

Abstract
Background
The alcohol dehydrogenases (ADH) are widely studied enzymes and the evolution of the mammalian gene cluster encoding these enzymes is also well studied. Previous studies have shown that the ADH1B*47His allele at one of the seven genes in humans is associated with a decrease in the risk of alcoholism and the core molecular region with this allele has been selected for in some East Asian populations. As the frequency of ADH1B*47His is highest in East Asia, and very low in most of the rest of the world, we have undertaken more detailed investigation in this geographic region.
Methodology/Principal Findings
Here we report new data on 30 SNPs in the ADH7 and Class I ADH region in samples of 24 populations from China and Laos. These populations cover a wide geographic region and diverse ethnicities. Combined with our previously published East Asian data for these SNPs in 8 populations, we have typed populations from all of the 6 major linguistic phyla (Altaic including Korean-Japanese and inland Altaic, Sino-Tibetan, Hmong-Mien, Austro-Asiatic, Daic, and Austronesian). The ADH1B genotyping data are strongly related to ethnicity. Only some eastern ethnic phyla or subphyla (Korean-Japanese, Han Chinese, Hmong-Mien, Daic, and Austronesian) have a high frequency of ADH1B*47His. ADH1B haplotype data clustered the populations into linguistic subphyla, and divided the subphyla into eastern and western parts. In the Hmong-Mien and Altaic populations, the extended haplotype homozygosity (EHH) and relative EHH (REHH) tests for the ADH1B core were consistent with selection for the haplotype with derived SNP alleles. In the other ethnic phyla, the core showed only a weak signal of selection at best.
Conclusions/Significance
The selection distribution is more significantly correlated with the frequency of the derived ADH1B regulatory region polymorphism than the derived amino-acid altering allele ADH1B*47His. Thus, the real focus of selection may be the regulatory region. The obvious ethnicity-related distributions of ADH1B diversities suggest the existence of some culture-related selective forces that have acted on the ADH1B region.
Introduction
Historically, the alcohol dehydrogenases (ADH) have been among the most widely studied sets of enzymes along with their genes. Alcoholism, a complex genetic disorder that affects a large proportion of people, has been known for some time to be strongly associated with variants of alcohol dehydrogenase [1]–[3]. Alcohol dehydrogenase plays a role not only in alcohol metabolism but also in many other metabolic pathways, and thus forms of the enzyme exist in many organs [4]–[8]. The human ADH gene cluster is located on chromosome 4q23-24, and the several genes are clearly related evolutionarily. Sequentially this ADH cluster contains ADH7, Class I ADH (1C, 1B, 1A), ADH6, ADH4, ADH5 [9], [10]. Hundreds of polymorphic sites have already been studied within the ADH cluster [11], [12]. Some polymorphic sites alter amino acids or lie in other functional regions; these can cause functional changes in the enzyme and result in different phenotypes [11], [13]–[20]. The Arg47His (rs1229984) polymorphism at ADH1B (previously ADH2) is a typical function-related polymorphism [21], [22]. The derived allele, coding for histidine, provides a well confirmed protection against alcoholism [23]–[29].
Global investigation of ADH1B diversity shows a strong geographic distribution [10], [30]. Some variants of ADH1B appear specifically in some geographic regions [10], [24], [31]–[33]. For instance, ADH1B*47His reaches very high frequencies almost exclusively in East Asian populations [10], [12], [34] while fairly high frequencies occur in West Asia and North Africa; in contrast the allele is rare to absent in the rest of the world [30]. This quite unusual geographic pattern argues for more detailed research. Linkage disequilibrium (LD) studies revealed evidence that the upstream region of ADH1B has been under positive selection in several East Asian populations such as Chinese, Koreans and Japanese, but the selective force remains unknown [12]. There are still unsolved issues in relating the high frequency of the functional variant to positive selection in East Asian populations. For example, in some Austronesian (AU) populations, the derived allele frequency of the functional polymorphism is very high, but there is no evidence by the REHH test showing that the functional allele ADH1B*47His underwent selection [12]. Thus, not only is the selective force unknown, but selection cannot explain the frequency of ADH1B*47His in all populations in which it is high. While absence of evidence in some populations may have been a power issue, further investigation is clearly needed.
Our previously studied populations belong to several ethnic phyla (same as linguistic phyla): the Korean-Japanese (KJ) subphylum of the Altaic phylum (Japanese, Koreans), the Chinese (also called Sinitic Han: SN) subphylum of the Sino-Tibetan phylum (Cantonese, Hakka and Minnam Taiwanese), the Taiwan subphylum of the AU phylum (Atayal, Amis), and the East Mon-Khmer subphylum of the Austro-Asiatic (AA) phylum (a small sample of Cambodians) [12]. These eight populations are all located in the coastal region. According to the complicated landforms, climates, ethnic distributions, and population histories in East Asia, this set of population samples is not sufficiently representative of East Asia, especially not for the populations in the western region of East Asia such as the Tibetan, Uigur, Mongol, etc., populations. Based on the geographic distribution of the ADH1B*47His allele [30], we do not believe that selection happened everywhere in East Asia, certainly not to the same effect. Therefore, we are working to improve coverage of other East Asian ethnic groups and geographic regions to reveal the true histories of ADH genes [30]. More detailed distributions of allele and haplotype frequencies will be essential for attempting to address the questions of when, where, and even how selection occurred, although the distributions alone will not allow definitive answers.
The cultural and ethnic diversity in East Asia is noticeable. For example, Daic (also called Tai-Kadai: TK) is the major phylum in the peninsula of Southeast Asia and southern East Asia. In northern East Asia, Altaic is the major phylum, but the KJ subphylum is an atypical branch. Inland Altaic (AT) subphyla such as Mongolian and Turkic are much more representative of Altaic. In the western side of East Asia, the Tibeto-Burman (TB) subphylum and the Hmong-Mien (HM) phylum are both dominant [35], [36]. Different ethnic phyla not only have different languages and religions but also completely different life styles [36], [37]. Almost all of those AT populations are nomandic tribes except Uigur, which switched to farming 1000 years ago [38], [39]. The northern tribes of TB are all mainly pastoralists. TK, HM, SN, KJ, and AU populations have very long histories of farming [40]–[42]. Most of the AA (mainly Mon-Khmer) groups were hunter-gatherers historically, and began farming very recently [43]. Different life styles might have led to differences in the impacts of any selective forces, and might have influenced the distribution of ADH allele and haplotype frequencies.
To determine the allele frequency distributions with greater geographic and ethnic precision, we have studied 24 more populations from different geographic areas of East Asia. These populations belong to different ethnic phyla and their major subphyla. The most important populations of TK, AT, HM, TB, and AA phyla are included (Figure 1A, 1B and Table 1). This type of sampling can minimize the bias of the geographic and ethnic distribution of the samples and the possible misreading of the results. With such data we may be able to identify selection on the relevant allele and to find relationships between haplotype pattern and selection, as much more genetic diversity may be found during this more comprehensive investigation in the focused East Asia region.
Figure 1. Locations of the populations and distributions of the ADH1B rs1229984 and rs3811801 derived allele frequencies.

Note: The map of part A showed the ethnic phyla in East Asia, and part B displayed the locations of the populations. Populations marked with stars were cited from literature [31], [34]. The codes of the star-marked populations are ISO639-3 codes. Populations shown by gray spots are previously published by our team [12]. The colorful spots are the populations collected in this study. Part C is the distribution of the derived allele frequency of rs1229984 (ADH1B*47His). Part D is the distribution of the regulatory region polymorphism rs3811801 derived allele frequency.
Table 1. General Information for the East Asian populations included in the analyses.
| Ethnic Phylum | Subphylum | Code | Population name | sample size | Country | Province | County | Long.(E) | Lat.(N) |
| Altaic | Turkic | KAZ* | Khazaks | 48 | China | Xinjiang | Balikun | 93.01 | 43.59 |
| Altaic | Turkic | YAK | Yakut | 51 | Russia | Saha | 124.20 | 62.07 | |
| Altaic | Turkic | UIG* | Uigur | 48 | China | Xinjiang | Turpan | 88.66 | 42.79 |
| Altaic | Mongolian | MVF* | Mongols | 75 | China | Inner Mongol | Shilingol | 116.07 | 43.95 |
| Altaic | Korean-Japanese | KOR | Koreans | 54 | S.Korea | 126.57 | 37.32 | ||
| Altaic | Korean-Japanese | JPN | Japanese | 47 | Japan | 139.49 | 35.38 | ||
| Sino-Tibetan | Tibeto-Burman | BQH* | BaimaDee | 42 | China | Sichuan | Pingwu | 104.53 | 32.41 |
| Sino-Tibetan | Tibeto-Burman | QMR* | Qiang | 40 | China | Sichuan | Mao | 103.85 | 31.69 |
| Sino-Tibetan | Tibeto-Burman | KHG* | Khamba Tibetan | 36 | China | Sichuan | Kangding | 101.96 | 30.05 |
| Sino-Tibetan | Tibeto-Burman | PHO* | Phunoi | 43 | Laos | Louang-Namtha | Louang-Namtha | 101.05 | 21.13 |
| Sino-Tibetan | Sinitic(Han Chinese) | WUU* | Wu Chinese | 53 | China | Shanghai | 121.37 | 31.11 | |
| Sino-Tibetan | Sinitic | HKA | Hakka Chinese | 41 | China | Taiwan | 121.05 | 24.20 | |
| Sino-Tibetan | Sinitic | CHT | Minnam Chinese | 50 | China | Taiwan | 120.31 | 23.31 | |
| Sino-Tibetan | Sinitic | CHS | Canton Chinese | 57 | USA | CA | San Francisco | 113.04 | 22.35 |
| Hmong-Mien | Hmongic | HMQ* | Black Hmong | 60 | China | Guizhou | Mashan | 109.80 | 27.88 |
| Hmong-Mien | Hmongic | MWW* | White Hmong | 60 | Laos | Huapuan | XamTai | 103.54 | 19.57 |
| Daic | Kam-Sui | KMC* | Kam | 74 | China | Guangxi | Sanjiang | 109.60 | 25.79 |
| Daic | Kam-Sui | LBC* | Laka | 98 | China | Guangxi | Jinxiu | 110.18 | 24.13 |
| Daic | Tai-Sek | CCX* | North Zhuang | 40 | China | Guangxi | Wuming | 108.28 | 23.17 |
| Daic | Tai-Sek | CCY* | South Zhuang | 30 | China | Guangxi | Chongzuo | 107.36 | 22.42 |
| Daic | Hlai | LIC* | Hlai | 59 | China | Hainan | Tongzha | 109.52 | 18.77 |
| Daic | Tai-Sek | LAO* | Lao | 117 | Laos | Cap.Vientiane | Sisattanak | 102.37 | 17.57 |
| Daic | Tai-Sek | SKB* | Saek | 57 | Laos | Khammouan | Boualapha | 104.55 | 17.28 |
| Austro-Asiatic | North Mon-Khmer | WBM* | Ava | 59 | China | Yunnan | Ximeng | 99.46 | 22.74 |
| Austro-Asiatic | North Mon-Khmer | KJG* | Khmu | 51 | Laos | LouangPrabang | Nambak | 102.33 | 20.27 |
| Austro-Asiatic | North Mon-Khmer | LBN* | Lamet | 42 | Laos | Louang-Namtha | Nale | 101.35 | 20.50 |
| Austro-Asiatic | Viet-Muong | BGL* | Bo | 52 | Laos | Bolikhamxai | Khamkheut | 105.09 | 18.08 |
| Austro-Asiatic | East Mon-Khmer | KUF* | Katu | 50 | Laos | Xekong | Thateng | 106.39 | 15.36 |
| Austro-Asiatic | East Mon-Khmer | LBO* | Laven | 47 | Laos | Xekong | Thateng | 106.35 | 15.34 |
| Austro-Asiatic | East Mon-Khmer | CBD | Cambodians | 25 | Cambodia | 104.55 | 11.33 | ||
| Austronesian | Malayo-Polynesian | HUQ* | Tsat | 52 | China | Hainan | Sanya | 109.27 | 18.17 |
| Austronesian | Atayalic | ATL | Atayal | 42 | China | Taiwan | Yilan | 121.74 | 24.76 |
| Austronesian | Paiwanic | AMI | Amis | 40 | China | Taiwan | Hualien | 121.60 | 23.98 |
| Austronesian | Malayo-Polynesian | MCR | Micronesians | 37 | Micronesia | 158.12 | 6.57 |
Note: *Those populations marked with stars are newly collected, and the triliteral codes are ISO639-3 codes [35]. Others are previously published populations with previously used codes [12]. San Francisco Chinese originally came from Jiangmen County, Guangdong, China; therefore, the location of CHS in the table is that of Jiangmen County.
Results
Distribution of ADH1B variants
We obtained the allele frequencies of 30 single nucleotide polymorphisms (SNPs) on the 24 new populations. The haplotype frequencies for all the 30 SNPs are given in Table S1 and allele frequencies are in ALFRED under the UIDs in Figure 2. The frequencies of the ADH1B functional variant, ADH1B*47His, and the variant at the ADH1B regulatory polymorphism rs3811801 (SNPs 9 and 11) were transformed into contour maps in Figure 1. The sharp borders across which the frequencies changed quickly are marked by bold lines in the map. The distributions of both variants show very clear clinal geographic patterns of east-west division. The sharp border for ADH1B*47His lies between the eastern part and western part of both the continental East and Southeast Asia, from the south end in Cambodia to the north end in Inner Mongol. The frequencies of ADH1B*47His are quite high to the east of this border. The rs3811801 derived allele shows a somewhat different distribution within the range of ADH1B*47His with a less sharp border and a similar higher frequency to the east.
Figure 2. Pattern of regions of high LD using HAPLOT and the default r2 algorithm.

Note: The codes of the non-East Asian populations are shown as NAS(Nasioi), KTY(Khanty), KMZ(Komi), FIN(Finns), SAM(Samaritans), DRU(Druze), ASH(Ashkenazi Jews). CN: New collected samples from China. LA: Newly collected samples from Laos. Both dbSNP numbers and ALFRED UID numbers are presented for the SNPs in the ADH region we typed.
Ethnically, the distributions of these two variants are also quite regular. The populations with high frequency of ADH1B*47His belong to TK, AU, SN, KJ, and HM. Those subphyla all have a long history of agriculture [40]–[42], while those populations with a low frequency of ADH1B*47His are all pastoral or hunting populations or began to farm recently. The TK and AU populations are excluded from among the populations with a high frequency of the rs3811801 (SNP 11) derived allele.
This non-synonymous SNP and the ADH1B promoter [44] polymorphism (SNP 11) have very high Fst values on a global scale [12]. The fixation index Fst, originally designed as the most inclusive measure of population substructure, is used here as a measure of allelic difference among the populations [10]. As the populations in East Asia are fairly similar to each other [45]–[47], the Fst values within the region are expected to be lower than those of the same SNPs globally. Using data on the East Asian populations listed in Table 1, we calculated the Fst values of all the SNPs as follows, numbered as in Figure 2: 1(.056), 2(.196), 3(.185), 4(.153), 5(.182), 6(.177), 7(.182), 8(.051), 9(.240), 10(.098), 11(.218), 12(.145), 13(.145), 14(.198), 15(.236), 16(.145), 17(.166), 18(.166), 19(.166), 20(.094), 21(.203), 22(.168), 23(.084), 24(.101), 25(.089), 26(.101), 27(.057), 28(.053), 29(.154), 30(.225). The Fst values among East Asian populations are much lower than the global values of the same SNPs, though the Fst of our focused SNPs 9 and 11 are still much higher than the global Fst mean of general SNPs (0.14). The Fst values of the Arg47His variant and the promoter variant of ADH1B are among the highest values, but we also see high values in the upstream region at ADH1C and even upstream of ADH1C.
Haplotypes and regions of high LD
Six-SNP haplotypes of the ADH1B upstream region (SNPs 6–11) were estimated and the haplotype frequencies of the Asian populations are displayed in Figure 3. The patterns of the East Asian populations are obviously different from the non-East Asian populations (Uralic and Afro-Asiatic). Haplotypes 1 and 2 have high frequencies in most of the East Asian populations, while they occurred at much lower frequency in ASH (Jews). The populations in the most southwest region of East Asia (WBM: Ava, KHG: Tibetan, and PHO: Phunoi Lolo) have the lowest frequencies of these two haplotypes.
Figure 3. Haplotype frequencies of the ADH1B gene region including the regulatory region.

Note: the SNPs in the haplotypes are rs2066701-rs2075633-rs4147536-rs1229984-6810842-rs3811801, corresponding to SNPs 6–11 in Figure 2. Phyla, PN: Papuan-New Guinean, AU: Austronesian, TK: Daic, AA: Austro-Asiatic, TB: Tibeto-Burman, HM: Hmong-Mien, SN: Sinitic Han, KJ: Korean-Japanese, AT: Altaic (inland), UR: Uralic, AF: Afro-Asiatic. The patterns of the non-East Asian phyla (UR, AF, PN) are quite different from those of East Asian phyla. The patterns of the phyla in East Asia can be classified into four groups as the colors shown in the left side bar. The frequency data for all haplotypes are in Table S1.
Among the East Asian populations, there are four types of haplotype patterns, and the classification shows an obvious ethnic correlation. The first group (Southeast) contains AU and TK, and haplotype 2 is the major haplotype of this group. The second group (Southwest) contains AA and TB, with the characteristic haplotype 3. Haplotype 1 has a frequency greater than 50% in the third group (Northeast) that contains HM, SN, and KJ. AT populations form the fourth group (Northwest). The haplotypes in group 4 are most diverse. Only haplotype 7 is a little richer than it is in the other groups. For those non-East Asian populations, the patterns also seem regular. Haplotypes 5 and 8 are frequent in Afro-Asiatic populations but absent in Uralic populations.
Regions of high LD across the whole region were displayed in Figure 2. The patterns of the ADH7 region and the region between Class I ADH and ADH7 (SNPs 17–30) are quite similar among the populations in East Asia, except for TB (BQH, QMR, KHG) with fewer regions of high LD and AU (ATL, AMI) with larger high LD regions. In contrast, the LD patterns of Class I ADH (SNPs 1–16) are quite different among the populations. It is interesting that only the Mongol (MVF) and Hmong (HMQ, MWW) have high LD extending to the region upstream of ADH1B (SNPs 9–11). In the two AU populations from Taiwan, high levels of LD encompass more SNPs than seen for the other populations; this pattern suggests they are quite young and/or have undergone considerable random genetic drift recently.
Population Comparison
A principal component analysis based on the ADH1B haplotype frequencies in Figure 3 was used to examine the data distribution among populations (Figure 4A). The first plot was constructed by principal components (PC) 1 and 2. In this plot, PC1 divides East Asian populations from non-East Asian populations. This confirms the overall genetic unity and distinctiveness of East Asia. In the second map PC2 divides the western part of East Asia from the eastern part with a sharp border. The distribution of PC2 is very similar to the distribution of ADH1B*47His, with the TK, HM, SN and KJ phyla in the east and the others in the west. The correlation between PC2 value and ADH1B*47His frequency is −0.971 (P<0.001), while the correlation between PC2 and longitude is −0.491 (P = 0.005). The distribution of PC3 is also quite geography-related. It divides southern phyla (TK, AA, AU) from the northern phyla. The correlation coefficient between PC3 and the latitude is −0.808 (P<0.001). Though PC2 and PC3 are significantly correlated with geography, they are more ethnic-related, as is shown in the second plot of Figure 4A. It is noticeable that those eastern populations such as LAO and MWW that have moved into the western area are still clustered with the eastern phyla. That suggests the distribution of this ADH1B haplotype (encompassing the 5′ half of the gene and the 5′ flanking region) must be related to the history of the ethnic phyla.
Figure 4. Principal Component Analysis plots.

Notes: The plots show the relationships among populations estimated by PCA. Plots in part A were based on the ADH1B haplotypes frequency data in Figure 3. Plots in part B used the haplotype frequency data of the whole ADH region in Figure 2. In part A, an ethnic related distribution is obvious, while in part B the distribution shows no strong distinct clusters corresponding to ethnicity.
To test if the ethnic-related distribution of ADH1B is common in the human genome, we applied principal component analysis (PCA) to the haplotype frequency data of the whole ADH region we typed. Results are shown in Figure 4B. The distribution of the PCs is much closer to the general genetic relationship (a supposed genetic relationship measured by the whole genome diversity) among populations than the ADH1B distribution. PC1 still divides East Asia from the rest of the world. Neither PC2 nor PC3 is related to ethnicities. The PCA result of the whole region is very different from that of the smaller ADH1B region. Therefore the ethnic-related distribution of the first PCA of ADH1B is uncommon. The distribution of the ADH1B upstream region diversity is related to ethnicity, but different from the distribution of the whole Class I ADH and ADH7 region diversity.
Positive Selection Test of ADH1B Core Haplotypes
As SNPs 9 to 11 are candidates for being related to function or having been positively selected, we chose this short region as the core for a test for selection. The allele frequencies of the core haplotypes are shown graphically in Figure 5A. The four-group classification of Figure 2 can still be observed in Figure 5A. There are eight haplotypes but only four are common in East Asia. Haplotype (2) is most common in the AU-TK group. Haplotype (3) reaches highest frequency in the HM-SN-KJ group. Uralic and Afro-Asiatic are obviously different. Though AT and Uralic phyla are neighbors and share much of their culture and customs, their haplotype patterns are quite different.
Figure 5. Frequencies and network of the core haplotypes (rs1229984-rs6810842-rs3811801).

Notes: Haplotype codes (1) to (8) are in the same system for both part A and part B of the figure. The sizes of the balls in the network of part B represent the rough relative frequencies of the haplotypes. The arrows are the most likely mutational relationships. The broken lines indicate possible historical recombinations. Haplotype (3) is more derived and presumably younger than haplotypes (1), (2), (4), and (6). Its high frequency in some populations suggests selection may have operated.
In Figure 5B, the relationships among the haplotypes are plotted in a network. Haplotype (1) is the ancestral haplotype based on sequence comparison with four other ape species. The functional mutation ADH1B*47His occurred in the step from (1) to (2). The promoter mutation occurred in the step from (2) to (3). The sizes of the circles in Figure 5B are roughly proportional to the haplotype frequencies in the different ethnic phyla. In the network, the frequencies of the younger haplotypes are expected to be lower. Actually, young haplotypes (5) to (8) are obviously less frequent than those old ones (1), (2) and (4). The only exception is haplotype (3) which is young but still reaches very high frequency in East Asia. Outside of East Asia, this haplotype is very rare. This indicates this haplotype has undergone strong genetic drift or positive selection in East Asia. This also focuses attention on the derived allele in the promoter region.
To test whether the high frequency of the young haplotype was caused by positive selection, we applied the EHH and REHH tests. The results are presented in Figure 6 and Figure S1 to scale along the chromosomal segment studied. If the EHH value decayed quickly or the REHH value did not rise to high levels with increasing distance from the core, there would be no evidence for selection. The EHH values of core haplotype (3), AGA, decayed more slowly than the other core haplotypes, indicating haplotype (3) may have been under positive selection. The REHH values of haplotype (3) also increased in the upstream direction. The REHH values of the Altaic populations including KJ were the highest among the ethnic phyla. The REHH values of two HM populations, BQH, CBD and SKB also rose to fairly high values. The high REHH values suggest the possible existence of positive selection; however, because of limitations of our sample sizes (∼50 individuals per population) only those core haplotypes of moderate frequency convey meaningful results. Some populations with high REHH values failed to have moderate haplotype frequencies, such as YAK, KAZ, CBD and SKB. Thus, we cannot draw a definite conclusion for these populations.
Figure 6. Extended Haplotype Homozygosity (EHH) and Relative Extended Haplotype Homozygosity(REHH) of Altaic, Han and Hmong populations.

Note: Colorful lines are data of core haplotype (3)AGA, and gray lines are data of other haplotypes. The data following the population codes are frequencies of the core haplotype in the populations. EHH and REHH of the other populations are in Figure S1.
We plotted all the REHH values for all core haplotypes at SNP 26 (around 90 kb from the core) in Figure 7A. In Altaic and some other populations, the REHH values of haplotype (3), AGA, were much higher than those of the other haplotypes. To test whether this haplotype might have been under positive selection in these populations, we simulated the population demography (Figure 7B). The population history model used in the simulation was based on the migration history reconstructed by other genetic studies (mtDNA [46] and Y-chromosome [47]) and the tremendous changes of the environment or the society in East Asia. The population size was at its lowest level at the peak of the ice age [48], and increased quickly when agriculture started. Archaeological evidence [41] confirms that agriculture started more than 8000 years ago in East Asia. If we assume a generation is about 20 years, agriculture started 400 generations ago. With this model, 10,000 populations were simulated, and the results of simulated REHH values were shown in Figure 7C together with the observed REHH values of haplotype AGA. Four lines (50, 75, 95 and 99 percentile) were drawn for visual comparison. All of the Altaic and HM populations are above the 95 percentile line. We also calculated the significance by a t-test after transforming our original result to achieve normality. The P values were transformed into the contour map in Figure 8A. There are 15 populations with a P value less than 0.05. The regions covered by these populations are marked in Figure 8B by a purple background. All of the Altaic populations and HM populations are included. Among four SN populations only HKA (Hakka) is excluded. BQH, SKB, CBD and KUF are also included. The sample size of CBD is rather small (25), and the core haplotype AGA frequency is low; therefore the REHH of CBD is not reliable. Fisher's Exact Test [49] on haplotype composition shows CBD can be combined with LBO, a nearby and similar population (P = 0.492). REHH of the combined population is 4.467, which suggests that the combined population shows no evidence of selection. Therefore the populations with significant evidence of selection are ethnic-specific with only four exceptions. The conclusion is that strong evidence of selection on the core haplotype AGA was found in Altaic and HM populations, and weak evidence of selection in SN.
Figure 7. REHH of observed and simulated populations.

Note: The colored dots are observed REHH data of core haplotype AGA both in chart A and C. In chart A, the observed REHH data shows that most of the REHH values of haplotype AGA are higher than those of the other haplotypes. Part B is the East Asian population history model determined by complicated factors. Six phases were defined with the effective population numbers and the generation numbers to present. Chart C indicates the REHH data simulated by the model in part B along with the data in Chart A. The lines in chart C are comparison borders of the simulated data. The observed REHH of haplotype AGA of all the Altaic and Hmong populations are above the 95% border, which is the evidence of positive selection.
Figure 8. Significance P values of the positive selection on the ADH1B gene in East Asia.

Note: The map of part A displayed the distribution of the significance P value of the positive selection on the ADH1B gene. Populations from most areas of East Asia have been significantly selected for except those in the southwest. In part B, the selection area was compared with the high frequency areas of two ADH1B SNPs. The dotted line encloses the region in which the ADH1B*47His frequency is >40%, and the broken line encloses the region in which the ADH1B rs3811801 frequency is >40%. The distributions of ADH1B*47His and selection area differ from each other, which indicates that they are unrelated. The high frequency area of rs3811801 derived allele is included in the selection area, indicating the possible effect of this allele in the selection on the ADH1B gene.
In the map of Figure 8, we can see that the geographic area with evidence of selection is quite different from the ADH1B*47His high frequency area. Correlation analyses were computed between the P values of the selection significance test and the derived allele frequencies of the SNPs in the core region. The results were SNP 9 (r = 0.100, P = 0.581), SNP 10 (r = 0.097, P = 0.590), SNP 11 (r = 0.522, P = 0.002). Only SNP 11 has significant P values for selection. SNP 11 (rs3811801) is located at −1761 bp, the promoter region of ADH1B [44]. Therefore the promoter region may be the actual region that has undergone selection. The alleles at this SNP may quantitatively alter ADH1B expression. However, since the derived allele at SNP 11 (rs3811801) occurs only on chromosomes with the ADH1B*47His allele, the cis-acting combination of the two alleles may have been the focus of selection.
Discussion
Population History and Ethnic-related Distribution of ADH1B
In all of the analyses based on the ADH1B region in this study, we saw ethnic relationships with allele and haplotype frequencies. Especially in Figure 4A of the PCA analysis, genetic clustering of populations matched very well with the ethnic phyla or subphyla. That is rather unusual, as we rarely see the distribution of alleles at a single autosomal locus matching finer structure of ethnic classification. There are many papers describing ethnic differences for certain loci, but those ethnic differences are among populations from different geographic regions of the world [50]–[52]. In our study on ADH1B, the eight ethnic phyla or subphyla included are all in East Asia. In the PCA, these eight groups can be distinguished very well by ADH1B data, but not by the whole Class I ADH and ADH7 region data. The ethnic-related patterns are only seen for the ADH1B region.
In East Asia, one hypothesis argues that most of the homologous populations in one ethnic phylum share a distinct common ancestor, which has some evidence from Y chromosome DNA [53]–[55]. As the paternal societies have lasted for a very long time in most of the East Asian populations, the Y chromosome had less chance to flow among populations than other chromosomes. Therefore the distribution of Y chromosome diversity can represent the original relationships among the ethnic phyla to a certain extent. On the other hand, two factors can change the ethnic-related genetic structure. Interactions among different ethnic phyla in East Asia have frequently occurred. Historical facts of interactions were often used to explain the different genetic structures revealed by autosomal [56] and mitochondrial [46] DNA diversity. At the same time, random genetic drift will change the frequency of a single locus in a certain population. Therefore most of the distributions of any single locus are not as ethnically related as the Y chromosome is. The ethnically related distribution of ADH1B cannot be explained only by random genetic drift. There can be two possible explanations. One is that some culture related incidence has maintained the special distribution. Another possible explanation is that different ADH1B haplotype patterns occurred at the founding of the different ethnic phyla and remained largely unchanged during population expansions and interactions.
A number of Y chromosome diversity studies showed that TB, SN, TK and AU phyla could be recognized by some ethnicity-specific haplogroups [53], [54], [57]. TB and SN subphyla belong to Sino-Tibetan phyla, and they both originated from the upper Yellow River area and shared a common ancestor [53]. TK and AU are two different ethnic phyla, but a great deal of evidence showed that they are very similar to each other [58]. Y-chromosome data also supports a common origin of TK and AU [57]. In our ADH1B data, TB and SN departed from each other, while TK and AU clustered closely. ADH1B data do not always match the hypothesized original relationships. Therefore the ethnic-related structure of ADH1B might not always be caused by the original differentiations of the ethnic phyla. Some cultural factors such as life styles may have influenced the special distribution of ADH1B, especially in the Sino-Tibetan phylum. Weak evidence of selection was seen in the SN subphylum. That implies the ADH1B haplotype pattern in SN may be due to selection, and the selective forces on ADH1B may be something like ethnic culture. Therefore, the high frequency of ADH1B*47His in TK and AU may have resulted from the original population differentiation, while that in SN may have resulted from the positive selection.
Cultural or Natural Force in the Selection of ADH1B Region
The frequencies of ADH1B*47His are high in AU, TK, HM, SN, and KJ, but ADH1B haplotype patterns of the AU-TK group are different from those of the other three subphyla. According to Figure 5, the haplotype AGG is responsible for the high frequency of ADH1B*47His in AU-TK group, while it was haplotype AGA in HM-SN-KJ group. And from the selection analyses only AGA showed evidence of having been selected. Therefore, we conclude that the high frequency of haplotype AGG in the AU-TK group most probably did not result from positive selection, but from the random drift that occurred in their common ancestral population. The populations in AU-TK group may have maintained the high frequency of haplotype AGG of their common ancestor since they diverged.
If we check the haplotype patterns of TB and SN, two members of the Sino-Tibetan phylum, in Figure 5, some similarities between them can be found. The AGA haplotype frequency with respect to the AGG haplotype frequency is high in each population. According to the history of Han Chinese, they derived from ancient Qiang population several thousand years ago [59]. QMR (modern Qiang) may be a very old population. There are even several individuals in QMR with the ATA haplotype that would need a quite long time to appear by rare recombination events. Therefore the haplotype pattern of QMR may be most similar to that of the Sino-Tibetan ancestor. The lower haplotype frequencies of AGG and AGA in the other TB populations might result from genetic drift, and the increase in SN might result from positive selection for which we found weak evidence. The TB and SN diverged more than 5000 years ago [53]. The SN populations moved to the east and started agriculture on the plain. Both the culture and environment changed. It is not easy to determine what might have been the selective force on the Sino-Tibetan populations.
Weakly or strongly, all of the Altaic (including AT and KJ) populations show evidence of selection. The ratio of the haplotype AGA frequency with respect to AGG in Altaic is highest among the ethnic phyla. This might be a characteristic of the original Altaic population. The highest significance level for the selection significance tests appeared in three eastern populations, MVF, KOR and JPN, but we are unaware of any social or environmental similarities among these populations. KOR and JPN are agriculture populations living by the sea, while MVF is a pastoral population on the highlands. As for the other populations with evidence of positive selection, HM phylum, KUF and SKB in the Southeast Asia, the environments and cultures are even more different from the Altaic phylum. Judging from the distribution of the “selected” populations, it is clear that climate and other environmental aspects differ. Therefore it is difficult to determine how nature has influenced the allele frequency. As the distribution is ethnic-related in some extent, some cultural force of selection is understandable, though we cannot determine the common cultural factors among the populations showing the strong evidence of selection. Perhaps cultural anthropologists will shed light on this problem.
The Selective Region and Allele
The distribution of populations showing evidence of selection at ADH1B is quite different from the high frequency distribution of the ADH1B*47His allele. The high significance levels for selection are only correlated with allele frequencies of the promoter SNP rs3811801, indicating that the ADH1B*47His allele has not been selected for in the absence of the derived promoter allele. Evolutionarily, the derived promoter allele at SNP 11, rs3811801, occurred on a chromosome with the ADH1B*47His allele; thus, the derived promoter allele appears together with the ADH1B*47His allele in most cases. Because the selected core haplotype AGA includes two SNPs with derived alleles, both with likely functional consequences, we cannot be sure if ADH1B*47His has been important in the positive selection on the ADH1B region. The core haplotypes with the promoter derived allele but without ADH1B*47His are very rare; therefore the sample is too small to test for positive selection. We can speculate that it is the combined effect of an increased activity of the enzyme caused by the ADH1B*47His allele and an increased quantity of the enzyme caused by the derived promoter allele. However, it is possible that the promoter region variant has no effect but is simply associated by chance with that chromosome that underwent selection in specific ethnic populations.
In previous case-control studies, it was found that the ADH1B*47His allele decreased the risk for alcoholism both in Asian and European populations. In the Asian populations [21]–[25] included in the alcoholism correlation studies, both derived allele frequencies of ADH1B*47His and the promoter polymorphism are high. But in the European populations [26], [27], [29], the promoter derived allele does not appear together with ADH1B*47His, implying that ADH1B*47His is solely responsible for the decrease in the risk for alcoholism. Others report that ADH1B*47His was not always related to alcoholism, especially in Taiwanese populations [32], [33], in which the frequency of the ADH1B promoter derived allele is very low as we revealed in our study. Some papers [28] also doubted the significance of the association between ADH1B*47His and alcoholism in European populations as the frequency of the ADH1B*47His is also low. Therefore the ADH1B*47His allele alone may not be important in changing the genetic structure of the populations.
However the decrease in the risk for alcoholism has not been argued to be the selective force, and our results argue that selection is not solely related to ADH1B*47His. The derived promoter allele may have led to the increase of the ADH enzyme expression, and then enhance the protection against some deleterious effects. The ADH variants are also related to some types of cancers and other serious diseases [4], [60]–[66]. Infectious disease is one of the plausible selective forces suggested by Goldman & Enoch [67]. Other diseases such as food poisoning can also have similar effects, and the populations will be susceptible if they happen to be partial to certain foods. This kind of pathology was reported in some populations in China [68]. In the case of ADH selection, Altaic and Hmong populations may have special food or other customs that increase the risk of a certain disease. The enhancement of the ADH enzyme caused by the ADH1B*47His may not be enough to protect the individuals from that disease, and an increased enzyme activity caused by the derived allele of the promoter variant would then be helpful. Our hypothesis is that ADH1B*47His can enhance the activity of ADH enzyme and the derived allele of the promoter variant of ADH1B can increase the quantity of the enzyme in the body; thus the protection against some related diseases will be stronger and the ADH1B haplotype will be selected. However, more case-control studies including both of the derived alleles are required. And we suggest these studies be conducted in the Daic or Austronesian populations, because there are more types of ADH1B core haplotypes and the linkage disequilibrium is weaker. Therefore, a false signal caused by the strong linkage disequilibrium can be avoided.
Materials and Methods
Populations
We collected samples of 24 populations (populations with stars behind their code in Table 1 and marked by colorful icons in Figure 1B) from six ethnic phyla. Among these populations, nine were collected from Laos, and 15 were from China. This is the first population genetics study in Laos. Laos is an inland country surrounded by all the other nations in the Peninsula. All types of populations in Peninsular Southeast Asia can be found in Laos. All individuals we collected are healthy adults without alcoholism or related disorders. Everyone signed the informed consent. Our study was approved by the Ethics Committee of the Chinese National Human Genome Center at Shanghai and the Human Investigations Committee at Yale University School of Medicine, and approved by the Laos government. The sample sizes are all large enough for meaningful frequency and haplotype estimates [69]. The population locations are also indicated in Table 1 by both county names and geographic coordinates, and also shown in the map of Figure 1B.
Table 1 also lists the information of populations previously studied [12] to give an overview of the ADH study in East Asia. These population samples are categorized both ethnically and geographically. Each phylum contains 2∼7 populations. HM is the smallest phylum, represented in our data by only two populations.
Sample Preparation
For the new populations blood samples were collected from finger tips of participants and kept dry on filter paper. A whole genome amplification (WGA) [70] was applied for each sample. A 3 mm2 piece of paper with blood was cut off and dipped into 5 µl water, lysed with 5 µl of alkaline lysis solution (400 mM KOH, 100 mM DTT, 10 mM EDTA) and incubated for 10 min on ice. The lysed liquid was neutralized with 5 µl neutralization solution(1 M HCl∶1 M Tris-HCl = 2∶3, pH 0.6). The neutralized mixture was used directly as template in WGA. The WGA cocktail is water 31.5 µl, 10×MDA buffer [Tris-HCl pH7.5 375 mM, KCl 500 mM, MgCl2 100 mM, (NH4)2SO4 50 mM] 5 µl, 10 mM dNTP 5 µl, 2 mM thiophosphate-modified random hexamer (NNNN*N*N, where* denotes a phosphorothioate) 1.25 µl, 2 mg/ml Rnase A 0.25 µl, template mixture 5 µl, 200 ng/µl φ29 polymerase 2 µl. Cocktail was mixed well and incubated at 31°C for 15 hours. After the reaction, the polymerase activity was inactivated at 75°C for 5 min. The product was spun down to get rid of the sediment, and the produced DNA was purified by alcohol precipitation before TaqMan reactions.
Markers
We chose 30 SNPs from the ADH cluster, covering Class I ADH (encompassing the ADH1B Arg47His polymorphism) and ADH7. These 30 SNPs have high heterozygosity in East Asia [71]. The dbSNP and ALFRED numbers of each SNP from the ADH clusters are listed in Figure 2. All SNPs were genotyped by the TaqMan method [72] using commercial assays and reagents (http://products.appliedbiosystems.com) except for the assay for ADH1B Arg47His, which we designed [30].
Analysis Methods
Allele frequencies were calculated directly by gene counting assuming two-allele codominant inheritance. Fst values [73] are calculated as 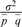 . Our Fst values were calculated across all 32 East Asian populations. Haplotypes of the 30 SNPs in ADH genes were estimated by PHASE2.1 [74], [75]. The population-specific patterns of regions of high LD were calculated and graphed (Figure 2) using HAPLOT [76] and the default Kidd r2 partition algorithm. The PCA [77] was initially applied based on the ADH1B haplotype frequency of 6 SNPs (rs2066701-rs2075633-rs4147536-rs1229984-rs6810842-rs3811801) centered on ADH1B Arg47His by SPSS13.0. Another PCA was based on frequencies of all available haplotypes of all the 30 SNPs we typed. The second PCA was expected to show the general population relationships with less functional effects, while the first one was supposed to be affected by the functional variant. SNP allele frequency and PCA maps were presented by Surfer8.0 .
. Our Fst values were calculated across all 32 East Asian populations. Haplotypes of the 30 SNPs in ADH genes were estimated by PHASE2.1 [74], [75]. The population-specific patterns of regions of high LD were calculated and graphed (Figure 2) using HAPLOT [76] and the default Kidd r2 partition algorithm. The PCA [77] was initially applied based on the ADH1B haplotype frequency of 6 SNPs (rs2066701-rs2075633-rs4147536-rs1229984-rs6810842-rs3811801) centered on ADH1B Arg47His by SPSS13.0. Another PCA was based on frequencies of all available haplotypes of all the 30 SNPs we typed. The second PCA was expected to show the general population relationships with less functional effects, while the first one was supposed to be affected by the functional variant. SNP allele frequency and PCA maps were presented by Surfer8.0 .
Analyses of empirical data suggest positive selection has operated on the ADH Class I cluster in East Asian populations [12] . Therefore, the Long Range Haplotype analysis [78] has been applied to test for a potential positive selection effect on these new populations. Pilot studies suggested that SNPs (9∼11: rs1229984-rs6810842-rs3811801) showed a signature of positive selection and therefore these three SNPs were selected as the core region. We used two values of Long Range Haplotype analysis to measure the selection, EHH and REHH [79].
EHH is defined as the probability that two randomly chosen chromosomes carrying a tested core haplotype are homozygous at all SNPs for the entire interval from the core region to the distance x. REHH is defined as the ratio of the EHH of the tested core haplotype to the EHH of the grouped set of core haplotypes at the region not including the tested core haplotype. In implementation, EHH is calculated as
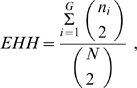 |
(1) |
where N is the total number of chromosomes/haplotype sequences, and G is the number of homozygous groups, with each group i having ni elements. If there are M chromosome groups, each with Ci chromosomes and an EHH value of EHHi, REHH is calculated as
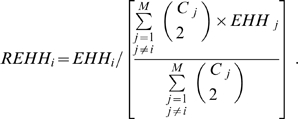 |
(2) |
Generally speaking, if a certain core haplotype with moderate to high frequency shows high EHH and REHH over a long distance, it will be considered as an effect of positive selection [78]–[80]. The REHH value of this core haplotype was further compared with numerous data points generated by simulation that assumes neutral evolution [81]. A pilot and cursory population history mode in our simulation was designed according to the migration history (out of Africa around 50,000 years ago and arrived in East Asia around 20,000 years ago), archaeological discoveries (agriculture stared in East Asia around 8,000 ago, and iron tools were used around 2,500 years ago) and the environmental changes (the Last Ice Age ended around 12,000 years ago) in East Asia [41], [52]. The simulated data were logarithmicly transformed to achieve normality for a T-test with REHH values of our core haplotype.
The network of the core region haplotypes was drawn by NETWORK4.201 [82]. The evolutionary relationships among the haplotypes were determined from the network given the identity of the ancestral allele.
Supporting Information
Haplotype frequencies of the ADH region in Asian populations
(0.73 MB XLS)
Extended Haplotype Homozygosity (EHH) and Relative Extended Haplotype Homozygosity(REHH) of southwest populations in East Asia. Note: Colorful lines are data of core haplotype (3)AGA, and gray lines are data of other haplotypes. The data following the population codes are frequencies of the core haplotype in the populations.
(0.31 MB TIF)
Acknowledgments
We especially thank the many individuals who volunteered to provide samples for this study. We also thank the many individuals who assisted in assembling the collection of population samples.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This research was funded primarily by US NIH grant AA009379 to KKK, and in part by USPHS grant GM057672 and NSF grant BCS0096588.
References
- 1.Agarwal DP, Goedde HW. Pharmacogenetics of alcohol metabolism and alcoholism. Pharmacogenetics. 1992;2:48–62. doi: 10.1097/00008571-199204000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Ramchandani VA, Bosron WF, Li TK. Research advances in ethanol metabolism. Pathol Biol (Paris) 2001;49:676–682. doi: 10.1016/s0369-8114(01)00232-2. [DOI] [PubMed] [Google Scholar]
- 3.Yoshida A, Hsu LC, Yasunami M. Genetics of human alcohol-metabolizing enzymes. Prog Nucleic Acid Res Mol Biol. 1991;40:255–287. doi: 10.1016/s0079-6603(08)60844-2. [DOI] [PubMed] [Google Scholar]
- 4.Triano EA, Slusher LB, Atkins TA, Beneski JT, Gestl SA, et al. Class I Alcohol Dehydorgenase is highly expressed in normal human mammary epithelium but not in invasive breast cancer: Implications for breast carcinogenesis. Cancer Research. 2003;63:3092–3100. [PubMed] [Google Scholar]
- 5.Dodd PR, Foley PF, Buckley ST, Eckert AL, Innes DJ. Genes and gene expression in the brain of the alcoholic. Addict Behav. 2004;29:1295–1309. doi: 10.1016/j.addbeh.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 6.Cheung C, Smith CK, Hoog JO, Hotchkiss SA. Expression and localization of human alcohol and aldehyde dehydrogenase enzymes in skin. Biochem Biophys Res Commun. 1999;261:100–107. doi: 10.1006/bbrc.1999.0943. [DOI] [PubMed] [Google Scholar]
- 7.Yao CT, Liao CS, Yin SJ. Human hepatic alcohol and aldehyde dehydrogenases: genetic polymorphism and activities. Proc Natl Sci Counc Repub China B. 1997;21(3):106–111. [PubMed] [Google Scholar]
- 8.Yin SJ, Liao CS, Wu CW, Li TT, Chen LL, et al. Human stomach alcohol and aldehyde dehydrogenases: comparison of expression pattern and activities in alimentary tract. Gastroenterology. 1997;112:766–775. doi: 10.1053/gast.1997.v112.pm9041238. [DOI] [PubMed] [Google Scholar]
- 9.HUGO Gene Nomenclature Committee. 2001. http://www.gene.ucl.ac.uk/nomenclature/ [Google Scholar]
- 10.Osier MV, Pakstis AJ, Soodyall H, Comas D, Goldman D, et al. A global perspective on genetic variation at the ADH genes reveals unusual patterns of linkage disequilibrium and diversity. Am J Hum Genet. 2002;71:84–99. doi: 10.1086/341290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edenberg HJ, Xuei X, Chen HJ, Tian H, Wetherill LF, et al. Association of alcohol dehydrogenase genes with alcohol dependence: a comprehensive analysis. Hum Mol Genet. 2006;15:1539–1549. doi: 10.1093/hmg/ddl073. [DOI] [PubMed] [Google Scholar]
- 12.Han Y, Gu S, Oota H, Osier MV, Pakstis AJ, et al. Evidence of Positive Selection on a Class I ADH Locus. Am J Hum Genet. 2007;80:441–456. doi: 10.1086/512485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eriksson CJ, Fukunaga T, Sarkola T, Chen WJ, Chen CC, et al. Functional relevance of human adh polymorphism. Alcohol Clin Exp Res. 2001;(5 Suppl ISBRA):157S–163S. doi: 10.1097/00000374-200105051-00027. [DOI] [PubMed] [Google Scholar]
- 14.Matsuo K, Hiraki A, Hirose K, Ito H, Suzuki T, et al. Impact of the Alcohol-Dehydrogenase (ADH) 1C and ADH1B polymorphisms on drinking behavior in nonalcoholic Japanese. Hum Mutat. 2007;28:506–510. doi: 10.1002/humu.20477. [DOI] [PubMed] [Google Scholar]
- 15.Mulligan CJ, Robin RW, Osier MV, Sambuughin N, Goldfarb LG, et al. Allelic variation at alcohol metabolism genes (ADH1B, ADH1C, ALDH2) and alcohol dependence in an American Indian population. Hum Genet. 2003;113:325–336. doi: 10.1007/s00439-003-0971-z. [DOI] [PubMed] [Google Scholar]
- 16.Chen CC, Lu RB, Chen YC, Wang MF, Chang YC, et al. Interaction between the functional polymorphisms of the alcohol-metabolism genes in protection against alcoholism. Am J Hum Genet. 1999;65:795–807. doi: 10.1086/302540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reich T, Edenberg HJ, Goate A, Williams JT, Rice JP, et al. Genome-wide search for genes affecting the risk for alcohol dependence. Am J Med Genet. 1998;81:207–215. [PubMed] [Google Scholar]
- 18.Saccone NL, Kwon JM, Corbett J, Goate A, Rochberg N, et al. A genome screen of maximum number of drinks as an alcoholism phenotype. Am J Med Genet B Neuropsychiatr Genet. 2001;96:632–637. doi: 10.1002/1096-8628(20001009)96:5<632::aid-ajmg8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 19.Long JC, Knowler WC, Hanson RL, Robin RW, Urbanek M, et al. Evidence for genetic linkage to alcohol dependence on chromosomes 4 and 11 from an autosome-wide scan in an American Indian population. Am J Med Genet. 1998;81:216–221. doi: 10.1002/(sici)1096-8628(19980508)81:3<216::aid-ajmg2>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 20.Uhl GR. Molecular genetic underpinnings of human substance abuse vulnerability: likely contributions to understanding addiction as a mnemonic process. Neuropharmacology. 2004;47(Suppl 1):140–147. doi: 10.1016/j.neuropharm.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 21.Thomasson HR, Crabb DW, Edenberg HJ, Li TK, Hwu HG, et al. Low frequency of the ADH2*2 allele among Atayal natives of Taiwan with alcohol use disorders. Alcohol Clin Exp Res. 1994;18:640–643. doi: 10.1111/j.1530-0277.1994.tb00923.x. [DOI] [PubMed] [Google Scholar]
- 22.Muramatsu T, Wang ZC, Fang YR, Hu KB, Yan H, et al. Alcohol and aldehyde dehydrogenase genotypes and drinking behavior of Chinese living in Shanghai. Hum Genet. 1995;96:151–154. doi: 10.1007/BF00207371. [DOI] [PubMed] [Google Scholar]
- 23.Higuchi S, Matsushita S, Murayama M, Takagi S, Hayashida M. Alcohol and aldehyde dehydrogenase polymorphisms and the risk for alcoholism. Am J Psychiatry. 1995;152:1219–1221. doi: 10.1176/ajp.152.8.1219. [DOI] [PubMed] [Google Scholar]
- 24.Shen YC, Fan JH, Edenberg HJ, Li TK, Cui YH, et al. Polymorphism of ADH and ALDH genes among four ethnic groups in China and effects upon the risk for alcoholism. Alcohol Clin Exp Res. 1997;21:1272–1277. [PubMed] [Google Scholar]
- 25.Osier M, Pakstis AJ, Kidd JR, Lee JF, Yin SJ, et al. Linkage disequilibrium at the ADH2 and ADH3 loci and risk of alcoholism. Am J Hum Genet. 1999;64:1147–1157. doi: 10.1086/302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borras E, Coutelle C, Rosell A, Fernandez-Muixi F, Broch M, et al. Genetic polymorphism of alcohol dehydrogenase in europeans: the ADH2*2 allele decreases the risk for alcoholism and is associated with ADH3*1. Hepatology. 2000;31:984–989. doi: 10.1053/he.2000.5978. [DOI] [PubMed] [Google Scholar]
- 27.Neumark YD, Friedlander Y, Thomasson HR, Li TK. Association of the ADH2*2 allele with reduced ethanol consumption in Jewish men in Israel: a pilot study. J Stud Alcohol. 1998;59:133–139. doi: 10.15288/jsa.1998.59.133. [DOI] [PubMed] [Google Scholar]
- 28.Hasin D, Aharonovich E, Liu XH, Mamman Z, Matseoane K, et al. Alcohol and ADH2 in Israel: Ashkenazis, Sephardics, and Recent Russian Immigrants. Am J Psychiatry. 2002;159:1432–1434. doi: 10.1176/appi.ajp.159.8.1432. [DOI] [PubMed] [Google Scholar]
- 29.Whitfield JB, Nightingale BN, Bucholz KK, Madden PA, Heath AC, et al. ADH genotypes and alcohol use and dependence in Europeans. Alcohol Clin Exp Res. 1998;22:1463–1469. [PubMed] [Google Scholar]
- 30.Li H, Mukherjee N, Soundararajan U, Tárnok Z, Barta C, et al. Geographically Separate Increases in the Frequency of the Derived ADH1B*47His Allele in Eastern and Western Asia. Am J Hum Genet. 2007;81:842–846. doi: 10.1086/521201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goedde HW, Agarwal DP, Fritze G, Meier-Tackmann D, Singh S, et al. Distribution of ADH2 and ALDH2 genotypes in different populations. Hum Genet. 1992;88:344–346. doi: 10.1007/BF00197271. [DOI] [PubMed] [Google Scholar]
- 32.Chen WJ, Loh EW, Hsu YP, Chen CC, Yu JM, et al. Alcohol-metabolising genes and alcoholism among Taiwanese Han men: independent effect of ADH2, ADH3 and ALDH2. Br J Psychiatry. 1996;168:762–767. doi: 10.1192/bjp.168.6.762. [DOI] [PubMed] [Google Scholar]
- 33.Chen WJ, Loh EW, Hsu YP, Cheng AT. Alcohol dehydrogenase and aldehyde dehydrogenase genotypes and alcoholism among Taiwanese aborigines. Biol Psychiatry. 1997;41:703–709. doi: 10.1016/S0006-3223(96)00072-8. [DOI] [PubMed] [Google Scholar]
- 34.Ma LL, Xue YL, Liu Y, Wang Z, Cui XB, et al. Polymorphism study of seven SNPs at ADH genes in 15 Chinese populations. Hereditas. 2005;142:103–111. doi: 10.1111/j.1601-5223.2005.01910.x. [DOI] [PubMed] [Google Scholar]
- 35.Gordon RG., Jr . Ethnologue: Languages of the World, Fifteenth edition. Dallas, Tex.: SIL International; 2005. Web version: http://www.ethnologue.com/. [Google Scholar]
- 36.Sinor D. Cambridge: Cambridge University Press; 1990. The Cambridge History of Early Inner Asia. [Google Scholar]
- 37.Roemer HR. Berlin: Klaus Schwarz Verlag; 2000. History of the Turkic Peoples in the Pre-Islamic Period. [Google Scholar]
- 38.Mackerras C. Canberra: Australian National University Press; 1972. The Uighur Empire According to the T'ang Dynastic Histories. [Google Scholar]
- 39.Liu ZX. Beijing: Minzu chubanshe; 1985. Weiwuerzu lishi:shang bian (History of the Uighur: Vol. 1). [Google Scholar]
- 40.Weng QH. A discussion of rice-growing origin in the Nanling Mountains, South China. Tropical Geography. 1998;18(1):72–80. [Google Scholar]
- 41.Gu WN. Agricultural Archeology in China its research development and main results. Agricultural Archaeology. 2001;21(1):1–16. [Google Scholar]
- 42.Tong EZ. Outlines of some topics on the origin of agriculture in Southeast Asia and South China. Agricultural Archaeology. 1984;4(2):29–38. [Google Scholar]
- 43.Baker C, Phongpaichit P. Bangkok: Chulalongkorn University; 2005. A History of Thailand. [Google Scholar]
- 44.Dannenberg LO, Chen HJ, Tian HJ, Edenberg HJ. Differential Regulation of the Alcohol Dehydrogenase 1B (ADH1B) and ADH1C Genes by DNA Methylation and Histone Deacetylation. Alcohol Clin Exp Res. 2006;30:928–937. doi: 10.1111/j.1530-0277.2006.00107.x. [DOI] [PubMed] [Google Scholar]
- 45.Tishkoff SA, Kidd KK. Implications of biogeography of human populations for ‘race’ and medicine. Nat Genet. 2004;36(11 Suppl):S21–7. doi: 10.1038/ng1438. [DOI] [PubMed] [Google Scholar]
- 46.Kong QP, Yao YG, Sun C, Bandelt HJ, Zhu CL, et al. Phylogeny of East Asian mitochondrial DNA lineages inferred from complete sequences. Am J Hum Genet. 2003;73:671–676. doi: 10.1086/377718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin L, Su B. Natives or immigrants: origin and migrations of modern humans in East Asia. Nat Rev Genet. 2000;1:126–133. doi: 10.1038/35038565. [DOI] [PubMed] [Google Scholar]
- 48.Shi YF, Cui ZJ, Li JJ. Beijing: Science Press; 1989. Quaternary glacier in Eastern China and the climate fluctuation. [Google Scholar]
- 49.Raymond M, Rousset F. An exact test for population differentiation. Evolution. 1995;49:1280–1283. doi: 10.1111/j.1558-5646.1995.tb04456.x. [DOI] [PubMed] [Google Scholar]
- 50.Inoue K, Asao T, Shimada T. Ethnic-related differences in the frequency distribution of genetic polymorphisms in the CYP1A1 and CYP1B1 genes in Japanese and Caucasian populations. Xenobiotica. 2000;30:285–295. doi: 10.1080/004982500237677. [DOI] [PubMed] [Google Scholar]
- 51.Charlton KE, Steyn K, Levitt NS, Zulu JV, Jonathan D, et al. Ethnic differences in intake and excretion of sodium, potassium, calcium and magnesium in South Africans. Eur J Cardiovasc Prev Rehabil. 2005;12:355–362. doi: 10.1097/01.hjr.0000170265.22938.d1. [DOI] [PubMed] [Google Scholar]
- 52.Issell BF, Maskarinec G, Pagano I, Gotay CC. Breast cancer treatment among women of different ethnicity in Hawaii. Cancer Invest. 2005;23:497–504. doi: 10.1080/07357900500201442. [DOI] [PubMed] [Google Scholar]
- 53.Su B, Xiao C, Deka R, Seielstad MT, Kangwanpong D, et al. Y chromosome haplotypes reveal prehistorical migrations to the Himalayas. Human Genetics. 2000;107:582–590. doi: 10.1007/s004390000406. [DOI] [PubMed] [Google Scholar]
- 54.Wen B, Li H, Lu DR, Song XF, Zhang F, et al. Genetic evidence supports demic diffusion of Han culture. Nature. 2004;431:302–305. doi: 10.1038/nature02878. [DOI] [PubMed] [Google Scholar]
- 55.Tajima A, Pan IH, Fucharoen G, Fucharoen S, Matsuo M, et al. Three major lineages of Asian Y chromosomes: implications for the peopling of east and southeast Asia. Hum Genet. 2002;110:80–88. doi: 10.1007/s00439-001-0651-9. [DOI] [PubMed] [Google Scholar]
- 56.Kim J, Verdu P, Pakstis AJ, Speed WC, Kidd JR, et al. Use of autosomal loci for clustering individuals and populations of East Asian origin. Hum Genet. 2005;117:511–519. doi: 10.1007/s00439-005-1334-8. [DOI] [PubMed] [Google Scholar]
- 57.Li H, Huang Y, Mustavich LF, Zhang F, Tan JZ, et al. Y chromosomes of Prehistoric People along the Yangtze River. Hum Genet. 2007;122:383–388. doi: 10.1007/s00439-007-0407-2. [DOI] [PubMed] [Google Scholar]
- 58.He P. On the Origin of Ethnic Groups Speaking Austronesian Languages and their Historical Relationship with the Ethnic Groups in South China. J Yunnan University for Nationalities. 2003;20:45–48. [Google Scholar]
- 59.Wang MK. Taipei: The Institute of Ethnology, Academia Sinica; 1999. Primordial History: Brothers Stories of the Qiang. Time, Memory and History. [Google Scholar]
- 60.Jia C, Liu T, Liu Z, Li M, Hu M. Joint effects of eNOS gene T-786C and ADH2 Arg47His polymorphisms on the risk of premature coronary artery disease. Thromb Res. 2007;120:679–684. doi: 10.1016/j.thromres.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 61.Seitz HK, Maurer B, Stickel F. Alcohol consumption and cancer of the gastrointestinal tract. Dig Dis. 2005;23:297–303. doi: 10.1159/000090177. [DOI] [PubMed] [Google Scholar]
- 62.Yokoyama A, Omori T. Genetic polymorphisms of alcohol and aldehyde dehydrogenases and risk for esophageal and head and neck cancers. Jpn J Clin Oncol. 2003;33:111–121. doi: 10.1093/jjco/hyg026. [DOI] [PubMed] [Google Scholar]
- 63.Homann N, Stickel F, Konig IR, Jacobs A, Junghanns K, et al. Alcohol dehydrogenase 1C*1 allele is a genetic marker for alcohol-associated cancer in heavy drinkers. Int J Cancer. 2006;118:1998–2002. doi: 10.1002/ijc.21583. [DOI] [PubMed] [Google Scholar]
- 64.Peters ES, McClean MD, Liu M, Eisen EA, Mueller N, et al. The ADH1C polymorphism modifies the risk of squamous cell carcinoma of the head and neck associated with alcohol and tobacco use. Cancer Epidemiol Biomarkers Prev. 2005;14:476–482. doi: 10.1158/1055-9965.EPI-04-0431. [DOI] [PubMed] [Google Scholar]
- 65.Tiemersma EW, Wark PA, Ocke MC, Bunschoten A, Otten MH, et al. Alcohol consumption, alcohol dehydrogenase 3 polymorphism, and colorectal adenomas. Cancer Epidemiol Biomarkers Prev. 2003;12:419–425. [PubMed] [Google Scholar]
- 66.Cichoz-Lach H, Partycka J, Nesina I, Celinski K, Slomka M, et al. Genetic polymorphism of alcohol dehydrogenase 3 in alcohol liver cirrhosis and in alcohol chronic pancreatitis. Alcohol Alcohol. 2006;41:14–17. doi: 10.1093/alcalc/agh225. [DOI] [PubMed] [Google Scholar]
- 67.Goldman D, Enoch MA. Genetic epidemiology of ethanol metabolic enzymes: a role for selection. World Rev Nutr Diet. 1990;63:143–160. doi: 10.1159/000418505. [DOI] [PubMed] [Google Scholar]
- 68.Chen JG, Zhu J, Parkin DM, Zhang YH, Lu JH, et al. Trends in the incidence of cancer in Qidong, China, 1978-2002. Int J Cancer. 2006;119:1447–1454. doi: 10.1002/ijc.21952. [DOI] [PubMed] [Google Scholar]
- 69.Fallin D, Schork NJ. Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am J Hum Genet. 2000;67:947–959. doi: 10.1086/303069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99:5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.The International HapMap Consortium. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 72.Livak KJ, Marmaro J, Todd JA. Towards fully automated genome-wide polymorphism screening. Nat Genet. 1995;9:341–342. doi: 10.1038/ng0495-341. [DOI] [PubMed] [Google Scholar]
- 73.Wright S. Evolution in Mendelian populations. Genetics. 1931;16:97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gu S, Pakstis AJ, Kidd KK. HAPLOT: a graphical comparison of haplotype blocks, tagSNP sets and SNP variation for multiple populations. Bioinformatics. 2005;21:3938–3939. doi: 10.1093/bioinformatics/bti649. [DOI] [PubMed] [Google Scholar]
- 77.Jöreskog KG. Factor analysis by least-square and maximum-likelihood method. In: Enslein K, Ralston A, Wilf RS, editors. Statistical Methods for Digital Computers, volume 3. New York: John Wiley & Sons, Inc; 1977. [Google Scholar]
- 78.Endo T, Ikeo K, Gojobori T. Large-scale search for genes on which positive selection may operate. Mol Biol Evol. 1996;13:685–690. doi: 10.1093/oxfordjournals.molbev.a025629. [DOI] [PubMed] [Google Scholar]
- 79.Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4(3):e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 82.Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Haplotype frequencies of the ADH region in Asian populations
(0.73 MB XLS)
Extended Haplotype Homozygosity (EHH) and Relative Extended Haplotype Homozygosity(REHH) of southwest populations in East Asia. Note: Colorful lines are data of core haplotype (3)AGA, and gray lines are data of other haplotypes. The data following the population codes are frequencies of the core haplotype in the populations.
(0.31 MB TIF)