
Figure 1.
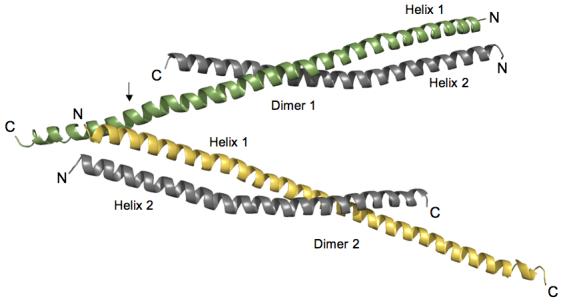
Orientation of HIP1 371-481 dimers in the crystal stabilizes the C-terminal region. Two dimers of HIP1 371-481 are arranged in an antiparallel “A” configuration. Helix 1 (green in Dimer 1) in each dimer shown is longer than Helix 2 (grey in Dimer 1) because of a crystal packing contact indicated by the arrow. At this point R380 and Y381 in Helix 1 of Dimer 2 (yellow) forms a pocket for D446 protruding from Helix 1 of Dimer 1 (green). The indicated lattice contact point faces away from the dimerization interfaces in the two dimers shown in the figure. The N- and C-termini of each helix are labeled N and C. The helices were rendered using PyMol [http://www.pymol.org].
Protein Expression and Purification: The plasmid encoding the N-terminal GST tagged HIP1 370-644 was kindly provided by the McPherson group. GST-HIP1 371-481 was made by altering the HIP1 cDNA to introduce a stop codon (TAG) at residue 482. Site-directed mutagenesis was performed according to the QuikChange mutagenesis protocol (Stratagene). The sequence was confirmed by DNA sequencing (IMBI, Indiana University) and then transformed into Rosetta 2 (DE3) pLysS cells (Novagen). The recombinant protein GST-HIP1 371-481 construct was expressed and purified as described previously 24 with some modifications. Briefly, GST-HIP1 371-481 was over-expressed at 37°C in M9 minimum medium. After reaching an O.D. 600 of 0.5 units, selenomethionine (50 μg/ml final concentration) and IPTG (100μg/ml final concentration) were added to the bacterial culture. Cells were then incubated for 16 h at 30°C before being harvested and frozen for use. Cell pellets were thawed on ice and then resuspended gently in 45 ml of PBS (10 mM Na2HPO4, 1.8 mM KH2PO4 (pH 7.3), 140 mM NaCl, 2.7 mM KCl), supplemented with 0.25 ml of 1 M DTT, 0.25 ml of protease inhibitor cocktail (Sigma), and 2 ml of PMSF (17.4 mg/ml in 2-propanol) and incubated on ice for ∼15min. After sonication, 2.5 ml of 20% (v/v) Triton X-100 was added and the lysate was incubated at room temperature by rotating for ∼30 min. The crude bacterial lysate was cleared by centrifugation (19 k, 4°C) for 15 min and then the supernatant was mixed with ∼5 ml of glutathione Sepharose 4B (Amersham) resin suspended in PBS. This slurry was rocked gently at room temperature for 2 h before being transferred into a small column. The packed column was washed with 50 ml of PBS supplemented with 0.25 ml of 1M DTT until no more background protein was detected by staining with Coomassie brilliant blue. The GST-HIP1 371-481 was eluted from the column with 50 ml of 3 mg/ml L-glutathione (sigma) in (PBS) at pH 8.0 and dialyzed against the same buffer. The GST tag was removed by rocking the protein with sequencing grade thrombin (Novagen) at 37°C for 3 h. The digested sample was then dialyzed against 10 mM HEPES (pH 8.5), 20 mM Imidazole, 2 mM tris(2-carboxyethyl)-phosphine, 1% (v/v) glycerol. The protein was passed through an anion-exchange column (POROS 20 HQ) equilibrated in buffer A (10 mM HEPES, 20 mM imidazole, 2 mM tris(2-carboxyethyl)-phosphine, 1% (v/v) glycerol pH 8.5). The target protein was eluted with a linear gradient of buffer B (10 mM HEPES (pH 8.5), 20mM imidazole, 2 mM tris(2-carboxyethyl)-phosphine, 1% (v/v) glycerol, 250 mM NaCl). We confirmed the single selenomethionine substitution by electrospray mass spectroscopy.
Crystallization and data collection: The protein was crystallized by the hanging drop method in reservoir buffer (15% PEG3350, 0.1 M succinate pH 7.0, 0.2 M potassium sodium tartrate, and 0.01 M NiCl2). The crystal grew in the tetragonal space group P43 (a=72.9, b=72.9, c=106.5, α=β=γ=90°), with two dimers in the asymmetric unit. The crystals were highly mosaic (∼2.9 degrees) along the c-axis. Exhaustive screening for alternative crystallization conditions or small molecule additives did not improve crystal quality. The crystals were looped out and quickly dipped in buffer containing ethylene glycol and then plunged in liquid nitrogen. The HIP 371-481 structure was solved by multi-wavelength anomalous dispersion (MAD). A 3-wavelength data set was obtained from a single crystal on beam line 4.2.2 at the Advanced Light Source, Lawrence Berkeley National Laboratory. The data were collected at 100 K in 1.0 degree oscillations using a NOIR-1 CCD detector. Wavelengths λ1 (1.0215Å) and λ2 (1.0213 Å) were at the peak and inflection, respectively, of the K-edge of selenium and λ3 (1.0373 Å) was a high energy remote. An inverse beam experiment was done at each wavelength, with E1=phi0 and E2=phi180 degrees, for a total oscillation of 90 degrees for each phi. The kappa angle was optimized to prevent overlaps in the long axis. Data were integrated and scaled using D*TREK.
Phasing and refinement: The MAD data was phased using a Bayesian approach 31 taking the high-energy remote (λ3) as the ‘native’ wavelength and the other two as ‘derivative’ wavelengths. The single selenium site in each monomer was found by SOLVE (http://www.solve.lanl.gov) and the experimental map was improved using RESOLVE 32. Model building was performed using O 33,34 the model was refined using CNS 34. Alternating rounds of positional, grouped B factor and simulated annealing were performed in reference to 2Fo-Fc and Fo- Fc maps and a bulk-solvent correction was applied near the end of refinement. The HIP1 371-481 structure refined against all the data from 30-2.8 Å with an R-factor of 26.5% and an Rfree of 32.4% at 2.8Å resolution.