

Abstract
Azidothymidine (AZT) is a reverse transcriptase (RT) inhibitor that efficiently blocks the replication of spumaretroviruses or foamy viruses (FVs). To more precisely elucidate the mechanism of action of the FV RT enzyme, we generated an AZT-resistant FV in cell culture. Biologically resistant virus was obtained for simian foamy virus from macaque (SFVmac), which was insensitive to AZT concentrations of 1 mM, but not for FVs derived from chimpanzees. Nucleotide sequencing revealed four non-silent mutations in the pol gene. Introduction of these mutations into an infectious molecular clone identified all changes to be required for the fully AZT-resistant phenotype of SFVmac. The alteration of individual sites showed that AZT resistance in SFVmac was likely acquired by consecutive acquisition of pol mutations in a defined order, because some alterations on their own did not result in an efficiently replicating virus, neither in the presence nor in the absence of AZT. The introduction of the mutations into the RT of the closely related prototypic FV (PFV) did not yield an AZT-resistant virus, instead they significantly impaired the viral fitness.
Keywords: Foamy virus, Reverse transcriptase, AZT resistance
Introduction
Foamy viruses (FVs) constitute one of two subfamilies of retroviruses and follow a unique replication pathway (for reviews, see Linial, 2007; Rethwilm, 2003, 2005). Aside from early studies the comparative analysis of the replication strategy between spuma- and orthoretroviruses has only recently focused on the biochemistry of the RT enzyme (Benzair et al., 1982, 1983; Boyer et al., in press, 2004; Kögel et al., 1995a, b; Liu et al., 1977; Rinke et al., 2002). Although the characterization of bacterially expressed PFV RT revealed many features common to all retroviruses (Boyer et al., in press, 2004; Kögel et al., 1995a, b), it was also shown that PFV RT is much more processive than orthoretroviral enzymes (Rinke et al., 2002). Furthermore, mutation of the active center of the PFV RT from YVDD to YMDD did not result in sensitivity to the antiretroviral drug 3TC, as in human immunodeficiency virus (HIV), but rendered the virus replication-deficient (Rinke et al., 2002). These studies indicated that there are similarities and differences in the biochemistry of the RT enzymes between orthoretroviruses and foamy viruses.
A major difference between ortho- and spumaretroviral RT enzymes consists in the nature of the precursor and the definite cleavage products of the Pol protein. While in orthoretroviruses, the enzymatic proteins are cleaved from a Gag-protease (PR)-Pol precursor into PR, RT/RNaseH, and integrase (IN), FV Pol cleavage products are processed from an authentic PR-Pol precursor and cleavage between the PR and RT/RNaseH subunits does not occur (for a review, see Linial and Eastman, 2003). Thus, the only observed end products of FV Pol precursor cleavage are the 85-kDa PR-RT/RNaseH and the 40-kDa IN (Flügel and Pfrepper, 2003). This structural feature of FV RT enzymes probably has an impact on the function that has not been elucidated in detail yet.
In addition to tenofovir, AZT (zidovudine) is the only RT-inhibiting drug, which is active against FVs and completely inhibits PFV replication in cell culture at a concentration as low as 5 μM (Lee et al., 2006; Moebes et al., 1997; Rosenblum et al., 2001). Since the understanding of the biochemistry of the FV RT would greatly profit from the characterization of an AZT-resistant variant and the understanding of the mechanism of AZT resistance in orthoretroviruses, namely, human immunodeficiency virus (HIV), would mutually profit from studying a distantly related virus, we attempted to generate and characterize AZT-resistant FVs.
Furthermore, based on homology predictions of the PFV and HIV RT enzyme structures, AZT resistance in PFV has been suggested to occur by alteration of certain residues that are known to confer AZT resistance to HIV-1 (Yvon-Groussin et al., 2001). We also wanted to investigate this possibility because experimental evidence for this theory does not exist.
Results
Generation of biologically AZT-resistant FV
To obtain AZT-resistant viruses we cultivated plasmid-derived PFV, the chimpanzee FV isolate (SFVcpz), and SFVmac virus stocks on the appropriate indicator cells and gradually raised the AZT concentration. Although we were successful in obtaining an SFVmac variant (SFVAZTres) that was able to replicate in medium containing 1 mM AZT, several attempts to similarly generate PFV or SFVcpz variants failed (data not shown).
Genotypic analysis of AZT-resistant SFVmac
The gag and pol genes of SFVAZTres were amplified by PCR and subjected to nucleotide sequence analysis. In comparison with the full-length SFVmac sequence, which was assembled from subgenomic SFVmac molecular clones (Genbank accession numbers: X58484 and M33561; see Mergia et al., 1990a, b), six nucleotide alterations leading to amino acid changes were found. Two of the changes were located in the gag open reading frame (ORF) and involved R535gag and G596gag, which were changed to Q and R, respectively. The presence of these gag mutations in the parental infectious SFVmac clone together with the pol mutations (as in pBK03QR-ITTK) did not influence the AZT susceptibility compared with an SFVmac clone having only the pol residues mutated (as in pBK04RG-ITTK) (Table 1). We therefore regarded the Gag ORF alterations as irrelevant for conferring AZT resistance and did not analyze them further.
Table 1.
Comparison of biologically selected AZT-resistant virus with molecularly cloned derivatives*
| Virus | D3 | D6 | D9 | D12 | D15 | D18 |
|---|---|---|---|---|---|---|
| SK29-KISE − AZT | + | +++ | ++++ | c.d. | n.d. | n.d. |
| SFVAZTres − AZT | + | +++ | ++++ | c.d. | n.d. | n.d. |
| BK03QR-ITTK − AZT | + | +++ | ++++ | c.d. | n.d. | n.d. |
| BK04RG-ITTK − AZT | + | +++ | ++++ | c.d. | n.d. | n.d. |
| SK29-KISE + AZT | (+) | (+) | (+) | − | − | − |
| SFVAZTres + AZT | + | + | + | ++ | +++ | ++++ |
| BK03QR-ITTK + AZT | + | + | + | ++ | +++ | ++++ |
| BK04RG-ITTK + AZT | + | + | + | ++ | +++ | ++++ |
105 BHK/LTR(SFVmac)lacZ cells were infected at a multiplicity of infection (MOI) of 0.001 with the respective viruses either in the absence (− AZT) or presence (+ AZT) of 50 μM AZT. The cultures were stained for blue cells at the indicated days after infection and the replication of virus was monitored. (+), <1% infected cells; +, 1–10% infected cells; ++, 10–25% of cells were infected; +++, 25–50% infected cells; ++++, more than 50% of the cell culture was infected; c.d., cell culture was destroyed; n.d., not done. SK29-KISE is the wild-type molecular clone-derived virus, SFVAZTres is the biologically selected AZT-resistant virus, BK03QR-ITTK and BK04RG-ITTK are viruses derived from molecular clones containing the two gag and four pol or only the four pol gene mutations, respectively.
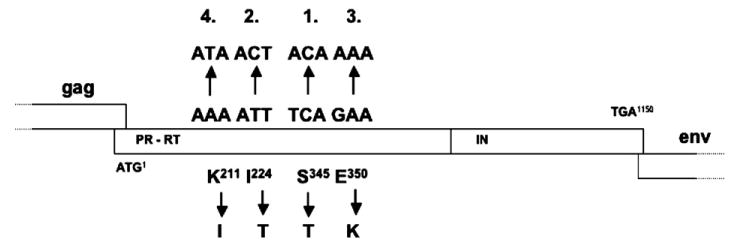
Four point mutations involving residues 211 (K211I), 224 (I224T), 345 (S345T), and 350 (E350K) were detected in pol (Fig. 1). On the nucleic acid level, all alterations leading to these amino acid changes required only the mutation of single nucleotides. The mutations identified were at codon 211 from AAA to ATA, at codon 224 from ATT to ACT, at codon 345 from TCA to ACA, and at codon 350 from GAA to AAA. One published complete genomic sequence of SFVmac already harbors a threonine at position 224 of the Pol protein (Kupiec et al., 1991). 224T would be more consistent with the other primate FV Pol proteins, which all harbor a threonine in this position (Fig. 1). However, the parental molecular clones pSFV-1 and pSK29-KISE code for an isoleucine at this position. Thus, the wild-type SFVmac may have a polymorphism at this site. Because we found I224T in SFVAZTres, we considered I224T as a mutation associated with AZT resistance.
Fig. 1.

Homology of Pol proteins of different FV and HIV-1. The RT regions of known FVs relevant to this study were aligned using the Genbank accession numbers for SFVmac (X58484 and M33561), PFV (Y07725), the chimpanzee isolate SFVcpz (U04327), orangutan FV (SFVora; AJ544579), African green monkey virus (SFVagm; M74895), FV from felines (FFV: AJ564746), bovines (BFV; U94514) and equines (EFV; AF20190).
Biologically selected SFVAZTres was compared with the parental virus and the virus derived from transfection of cells with molecular cloned variants bearing all four pol gene mutations. We observed only slight differences in the development of cell-free virus titers between the molecularly cloned and uncloned resistant viruses in the absence or presence of AZT (Table 1). This indicated that the four mutations identified in pol are sufficient to confer AZT resistance to SFVmac.
Analysis of reconstituted SFVmac molecular clones
To investigate which of the pol gene mutations was responsible for the resistant phenotype of SFVmac, we generated a series of full-length molecular clones in which all possible combinations of the four mutations found in SFVAZTres were represented (Fig. 2).
Fig. 2.
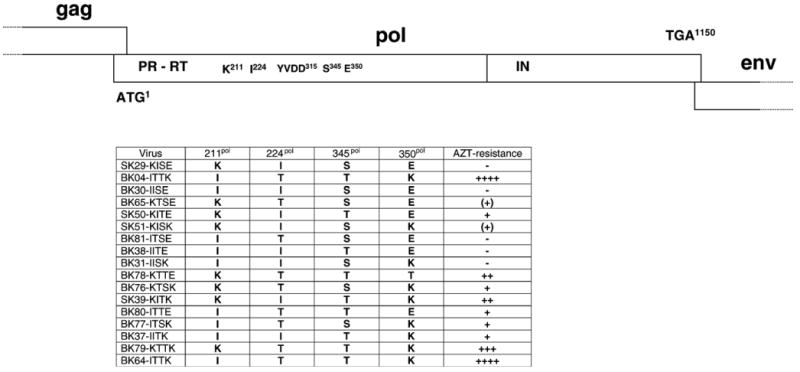
Genome organization of the wild-type and mutant FV Pol open reading frames. The pol gene of SFVmac is shown in the upper panel with the relative location of the RT active center and the four amino acid residues associated with AZT resistance. The SFVmac mutants representing all possible combinations of the four mutations are shown in the lower panel together with the approximate levels of AZT resistance deduced from Table 2. The plasmids pBK04-ITTK and pBK64-ITTK are identical. Plasmid pBK04-ITTK was made by exchanging a pol gene fragment containing the four mutations and amplified from DNA of cell cultures infected with SFVAZTres for a corresponding fragment of pSK29-KISE and pBK64-ITTK was made by in vitro mutagenesis. M108 is a PFV mutant with the changed residues found in SFVAZTres.
To analyze whether the introduced mutations affected the stability or other properties of SFVmac Pol protein, we first investigated the protein expression following transient transfection of 293T cells with the recombinant plasmids in cellular lysates. As shown in Fig. 3, all mutants were able to express Gag and Pol proteins to approximately the same level as non-mutant virus.
Fig. 3.

SFVmac protein expression. The expression of Gag (pr71/p68) and Pol (pr127) proteins was analyzed by immunoblot with polyclonal rabbit antisera in lysates from cells transfected with the indicated plasmids.
AZT resistance of SFVmac mutants
After demonstrating that the altered Pol proteins are efficiently expressed 293T cells were transfected with the parental molecular clone or the variants shown in Fig. 2 and analyzed on the appropriate indicator cells. Due to the peculiar replication strategy of FVs, namely, reverse transcription taking place already in virus producing cells (Moebes et al., 1997; Yu et al., 1999), the production of virus after transfection of cells and the analysis of cell-free viral titers were performed in the absence or presence of AZT. As shown in Table 2, we observed pronounced differences in the replication competence of the various mutants that argue for a sequential acquisition of SFVmac pol gene mutations for replication in the presence of AZT.
Table 2.
AZT-sensitivity of mutant viruses*
| Virus | 0.0 μM | 0.5 μM | 5.0 μM | 50 μM |
|---|---|---|---|---|
| SK29-KISE | 100% | 0.7±0.5% | <0.1% | <0.1% |
| BK04-ITTK | 117.0±18.4% | 84.3±27.8% | 71.0±12.7% | 34.9±12.9% |
| BK30-IISE | 1.2±1.2% | <0.1% | <0.1% | <0.1% |
| BK65KTSE | 146.5±21.4% | 1.2±0.9% | <0.1% | <0.1% |
| SK50-KITE | 40.0±16.8% | 5.6±3.2% | 1.1±1.0% | <0.1% |
| SK51-KISK | 31.7±8.8% | 1.8±0.8% | <0.1% | <0.1% |
| BK81-ITSE | 2.5±0.6% | <0.1% | <0.1% | <0.1% |
| BK38-IITE | <0.1% | <0.1% | <0.1% | <0.1% |
| BK31-IISK | 23.0±7.9% | 0.6±0.5% | <0.1% | <0.1% |
| BK78-KTTE | 86.7±20.5% | 18.7±12.9% | 0.5±0.3% | <0.1% |
| BK76-KTSK | 175.5±33.9% | 8.1±4.5% | <0.1% | <0.1% |
| SK39-KITK | 20.6±5.5% | 10.8±7.5% | 3.0±2.1% | 1.0±0.6% |
| BK80-ITTE | 7.3±3.6% | 4.0±2.7% | 1.0±0.8% | 0.3±0.2% |
| BK77-ITSK | 33.6±11.3% | 3.8±3.0% | 0.9±0.5% | <0.1% |
| BK37-IITK | 8.6±2.7% | 5.9±3.0% | 2.8±1.3% | 2.0±0.8% |
| BK79-KTTK | 138.2±21.8% | 86.9±11.5% | 16.4±10.9% | 1.4±0.4% |
| BK64-ITTK | 113.0±19.2% | 79.3±20.1% | 68.8±18.8% | 31.3±20.6% |
| HSRV2 | 100% | 1.1±0.9% | 0.4±0.1% | <0.1% |
| M108 | 1.3±0.1% | 0.5±0.1% | 0.4±0.1% | <0.1% |
293T cells were transiently transfected with 10 μg plasmid DNA either in the absence or presence of the concentrations of AZT indicated. The viral titers in the cell-free supernatants were determined on the appropriate indicator cells and are expressed as values relative to SK29-KISE (for SFVmac mutants) or HSRV2 (for the PFV mutant) in the absence of AZT and arbitrarily set to 100%. This corresponded to cell-free titers of 7 × 104 in the case of SFVmac and 6.3 × 104 for PFV.
The BK38-IITE virus, for instance, which bears the two mutations leading to K211I and S345T, did not replicate either in the absence or presence of AZT. Replication was also severely limited in the BK30-IISE, BK31-IITK, and BK81-ITSE viruses, whereas BK37-IISK, BK77-ITSK, BK80-ITTE, SK39-KITK, and SK51-KISK were moderately impaired to replicate irrespective of the AZT concentrations. All these mutants have K211 changed to isoleucine in various combinations (the BK viruses) or E350 modified to lysine (the SK viruses). It is therefore unlikely that either of these changes occurred first under drug selection. The only single mutation that was found to result in a moderate drug resistance was that leading to S345T (SK50-KITE). While conferring partial AZT resistance, this mutation weakened the virus to replicate in its absence (Table 2). Subsequent acquisition of I224T, in addition to S345T (BK78-KTTE), resulted in an enhancement of viral fitness and replication predominantly at low AZT concentration.
Alternatively, threonine may be the natural residue at position 224 in wild-type SFVmac (as discussed above), in which case the BK78-KTTE virus would be the first variant to emerge during AZT-induced selective pressure. Since the K211I variant greatly decreased replication, if it occurred before E350K (BK80-ITTE in Table 2), BK79-KTTK was probably the next virus to emerge. BK79-KTTK was able to replicate at 5 μM AZT and strongly enhanced replication without the drug. Finally, the resistant virus acquired the mutation leading to K211I in addition to I224T, S345T, and E350K (BK64-ITTK). Compared to BK79-KTTK the replication ability of BK64-ITTK in the absence of AZT was slightly reduced, however, it was greatly enhanced at higher drug concentrations (Table 2).
Attempts to create an AZT-resistant PFV
The introduction of the mutations leading to R211I, S345T, and E350K in M108 of PFV (residue 224 is already a threonine; see Fig. 1) did not lead to AZT resistance and, compared to the parental virus (HSRV2), severely reduced the viral fitness in the absence of AZT (Table 2). However, we cannot formally exclude the possibility that only one or two alterations of these residues might induce AZT resistance to PFV, although we regard this as very unlikely.
Discussion
The results shown here suggest that AZT resistance in SFVmac was acquired by sequential acquisition of mutations in the pol gene in the order: wild-type virus ->S345T ->I224T ->E350K ->K211I (Fig. 4). However, alternative scenarios are also possible, although less likely. In particular, the position in the order of events of the mutation leading to I224T appears variable (see above). The fully resistant virus was found to be able to replicate at 1 mM AZT. As shown in Table 2 the BK64-ITTK virus replicated only to approximately one-third the level of wild-type virus. Thus, the acquisition of the mutations leading to drug resistance was accompanied by a moderate reduction of viral fitness.
Fig. 4.
Most likely order of mutagenic events resulting in AZT-resistant SFVmac.
The inability to obtain AZT-resistant PFV or chimpanzee foamy virus by gradually raising the drug concentration in the cell culture medium was surprising. Because SFVmac and PFV derived from our molecular clones replicate to approximately the same extracellular viral titers (Table 2), we regard it very unlikely that significant differences in the replication kinetics between the two parental viruses are responsible for our futile attempts in generating biologically drug-resistant PFV. Both viruses, SFVmac and PFV, have been amplified in cell culture for years. Although FVs are known to be genetically extremely stable (Switzer et al., 2005; Thümer et al., 2007), the cell culture-adapted virus does not necessarily reflect the wild-type situation. However, SFVcpz was molecular cloned approximately 1 month after virus isolation (Herchenröder et al., 1994, 1995). This questions significant changes in the FV RT sequence upon cell culture replication and points to differences between SFVmac and FVs of the higher primates in the ability to respond to AZT drug selection.
In addition, the introduction of the mutations conferring AZT resistance to SFVmac into the infectious PFV molecular clone also did not result in a drug-resistant virus. These mutations resulted in an approximately 70% reduction in viral fitness of SFVmac (Table 2). The reduction was much more substantial when the same mutations were introduced into PFV (Table 2). This may indicate that PFV is, per se, able to mutate into an AZT-resistant variant, but that the resistant virus lost the ability to replicate either in the presence or absence of AZT. The reason for this currently remains unknown. Answers may emerge when comparative biochemical analyses of PFV, SFVmac, and SFVAZTres RT enzymes and structural information become available.
In the HIV system, AZT resistance develops in vivo at least under the condition of monotherapy very quickly by consecutive mutations in the pol gene, while there exist only a few reports on cell culture selection of AZT-resistant HIV from wild-type virus (Dianzani et al., 1992; Gao et al., 1992; Kellam et al., 1994; Larder et al., 1991, 1989; Larder and Kemp, 1989; Smith et al., 1987). Nothing is known yet about the in vivo development of AZT resistance in FV infections. The biochemical and molecular bases of AZT resistance in HIV-1 and HIV-2 have been thoroughly studied. In each virus, a different mechanism dominates in the acquisition of AZT drug resistance (Boyer et al., 2006). Although HIV-1 preferentially excises AZT from the growing DNA chain, resistant HIV-2 was reported to discriminate between the inhibitor AZTTP and TTP during incorporation (Boyer et al., 2006). Thus, even closely related viruses can make use of different strategies to develop drug resistance. The homology between HIV-1 and HIV-2 RTs is around 60% (Boyer et al., 2006), while it is approximately 90% for PFV and SFVmac (Kupiec et al., 1991). Therefore, only subtle differences in the primary amino acid composition of SFVmac and PFV RT enzymes are responsible for the inability to generate an AZT-resistant PFV.
The analyses of FV RT enzymes revealed similarities with as well as differences to orthoretroviral RT enzymes. Our study shows that differences even exist within the rather homogenous FV subfamily of retroviruses. In this respect, it would be interesting to know whether the conversion of the active center of the FV RT enzyme from YVDD to YMDD, which led to a replication-deficient PFV (Rinke et al., 2002), would be tolerated by SFVmac.
Furthermore, our results do not indicate that the residues actually involved in FV AZT resistance, are those thought previously (Yvon-Groussin et al., 2001). In particular, the residues I182, D209, V343, and K347 were modelled to be involved in PFV AZT resistance. The results presented here do not support this view, since (i) the residues identified to confer AZT resistance to SFVmac are different and (ii) AZT-resistant PFV or SFVcpz could not be generated at all.
Materials and methods
Cells and viruses
BHK/LTR(SFVmac)lacZ (Roy et al., 2003), BHK/LTR(PFV)lacZ (Schmidt and Rethwilm, 1995) indicator cells, and 293T cells (DuBridge et al., 1987) were cultivated as described (DuBridge et al., 1987; Roy et al., 2003; Schmidt and Rethwilm, 1995). After transient transfection of cells with the proviral plasmids pSFV-1 (Mergia and Wu, 1998), pcHSRV2 (Moebes et al., 1997), or pSFVcpz (Herchenröder et al., 1995) viruses were cultivated on the appropriate indicator cells which were stained for β-Gal as reported (Schmidt and Rethwilm, 1995). AZT (Glaxo) was added to the culture medium at the concentrations indicated in the figures and tables.
To determine the susceptibility to AZT, virus was produced by transient transfection (48 h) of 293T cells using calcium phosphate coprecipitation with 10 μg of plasmid DNA in the absence or presence of the drug (Moebes et al., 1997). Gene expression was induced using Na-butyrate at a final concentration of 10 mM for 8 h (Heinkelein et al., 1998). The virus titer in the cell-free supernatant was determined on indicator cells in the absence or presence of AZT as described (Moebes et al., 1997). All virus titrations were performed at least three times. Virus stocks derived from molecular clones were abbreviated with the plasmid name lacking the “p” or “pc”.
Molecular cloning
The plasmid pSK29-KISE was obtained by treatment of partially KpnI-digested pSFV-1 (Mergia and Wu, 1998) with T4 polymerase. This eliminated the KpnI restriction site in the polylinker of the vector backbone.
After amplification and nucleotide sequencing of the complete gag and pol genes of the biological resistant SFVmac, a 3.4-kb XhoI/EcoRI fragment harboring the region with the identified mutations was amplified and inserted into the full-length proviral clone via a subclone containing a 8.2 kb XbaI/KpnI fragment. Mutations were introduced into the XhoI/EcoRI subclone by recombinant PCR following the method of Higuchi (1990) and then introduced back into the full-length clone as described above. To ease the identification of amino acid changes in Gag and Pol, relevant residues are indicated after the plasmid name.
Because PFV already bears a threonine at position 224 of its pol gene (see below), we constructed a triple mutant (M108) of pcHSRV2 (Moebes et al., 1997) that contains R211I, S345T, and E350K. PFV pol gene mutagenesis was carried out by recombinant PCR (Higuchi, 1990) on a subcloned 1.83-kb PacI/SwaI fragment of pcHSRV2 before reinsertion into the full-length molecular clone.
For exclusion of inadvertent nucleotide exchanges all PCR-generated fragments were sequenced on the level of the full-length molecular clones. A detailed description of primers used for mutagenesis, amplification, and sequencing can be found online at http://viminfo.virologie.uni-wuerzburg.de/onlinematerial/Kretzschmar.pdf.
The complete gag and pol open reading frames (ORFs) of SFVmac were amplified separately and inserted into the vectors pRSET-A (Invitrogen) and pET28c (Novagen), respectively. The pET28c-SFVpol plasmid was used to delete the integrase gene via PCR in order to obtain a PR-RT subclone. A protease active site mutant (D24A) was created thereof by overlap PCR and transformed into the E. coli strain Rosetta DE3 (Novagen) for expression and purification of a PR (D24A)-RT fusion protein. Bacterial proteins were induced and purified via the C-terminal 6xHis-tag as described (Imrich et al., 2000) and used to generate polyclonal rabbit antisera at a commercial facility.
Immunoblotting
After transfection of 293T cells with proviral constructs, Gag and Pol protein expression in intracellular lysates was analyzed by immunoblotting with the rabbit Gag and Pol antisera as described previously (Heinkelein et al., 1998; Imrich et al., 2000).
Acknowledgments
We thank the DFG (Re 627/7-1, Re 627/8-1, Wo 630/7-1, and SFB479) for financial support.
References
- Benzair AB, Rhodes-Feuillette A, Emanoil-Ravicovitch R, Peries J. Reverse transcriptase from simian foamy virus serotype 1: purification and characterization. J Virol. 1982;44:720–724. doi: 10.1128/jvi.44.2.720-724.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzair AB, Rhodes-Feuillette A, Emanoil-Ravicovitch R, Peries J. Characterization of RNase H activity associated with reverse transcriptase in simian foamy virus type 1. J Virol. 1983;47:249–252. doi: 10.1128/jvi.47.1.249-252.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer PL, Stenbak CR, Clark PK, Linial ML, Hughes SH. Characterization of the polymerase and RNase H activities of human foamy virus reverse transcriptase. J Virol. 2004;78:6112–6121. doi: 10.1128/JVI.78.12.6112-6121.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer PL, Srafianos SG, Clark PK, Arnold E, Hughes SH. Why do HIV-1 and HIV-2 use different pathways to develop AZT resistance? PLoS Pathog. 2006;2:e10. doi: 10.1371/journal.ppat.0020010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer PL, Stenbak C, Hoberman D, Linial M, Hughes SH. Fidelity of the prototype primate foamy virus (PFV) RT compared to HIV-1 RT. Virology. doi: 10.1016/j.virol.2007.05.034. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianzani F, Antonelli G, Turriziani O, Dong G, Capobianchi MR, Riva E. In vitro selection of human immunodeficiency virus type 1 resistant to 3′-azido-3′-deoxythymidine. Antivir Res. 1992;18:39–52. doi: 10.1016/0166-3542(92)90004-o. [DOI] [PubMed] [Google Scholar]
- DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human cells by using Epstein–Barr virus shuttle system. Mol Cell Biol. 1987;7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flügel RM, Pfrepper KI. Proteolytic processing of foamy virus gag and pol proteins. Curr Top Microbiol Immunol. 2003;277:63–88. doi: 10.1007/978-3-642-55701-9_3. [DOI] [PubMed] [Google Scholar]
- Gao Q, Gu Z, Parniak MA, Li Y, Wainberg MA. In vitro selection of variants of human immunodeficiency virus type 1 resistant to 3′-azido-3′-deoxythymidine and 2′,3′-dideoxyinosine. J Virol. 1992;66:12–19. doi: 10.1128/jvi.66.1.12-19.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinkelein M, Schmidt M, Fischer N, Moebes A, Lindemann D, Enssle J, Rethwilm A. Characterization of a cis-acting sequence in the Pol region required to transfer human foamy virus vectors. J Virol. 1998;72:6307–6314. doi: 10.1128/jvi.72.8.6307-6314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herchenröder O, Renne R, Loncar D, Cobb EK, Murthy KK, Schneider J, Mergia A, Luciw PA. Isolation, cloning, and sequencing of simian foamy viruses from chimpanzees (SFVcpz): high homology to human foamy virus (HFV) Virology. 1994;201:187–199. doi: 10.1006/viro.1994.1285. [DOI] [PubMed] [Google Scholar]
- Herchenröder O, Turek R, Neumann-Haefelin D, Rethwilm A, Schneider J. Infectious proviral clones of chimpanzee foamy virus (SFVcpz) generated by long PCR reveal close functional relatedness to human foamy virus. Virology. 1995;214:685–689. doi: 10.1006/viro.1995.0086. [DOI] [PubMed] [Google Scholar]
- Higuchi R. Recombinant PCR. In: Innis MA, Gelfand DH, White TJ, editors. PCR Protocols, A Guide to Methods and Applications. Academic Press; San Diego, CA: 1990. pp. 177–183. [Google Scholar]
- Imrich H, Heinkelein M, Herchenröder O, Rethwilm A. Primate foamy virus Pol proteins are imported into the nucleus. J Gen Virol. 2000;81:2941–2947. doi: 10.1099/0022-1317-81-12-2941. [DOI] [PubMed] [Google Scholar]
- Kellam P, Boucher CAB, Tijnagel JMGH, Larder BA. Zidovudine treatment results in the selection of human immunodeficiency virus type 1 variants whose genotypes confer increasing levels of drug resistance. J Gen Virol. 1994;75:341–351. doi: 10.1099/0022-1317-75-2-341. [DOI] [PubMed] [Google Scholar]
- Kögel D, Aboud M, Flügel RM. Molecular biological characterization of the human foamy virus reverse transcriptase and ribonuclease H domains. Virology. 1995a;213:97–108. doi: 10.1006/viro.1995.1550. [DOI] [PubMed] [Google Scholar]
- Kögel D, Aboud M, Flügel RM. Mutational analysis of the reverse transcriptase and ribonuclease H domains of the human foamy virus. Nucleic Acids Res. 1995b;23:2621–2625. doi: 10.1093/nar/23.14.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupiec JJ, Kay A, Hayat M, Ravier R, Peries J, Galibert F. Sequence analysis of the simian foamy virus type 1 genome. Gene. 1991;101:185–194. doi: 10.1016/0378-1119(91)90410-d. [DOI] [PubMed] [Google Scholar]
- Larder BA, Kemp SD. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT) Science. 1989;246:1155–1158. doi: 10.1126/science.2479983. [DOI] [PubMed] [Google Scholar]
- Larder BA, Darby G, Richman DD. HIV with reduced sensitivity to zidovudine (AZT) isolated during prolonged therapy. Science. 1989;243:1731–1734. doi: 10.1126/science.2467383. [DOI] [PubMed] [Google Scholar]
- Larder BA, Coates KE, Kemp SD. Zidovudine-resistant human immunodeficiency virus selected by passage in cell culture. J Virol. 1991;65:5232–5236. doi: 10.1128/jvi.65.10.5232-5236.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Ye F, Tarantal AF. Comparison of growth and differentiation of fetal and adult rhesus monkey mesenchymal stem cells. Stem Cells Dev. 2006;15:209–220. doi: 10.1089/scd.2006.15.209. [DOI] [PubMed] [Google Scholar]
- Linial M. Foamy viruses. In: Knipe DM, Howley PM, editors. Fields Virology. 5th. Vol. 2. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2245–2262. [Google Scholar]
- Linial ML, Eastman SW. Particle assembly and genome packaging. Curr Top Microbiol Immunol. 2003;277:89–110. doi: 10.1007/978-3-642-55701-9_4. [DOI] [PubMed] [Google Scholar]
- Liu WT, Natori T, Chang KS, Wu AM. Reverse transcriptase of foamy virus. Purification of the enzymes and immunological identification. Arch Virol. 1977;55:187–200. doi: 10.1007/BF01319905. [DOI] [PubMed] [Google Scholar]
- Mergia A, Wu M. Characterization of provirus clones of simian foamy virus type 1. J Virol. 1998;72:817–822. doi: 10.1128/jvi.72.1.817-822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergia A, Shaw KES, Lackner E, Luciw PA. Relationship of the env genes and the endonuclease domain of the pol genes of simian foamy virus type 1 in human foamy virus. J Virol. 1990a;64:406–410. doi: 10.1128/jvi.64.1.406-410.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergia A, Shaw KES, Pratt-Lowe E, Barry PA, Luciw PA. Simian foamy virus type 1 is a retrovirus which encodes a transcriptional transactivator. J Virol. 1990b;64:3598–3604. doi: 10.1128/jvi.64.8.3598-3604.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moebes A, Enssle J, Bieniasz PD, Heinkelein M, Lindemann D, Bock M, McClure MO, Rethwilm A. Human foamy virus reverse transcription that occurs late in the viral replication cycle. J Virol. 1997;71:7305–7311. doi: 10.1128/jvi.71.10.7305-7311.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rethwilm A. The replication strategy of foamy viruses. Curr Top Microbiol Immunol. 2003;277:1–26. doi: 10.1007/978-3-642-55701-9_1. [DOI] [PubMed] [Google Scholar]
- Rethwilm A. Foamy viruses. In: Mahy BWJ, ter Meulen V, editors. Topley & Wilson's Microbiology and Microbial Infections—Virology. 10th. Vol. 2. Hodder Arnold; London: 2005. pp. 1304–1321. [Google Scholar]
- Rinke CS, Boyer PL, Sullivan MD, Hughes SH, Linial ML. Mutation of the catalytic domain of the foamy virus reverse transcriptase leads to loss of processivity and infectivity. J Virol. 2002;76:7560–7570. doi: 10.1128/JVI.76.15.7560-7570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum LL, Patton G, Grigg AR, Frater AJ, Cain D, Erlwein O, Hill CL, Clarke JR, McClure MO. Differential susceptibility of retroviruses to nucleoside analogues. Antivir Chem Chemother. 2001;12:91–97. doi: 10.1177/095632020101200202. [DOI] [PubMed] [Google Scholar]
- Roy J, Rudolph W, Juretzek T, Gärtner K, Bock M, Herchenröder O, Lindemann D, Heinkelein M, Rethwilm A. Feline foamy virus genome and replication strategy. J Virol. 2003;77:11324–11331. doi: 10.1128/JVI.77.21.11324-11331.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Rethwilm A. Replicating foamy virus-based vectors directing high level expression of foreign genes. Virology. 1995;210:167–178. doi: 10.1006/viro.1995.1328. [DOI] [PubMed] [Google Scholar]
- Smith MS, Brian EL, Pagano JS. Resumption of virus production after human immunodeficiency virus infection of T lymphocytes in the presence of azidothymidine. J Virol. 1987;61:3769–3773. doi: 10.1128/jvi.61.12.3769-3773.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Switzer WM, Salemi M, Shanmugan V, Gao F, Cong M, Kulken C, Bhullar V, Beer BE, Vallet D, Gautler-Hlon A, Tooze Z, Villinger F, Holmes EC, Heneine W. Ancient co-speciation of simian foamy viruses and primates. Nature. 2005;434:376–380. doi: 10.1038/nature03341. [DOI] [PubMed] [Google Scholar]
- Thümer L, Rethwilm A, Holmes EC, Bodem J. The complete nucleotide sequence of a New World simian foamy virus. Virology. doi: 10.1016/j.virol.2007.07.018. in press. [DOI] [PubMed] [Google Scholar]
- Yu SF, Sullivan MD, Linial ML. Evidence that the human foamy virus genome is DNA. J Virol. 1999;73:1565–1572. doi: 10.1128/jvi.73.2.1565-1572.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvon-Groussin A, Mugnier P, Bertin P, Grandadam M, Agut H, Huraux JM, Calvez V. Efficacy of dideoxynucleosides against human foamy virus and relationship to its reverse transcriptase amino acid sequence and structure. J Virol. 2001;75:7184–7187. doi: 10.1128/JVI.75.15.7184-7187.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]