence of two repeating units. The sialic acid residue was not affected by the depolymerization process as evidenced by its H-3 resonances. The above NMR results clearly demonstrated that these oligosaccharide products had the same basic sequence as the parent polymer.
Figure 2.

1H NMR spectra of type II GBS native polysaccharide (spectrum PS) and the saccharides of fractions 3 and 2 from Fig. 1 (spectra 3 and 2). The polysaccharide repeating unit consists of six hexoses as shown by six anomeric resonances at 4.90 (one proton), 4.84 (one proton), 4.66 (one proton), and 4.50 (two protons) ppm and a sialic acid as shown by its H-3 resonances. Spectrum 3 is nearly identical to spectrum PS, except for some differences resulting from the terminal residue, indicating that saccharide if has the same structure as the polymer. Spectrum 2 represents two repeating units, including 11 hexoses with 11 anomeric signals at 4.90 (2 signals), 4.84 (2 signals), 4.76 (1 proton), 4.66 (1 proton), 4.62 (1 proton), and 4.50 (4 proton) ppm, respectively, two sialic acids, and one aldonic acid. Note that the sialic acid residue was retained in all fragments as evidenced by its H-3 resonances.
Depolymerization of the Type III GBS Polysaccharide.
The type III GBS polysaccharide consists of a pentasaccharide repeating unit (Structure 3 in Scheme) composed of Glc, galactose, and GlcNAc residues linked through β-d-configuration and a sialic acid residue at the end of the side chain linked through an α-ketosidic linkage (9). The oligosaccharides generated from ozonolytic degradation of the polymer were separated on a Superdex 75 column, which resolved four major peaks (Fig. 3). Fractions 2–5 corresponded to oligosaccharides containing two to five copies of the type III repeats, respectively; this conclusion was drawn from comparing their elution volumes with those of the standard type III GBS oligosaccharides of one to five repeating units. The elution of these saccharides in distinct peaks showed again that the depolymerization was very selective.
Figure 3.

Elution profile on a Superdex 75 column of 28 mg of oligosaccharides generated from type III GBS polysaccharide after 22 h of ozonolysis. As judged by column calibration using standards obtained by glycosidase digestion of type III GBS polysaccharide, fractions 2–5 correspond to saccharides of two to five repeating units, with average molecular sizes of 2.0, 3.1, 4.1, and 5.1 kDa, respectively.
Oligosaccharides isolated from each of these fractions were analyzed by NMR spectroscopy. Fig. 4 shows the 1H NMR spectra of the native type III GBS polysaccharide and the oligosaccharides of fractions 3, 2, and 1 from Fig. 3, respectively. The native polysaccharide (Fig. 4, spectrum PS) gave rise to four anomeric proton resonances between 4.4 and 4.8 ppm from the four hexose residues and a pair of characteristic H-3 resonances at 2.77 and 1.80 ppm from the sialic acid. The spectra of saccharides from fractions 5 and 4 (Fig. 3) were identical (data not shown) to that of the polymer, indicating that they had the same repeating unit as the parent polymer. The compositional analysis (as measured by GC/MS) and NMR spectra of fractions 3 and 2 (Fig. 4, spectra 3 and 2) were nearly identical to that of the polysaccharide except for the terminal aldonic acid residue. Fractions 2 and 1 were different oligomers as revealed by their NMR spectra and compositional analysis. In spectra 3, 2, and 1, the resonances at ∼3.97 ppm (typically due to H-4 of galactose) are different from that of the polymer, and the resonance at 3.35 ppm that is due to H-2 of the Glc residue progressively decreases. Fraction 1 in Fig. 3 showed a very different spectrum (Fig. 4, spectrum 1) because the oligosaccharides are smaller than two repeating units and no longer have the same repeating unit as the parent polymer, and the anomeric resonance of GlcNAc at 4.73 ppm (24) disappears. The differences in these spectra suggest that after prolonged ozonolysis the galactose, Glc, and GlcNAc residues have been oxidized by ozone. The GlcNAc residue is affected only in the smallest fragment. These changes were confirmed by the GC/MS analyses. The sialic acid was retained in all these fragments as revealed by its H-3 resonances.
Figure 4.

1H NMR spectra of type III GBS polysaccharide (spectrum PS) and the oligosaccharides of fractions 3, 2, and 1 from Fig. 3 (spectra 3–1). The repeating unit contains five residues, as shown by four anomeric signals between 4.4 and 4.8 ppm, and a pair of sialic acid H-3 signals. Spectra 3 and 2 are nearly identical to spectrum PS, indicating that they have the same basic structure as the polymer. In spectra 3–1, the galactose H-4 resonances were shifted to the right and the intensity of Glc H-2 resonances decreased progressively, and the GlcNAc H-1 signal disappeared in spectrum 1; these differences suggest that galactose, Glc, and GlcNAc were affected by ozonolysis.
Kinetics.
We examined the effect of ozonolysis for various periods on two lots of type III GBS polysaccharides with average molecular mass of 115 and 61 kDa, respectively (Fig. 5). In both cases, the average size of saccharides decreased very rapidly in the beginning of the degradation, but more slowly when the size reached about 20 kDa. Unlike some physical degradation methods such as ultrasonic degradation, however, there is no limiting molecular weight, and very small oligosaccharides have been obtained after prolonged ozonolysis. The molecular size distribution of the products remains narrow throughout the degradation process. The above kinetics is consistent with a random depolymerization mechanism, in which the reaction takes place at a particular type of linkage but randomly throughout the polysaccharide. Ozone attacks the polymer, progressively decreasing the molecular size, rather than initially producing small oligosaccharides. Ozone reacts with glycosides quantitatively; therefore, the desired size of saccharides may be obtained simply by controlling such conditions as the reaction time and the rate at which ozone passes through the reaction mixture. As demonstrated above, longer reaction time and consumption of more ozone result in smaller saccharides. Controlled cleavage of polysaccharides thus results in saccharides of desired size with narrow size-distribution.
Figure 5.

Average molecular size of products (Mr) vs. reaction time from depolymerizing 2 lots of type III GBS polysaccharides. The size of the products decreased very rapidly in the beginning but more slowly when it reached 20 kDa.
Cleavage Site and Reaction Rate.
Each of the GBS polysaccharides investigated contains several β-d-aldosides, including Glc, galactose, and GlcNAc, that should be amenable to ozonolysis. To determine whether there was only a single reactive linkage, we analyzed the composition of saccharide fractions from the type II and III GBS polysaccharides after conversion to alditol acetates and analysis by GC/MS. For the type II saccharides, we found a constant ratio of galactose to Glc in all fractions, but a progressively decreasing ratio of GlcNAc to Glc only in the small oligosaccharide fractions (Fig. 1, fractions 3–1). This finding indicated that in the type II polysaccharide GlcNAc was the most reactive residue. In contrast, for the type III saccharides, the ratio of the component monosaccharides was relatively constant for saccharides of more than three repeating units. For oligosaccharides of fewer than three repeating units, both galactose and GlcNAc decreased at a similar rate to each other when compared with Glc. Our results showed that, in some cases, as with the type II polysaccharide, one residue or linkage was more reactive to ozone than the others. The selective cleavage at this most reactive linkage resulted in saccharides with highly conserved structure. In other cases, as with the type III polysaccharide, more than one linkage reacted, but ozonolysis still resulted in polysaccharides of smaller molecular size than the native molecule, but retaining the intact repeating unit structure.
The reaction of ozone with β-d-glycopyranosides proceeds under strong stereoelectronic control. It is clear that there is a strong relationship between the conformation of glycosides and their reactivity to ozone. The preferred conformer must have on each oxygen atom a lone pair orbital oriented trans-antiperiplanar to the C—H bond of the acetal function of glycosides. Many conformations are possible for a β-d-glycosidic linkage (Fig. 6), but only two of them (conformations A and E) have the required orbital orientation. The ozonolysis rate is, therefore, determined by the initial conformation of a glycoside. For glycosides with the preferred conformation in the initial state, the reaction is expected to occur readily; for other glycosides, oxidation should be faster at residues with initial conformation closer to the preferred one.
Figure 6.

Conformations around the glycosidic bond of a β-d-glycoside. The center C in the back is C-1. Ys are the lone pair orbitals of O-1. Of all possible conformations, only conformations A and E have one of their lone orbitals (Y) oriented perfectly antiperiplanar to the C1—H1 bond and are thus preferred in ozonolysis.
In a polysaccharide with different linkages, all linkages may be of different conformation. A molecular modelling of the conformation of type III GBS oligosaccharides (24) showed that all the dihedral angles along the glycosidic bond, which determines the conformation of the linkage, differ from each other. This difference in conformation around the glycosidic bond results in different reaction rates even for the same aldosyl residue. A difference in susceptibility to ozone of the two galactose residues in type III GBS polysaccharide is suggested by the decreasing content of galactose in the small oligosaccharides, whereas the sialic acid content in these oligosaccharides remained constant. The sialic acid is linked on the side chain to one of the two galactose residues of the repeating unit, and oxidation of this side-chain galactose would have resulted in loss of sialic acid. Since sialic acid remained intact even in the smallest oligosaccharides, it was the backbone galactose that was oxidized, while the side-chain galactose was nonreactive. The difference in reactivity of the two galactose residues in the repeating unit of type III GBS polysaccharides most likely originates from their conformation. In our conformational study of the type III polysaccharide (unpublished data), we found that the galactose-sialic acid side chain is very flexible, with many possible conformations for the side chain galactose. The flexibility of this galactose residue may also contribute to its lower reactivity to ozone, because it does not spend much time in an orientation in which the stereoelectronics favor cleavage. Because polysaccharides are not strictly rigid molecules, one would expect all the β-d-glycosides to be oxidized eventually under prolonged ozonolysis. In fact, 44-h ozonolysis of the type III GBS polysaccharide sample affected all linkages, and the polymer was completely decomposed into very small fragments.
In conclusion, we found that ozone can preferentially oxidize β-d-glycosidic linkages in a polysaccharide, and the cleavage of the most reactive linkage under limited ozonolytic conditions affords a method for generating oligosaccharide fragments of various repeat lengths with predominantly the same repeating-unit structure. This ozonolysis method may be used to depolymerize any polysaccharide that contains β-d-aldosidic linkages and is especially valuable for depolymerizing polysaccharides that contain acid-labile groups such as sialic acids or pyruvyl acetals.
Acknowledgments
We thank April Blodgett and Dr. Jean Lee for the isolation and purification of GBS and S. aureus polysaccharides and Barbara Reinap for technical assistance. This work is supported in part by Grants AI75326 and AI39576 and Merit Award AI23339 to D.L.K. from the National Institute of Allergy and Infectious Diseases.
ABBREVIATION
- GBS
group B Streptococcus
References
- 1.Anderson P W, Pichichero M E, Stein E C, Porcelii S, Betts R F, Connuck D M, Korones D, Insel R A, Zahradnik J M, Ebby R. J Immunol. 1989;142:2464–2468. [PubMed] [Google Scholar]
- 2.Black S B, Shinefield H R, Fireman B, Hiatt R, Polen M, Vittinghoff E the Northern California Kaiser Permanente Study Center Pediatrics Group. Pediatr Infect Dis. 1991;10:97–104. doi: 10.1097/00006454-199102000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Baker C J, Edwards M S. In: Infectious Diseases of the Fetus and Newborn Infant. Remington J S, Klein J O, editors. Philadelphia: Saunders; 1990. pp. 742–811. [Google Scholar]
- 4.Ferrieri P. Rev Infect Dis. 1990;12:S394–S400. doi: 10.1093/clinids/12.supplement_4.s394. [DOI] [PubMed] [Google Scholar]
- 5.Paoletti L C, Kasper D L, Michon F, DiFabio J, Jennings H J, Tosteson T D, Wessels M R. J Clin Invest. 1992;89:203–209. doi: 10.1172/JCI115564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wessels M R, Munoz A, Kasper D L. Proc Natl Acad Sci USA. 1987;84:9170–9174. doi: 10.1073/pnas.84.24.9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paoletti L C, Kasper D L, Michon F, DiFabio J, Holme K, Jennings H J, Wessels M R. J Biol Chem. 1990;265:18278–18283. [PubMed] [Google Scholar]
- 8.Jennings H J, Rosell K G, Katzenellenbogen E, Kasper D L. J Biol Chem. 1983;258:1793–1798. [PubMed] [Google Scholar]
- 9.Wessels M R, Pozsgay V, Kasper D L, Jennings H J. J Biol Chem. 1987;262:8262–8267. [PubMed] [Google Scholar]
- 10.Kogan G, Uhrin D, Brisson J R, Paoletti L C, Blodgett A E, Kasper D L, Jennings H J. J Biol Chem. 1996;271:8786–8790. doi: 10.1074/jbc.271.15.8786. [DOI] [PubMed] [Google Scholar]
- 11.BeMiller J N. Adv Carbohydr Chem. 1967;22:25–108. doi: 10.1016/s0096-5332(08)60151-4. [DOI] [PubMed] [Google Scholar]
- 12.Angyal S J, James K. Aust J Chem. 1970;23:1209–1215. [Google Scholar]
- 13.Lindberg B, Lonngren J, Svensson S. Adv Carbohydr Chem Biochem. 1975;31:185–240. [Google Scholar]
- 14.Pazur J H. In: Carbohydrate Analysis: A Practical Approach. Chaplin M F, Kennedy J F, editors. New York: IRL; 1994. pp. 93–100. [Google Scholar]
- 15.Hollingsworth R I. Biotechnol Annu Rev. 1996;2:281–291. doi: 10.1016/s1387-2656(08)70014-0. [DOI] [PubMed] [Google Scholar]
- 16.Deslongchamps P, Moreau C. Can J Chem. 1971;49:2465–2467. [Google Scholar]
- 17.Deslongchamps P, Moreau C, Frehel D, Atlani P. Can J Chem. 1972;50:3402–3404. [Google Scholar]
- 18.Deslongchamps P, Atlani P, Frehel D, Malaval A, Moreau C. Can J Chem. 1974;52:3651–3664. [Google Scholar]
- 19.Deslongchamps P, Moreau C, Frehel D, Chenevert R. Can J Chem. 1975;53:1204–1211. [Google Scholar]
- 20.Paoletti L C, Johnson K D. J Chromatogr A. 1995;705:363–368. doi: 10.1016/0021-9673(95)00295-x. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Hollingsworth R I. Carbohydr Res. 1994;260:305–317. doi: 10.1016/0008-6215(94)84048-2. [DOI] [PubMed] [Google Scholar]
- 22.Moreau M, Richards J C, Fournier J M, Byrd R A, Karakawa W W, Vann W F. Carbohydr Res. 1990;201:285–297. doi: 10.1016/0008-6215(90)84244-o. [DOI] [PubMed] [Google Scholar]
- 23.Tzianabos A O, Pantosti A, Baumann H, Brisson J R, Jennings H J, Kasper D L. J Biol Chem. 1992;267:18230. [PubMed] [Google Scholar]
- 24.Brisson J R, Uhrinova S, Woods R J, Zwan M, Jarrell H C, Paoletti L C, Kasper D L, Jennings H J. Biochemistry. 1997;36:3278–3292. doi: 10.1021/bi961819l. [DOI] [PubMed] [Google Scholar]