

Summary
Molecular mechanisms regulating the development of physiological and behavioral tolerance to cannabinoids are not well understood. Two cellular correlates implicated in the development and maintenance of tolerance are CB1 cannabinoid receptor internalization and uncoupling of receptor signal transduction. Both processes have been proposed as mediators of tolerance because of observations that chronic Δ9-tetrahydrocannabinol (THC) treatment causes both region-specific decreases in CB1 receptors and G-protein coupling in the brain. To determine the balance of these two processes in regulating CB1 receptor signaling during sustained receptor stimulation, we evaluated the parameters affecting ERK1/2 MAP kinase activity in HEK293 cells stably expressing CB1 receptors. CB1 receptor stimulation by the potent CB1 receptor agonist, CP 55,940 transiently activated ERK1/2. To determine if CB1 receptor desensitization or internalization was responsible for the transient nature of ERK1/2 activation, we evaluated ERK1/2 phosphorylation in HEK293 cells expressing a desensitization-deficient CB1 receptor (S426A/S430A CB1). Here, the duration of S426A/S430A CB1 receptor-mediated activation of ERK1/2 was markedly prolonged relative to wild-type receptors, and was dynamically reversed by SR141716A. Interestingly, the S426A/S430A CB1 receptor was still able to recruit βarrestin-2, a key mediator of receptor desensitization, to the cell surface following agonist activation. In contrast to a central role for desensitization, pharmacological and genetic approaches suggested CB1 receptor internalization is dispensable in the transient activation of ERK1/2. This study indicates that the duration of ERK1/2 activation by CB1 receptors is regulated by receptor desensitization and underscores the importance of G-protein uncoupling in the regulation of CB1 receptor signaling.
Keywords: cannabinoids, MAP kinase, desensitization, internalization, phosphorylation, βarrestin
1. Introduction
Most CNS effects of cannabinoids, the principal psychoactive ingredients of marijuana and their endogenous counterparts, are mediated by the CB1 cannabinoid receptor. It is well established that the CB1 receptor couples in an inhibitory manner to voltage-dependent Ca2+ channels and adenylyl cyclase and activates G-protein-regulated inwardly rectifying K+ (GIRK) channels (Howlett et al., 2002; Howlett and Fleming, 1984; Mackie and Hille, 1992; Mackie et al., 1995). In vitro studies have demonstrated that cannabinoids can also induce the activation of mitogen-activated protein (MAP) kinases of the extracellular signal-regulated kinase (ERK) superfamily (Bouaboula et al., 1995). Consistent with these findings, acute THC administration has been reported to increase CB1 receptor-mediated ERK1/2 activation in the dorsal striatum, nucleus accumbens, and hippocampus (Derkinderen et al., 2003; Valjent et al., 2001), while chronic THC treatment has been shown to increase ERK1/2 protein levels in the prefrontal cortex and hippocampus in vivo (Rubino et al., 2004).
Chronic administration of cannabinoids leads to the rapid development of tolerance. This tolerance is accompanied by changes in CB1 receptor number and/or function (Bass et al., 2004; Breivogel et al., 2003; Gonzalez et al, 2005; Sim-Selley et al., 2003) and the inhibition of cannabinoid-mediated plasticity in different brain regions (Hoffman et al., 2003; Mato et al., 2004). The uncoupling of G-proteins from their cognate receptor, or desensitization, terminates receptor signaling and has been implicated as one of the cellular adaptations underlying tolerance (Martin et al., 2004). G-protein-coupled receptors (GPCRs) can be desensitized following agonist activation through phosphorylation by GPCR kinases (GRKs) and subsequent βarrestin binding (Gainetdinov et al., 2004). The physical interaction of βarrestin with the phosphorylated receptor reduces the affinity of the GPCR for G-proteins, likely via a mechanism involving steric hindrance, thereby attenuating signaling (Dewire et al., 2007). Previous studies have found that the βarrestin-2 isoform of βarrestin and GRK3 are capable of desensitizing CB1 receptor-mediated activation of GIRK channels and inhibition of neurotransmission (Jin et al., 1999; Kouznetsova et al., 2002). Furthermore, serines 426 and 430 in the proximal carboxy-terminus of CB1 are the likely GRK3 phosphorylation sites underlying βarrestin-2 mediated CB1 receptor desensitization of GIRK activation (Jin et al., 1999).
Following binding of agonist and activation, CB1 receptors internalize via clathrin-coated pits. However, the details and regulation of this process are now only emerging (Coutts et al., 2001; Hsieh et al., 1999; Leterrier et al., 2004; McDonald et al., 2007; Rinaldi-Carmona et al., 1998). Specifically, while βarrestin-2 is a multi-functional protein (Dewire et al., 2007), whose binding has also been proposed to regulate many forms of receptor endocytosis (Claing et al., 2002), its involvement in CB1 receptor internalization remains unknown.
In these studies we used HEK293 cells stably expressing CB1 to investigate the relative contributions of CB1 receptor desensitization and internalization to the time course of ERK1/2 phosphorylation following CB1 receptor activation. We found that CB1 receptor desensitization primarily determines the time course of ERK1/2 activation. In contrast, CB1 internalization is required for neither the onset nor the decay of ERK1/2 phosphorylation.
2. Materials and Experimental Methods
2.1 Reagents and drugs
CP 55,940 ((−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl) cyclohexanol) and SR141716A (4-Chlorophenyl)-1-(2,4-dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide) were provided by the National Institute of Drug Abuse Drug Supply Program (RTI, Research Triangle Park, NC). Concanavalin A, was purchased from Vector Laboratories (Burlingame, CA). Pertussis toxin and cholera toxin were purchased from List Biological Laboratories, Inc. (USA).
2.2 Generation of mutant CB1 receptor constructs
The CB1 receptor mutant, S426A/S430A, was made using a modified QuikChange PCR strategy (Stratagene, La Jolla, CA) to introduce serine to alanine point mutations. Full-length rat CB1, with an amino-terminal pplssHA (prolactin signal sequence and hemagglutinin epitope) tag (Andersson et al., 2003) in pcDNA3.0 (Invitrogen, Carlsbad, CA), was used as a template with the following primers: forward 5′ccgcacagcctcttgacaacgccatgggggacgcagactgcctgc-3′ and reverse 5′-gcaggcagtctgcgtcccccatggcgttgtcaagaggctgtgcgg-3′. A silent mutation was inserted (the 14th nucleotide) to remove the XbaI site for screening purposes. Integrity of the coding region for all constructs was confirmed by sequencing. The D164N CB1 receptor mutant was generated by overlapping extension PCR using full-length rat CB1 in pcDNA3.0 as template (Roche et al., 1999).
2.3 Cell culture and transfection
HEK293 cells were grown in DMEM containing 10% fetal bovine serum and penicillin/streptomycin (GIBCO, Carlsbad, CA) at 37°C in 5% CO2. Transfections were performed in 35-mm dishes with 2 μg of the appropriate plasmid and Lipofectamine 2000 (Invitrogen, Carlsbad, CA), following the manufacturer’s protocol. Stable cell lines were generated by selection in Geneticin (G418; Invitrogen, Carlsbad, CA). G418-resistant colonies were evaluated for the surface expression of CB1 by live cell immunostaining using an antibody (Covance, Berkeley, CA) directed towards the amino-terminal, extracellular HA epitope tag and a fluorescein isothiocyanate (FITC) secondary antibody (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Clones expressing uniform and moderate to high levels of CB1 were expanded and used for the subsequent experiments as previously described (Brown et al., 2002; Hsieh et al., 1999).
2.4 Quantitative internalization assay
HEK293 cells stably expressing rat CB1 receptors (wild-type or with S426A/S430A or D164N mutations) were seeded onto poly-D-lysine-coated 96-well plates (Corning, Corning, NY) and grown until 90% confluent in DMEM containing 10% fetal bovine serum and penicillin/streptomycin at 37°C in 5% CO2. Prior to drug treatment, plates were washed once in HEPES-buffered saline (HBS; 130 mM NaCl, 5.4 mM KCl, 1.8 mM MgCl2, and 10 mM HEPES, pH 7.5) containing 1.0 mg/ml bovine serum albumin. Cells were then incubated with treatment supplied in HBS containing 1.0 mg/ml bovine serum albumin and incubated at 37°C for the specified times. In all cases, the matching concentration of vehicle (DMSO) was included in control experiments. At the end of the drug treatment, the plate was placed on ice, the buffer was removed, and cells were immediately fixed with 4% paraformaldehyde for 30 min at room temperature. The integrity of the plasma membrane was evaluated following fixation using antibodies directed against both the amino- and carboxy-terminus of CB1 (with and without the addition of detergent). Paraformaldehyde itself did not cause substantial permeablization in this cell model (data not shown). Following fixation, cells were washed 5X for 30 min in phosphate-buffered saline (PBS; 137 mM NaCl, 10 mM NaH2 PO4, 2.7 mM KCl, pH 7.4) (without detergent to avoid membrane permeabilization) and blocked for 90 min in LI-COR Odyssey Blocking Buffer® (LI-COR Biosciences, Lincoln, Nebraska) at room temperature with gentle rocking. After blocking, cells were incubated overnight at 4°C with a monoclonal anti-HA antibody (1:100; Covance, Berkeley, CA). The cells were washed 5X the following day in Tris-buffered saline containing 0.05% Tween-20 (TBST; 137 mM NaCl, 10 mM Tris, 0.05% Tween-20, pH 7.4) for 30 min. Bound primary antibody was detected with an IRDye 800 conjugated anti-mouse IgG (1:800; Rockland Immunochemicals, Inc., Gilbertsville, PA). Plates were washed 5X in TBST, dried, and the immunocomplex was quantified on a LI-COR Odyssey following the manufacturer’s recommendations.
2.5 Quantitative Measurement of ERK1/2 Phosphorylation
HEK293 cells stably expressing either wild-type or modified CB1 receptors were seeded onto poly-D-lysine-coated 96-well plates, grown until 90% confluent at 37°C in 5% CO2. The cells were serum-starved overnight in DMEM containing only penicillin/streptomycin prior to the experiment. Drug treatments were performed as described in the preceding section. Following drug treatment, cells were fixed with 4% paraformaldehyde on ice for 15 min and then at room temperature for an additional 45 min. Cell membranes were permeabilized with 100% methanol at −20°C for ≥ 20 min and wells were blocked for 1.5 hr in Tris-buffered saline (TBS; 137 mM NaCl, 10 mM Tris, pH 7.4) containing 5.0 mg/ml bovine serum albumin. Following blocking, cells were incubated overnight at 4°C with a phospho-ERK1/2-specific antibody (1:200; Cell Signaling Technologies Inc., Danvers, MA). The primary antibody was removed with 5X washes over 1.5 hr in TBST and detected with an IRDye 800 conjugated anti-rabbit IgG (1:1000; Rockland, Gilbertsville, PA). Plates were washed 5X for 1.5 hr in TBST, dried and immunofluorecence was quantified on the LI-COR Odyssey.
2.6 Immunocytochemistry and fluorescent imaging
Immunostaining of cells stably expressing either the wild-type, S426A/S430A, or D164N CB1 receptors was performed using a previously described method to detect surface and intracellular receptors (Hsieh et al., 1999; Kearn et al., 2005). Briefly, cells were grown on poly-D-lysine-coated glass coverslips, washed in HBS and incubated with the specified drug diluted in HBS solution containing 0.2 mg/ml bovine serum albumin. After drug treatment, cells were fixed with 4% paraformaldehyde for 20 min, washed 2X with phosphate-buffered saline (PB; 10 mM NaH2PO4, 2.7 mM KCl, pH 7.4), and 3X with PBS. Cells were blocked for 1 hr in PBS containing 5% donkey serum and 0.1% saponin (to permeabilize cell membranes) at room temperature. After blocking, cells were incubated overnight at 4°C with a monoclonal mouse anti-HA antibody (1:1000; Covance, Berkeley, CA). Following 5X washes in PBS, immunoreactivity was detected with a FITC anti-mouse IgG secondary antibody (1:150; Jackson ImmunoResearch Laboratories Inc., West Grove, PA). The coverslips were washed 2X in PBS, 2X PB, 1X in water, dried, mounted in Vectashield (Vector Laboratories, Burlingame, CA), and imaged on a Leica SP1/MP confocal microscope. All images (1024 X 1024 pixels) were obtained by the use of a 100X objective.
For immunocytochemical phospho-ERK1/2 detection, HEK293 cells were treated the same as above through the fixation step. However, cells were then permeabilized with 100% methanol for ≥ 30 minutes at −20°C and then incubated overnight with a rabbit polyclonal p-ERK1/2 antibody (1:500; Cell Signaling Technology, Danvers, MA) diluted in a TBS solution containing 5.0 mg/ml BSA. A FITC anti-rabbit IgG antibody (1:150; Jackson ImmunoResearch Laboratories Inc., West Grove, PA) was used to detect the immunocomplex the following day. All confocal images of the phospho-ERK1/2 immunocomplex were acquired with the same instrument settings.
For βarrestin-2 recruitment studies, HEK293 cells were transiently transfected with constructs encoding either wild type or S426A/S430A CB1 receptors and an RFP-tagged βarrestin-2 fusion protein. GFP-tagged βarrestin-2 (a gift from Dr. Marc Caron) was modified using standard molecular biology techniques to exchange the GFP for RFP (Campbell et al., 2002). The cells were plated onto coverslips the same day and the experiment was performed 24 hours post-transfection. CB1 receptor expression was detected using the above-mentioned HA antibody and protocol.
2.7 Statistical analysis
In the internalization studies, the percentage of cell surface receptors remaining was determined by first calculating the mean integrated intensity values for each drug concentration. Background subtraction was not taken into account for individual experiments. However, non-specific binding was determined to be 5–10% for antibodies tested during the assay development phase. Final values were normalized to the mean integrated intensity values for the no drug treated controls and are represented as the mean ± SEM. Extent of internalization curves were fit by nonlinear regression. The half-life of the decay (t1/2) was calculated by fitting the data to a single-phase exponential decay model. In the ERK1/2 phosphorylation studies, percent maximal response for ERK1/2 activation was calculated by defining basal as the value at time zero and setting the maximal peak to 100%. The statistical significance with respect to the extent of internalization and maximal ERK1/2 activation was determined with an unpaired student’s t-test (**p<0.01, *p<0.05). To quantify βarrestin-2 recruitment, the membrane to cytosolic ratio of fluorescence in the 561 nm channel (obtained using the 100X objective with 2X zoom), was calculated by region-specific analysis of averaged integrated intensity values using MetaMorph software (Molecular Devices, Downingtown, PA). Co-detection of RFP and FITC fluorescence was done by excitation with 488 and 561 nm lasers. These data are represented as the mean AFU ± SEM. Statistical significance with respect to the membrane:cytosolic ratios was determined with an unpaired student’s t-test (**p<0.01, *p<0.05). All graphs and statistical analyses were generated using GraphPad Prism 4.0 software (Hearne Scientific Software, Chicago, IL).
3. Results
3.1 Mutation of serines 426 and 430 of the CB1 receptor prolong the time course of ERK1/2 activation in a SR141716A reversible manner
To test the hypothesis that putative GRK phosphorylation sites (serines 426 and 430) (Jin et al., 1999) in the proximal carboxy-terminus of the CB1 receptor determine the rate of ERK1/2 inactivation, the kinetics of ERK1/2 phosphorylation were evaluated following agonist stimulation of wild-type or S426A/S430A CB1 receptors stably expressed in HEK293 cells. A maximally efficacious concentration (100 nM) of the CB1 agonist, CP 55,940, was used for subsequent experiments (EC50 = 0.97 nM ± 0.06 for ERK1/2 activation at 7 min; data not shown). CP 55,940 stimulation of wild-type CB1 resulted in the transient activation of ERK1/2 with peak activation at 5 min (Fig. 1A). ERK1/2 phosphorylation then rapidly decayed (t1/2= 4.9 min) to near basal levels. While both wild-type and S426A/S430A CB1 receptors activated ERK1/2 to a similar extent (2–3 fold activation over basal; data not shown), there was little decay (71 ± 4% of peak at 15 min) in ERK1/2 phosphorylation with S426A/S430A CB1 receptor stimulation (Fig. 1A, Bf) relative to the wild-type receptor (33 ± 5% of peak at 15 min). Significantly, the sustained ERK1/2 activation was very long lasting in S426A/S430A CB1 expressing cells (71 ± 4% of peak at 45 min for S426A/S430A CB1 versus 20 ± 3% at 45 min for wild-type CB1). This prolonged ERK1/2 activation was observed in eight different monoclonal cell populations expressing S426A/S430A CB1 receptors (data not shown) and is not due to an increase in the constitutive activity of the modified receptors. Treatment with an inverse agonist, 1 μM SR141716A, does not decrease basal ERK1/2 activation in cells expressing either wild-type of S426A/S430A CB1 receptors (107 ± 1% of peak for S426A/S430A CB1 versus 100 ± 3% for wild-type CB1 following a 30 min incubation with SR1; data not shown). Additionally, the time course of ERK1/2 activation following 3 μM Δ9-THC treatment is similar in cells expressing either wild-type or S426A/S430A CB1 receptors (data not shown). Since these residues are required for homologous desensitization of GIRK activation by CB1 (Jin et al., 1999), this result suggests that CB1 receptor desensitization modulates the rapid decay of ERK1/2 phosphorylation. To evaluate if ERK1/2 phosphorylation was dependent upon CB1 receptor activation, we pretreated cells expressing wild-type or S426A/S430A CB1 receptors with 1μM of the CB1 antagonist, SR141716A and then applied 100 nM of CP 55,940 in the presence of 1μM SR141716A (Fig. 1Bg,h). SR141716A treatment prevented ERK1/2 phosphorylation indicating it is a CB1 receptor-dependent process.
Fig. 1. The sustained ERK1/2 phosphorylation seen with S426A/S430A CB1 is mediated by continued agonist-induced activation.

(A) Quantitative detection of phospho-ERK1/2. HEK293 cells stably expressing either wild-type or S426A/S430A CB1 receptors were treated with 100 nM CP 55,940 for the indicated times and the time course of ERK1/2 activation (measured by ERK1/2 phosphorylation) was determined (see Methods). The S426A/S430A CB1 receptor-mediated activation of ERK1/2 is significantly prolonged. Data are mean ± SEM and were collected from 5 or more experiments performed in duplicate. **p<0.01 compared with individual wild-type time points by unpaired t-test. Arrow indicates where SR1 was added in (C). (B) Immunocytochemical detection of phospho-ERK1/2 in HEK293 cells stably expressing either wild-type or S426A/S430A CB1. Cells were treated with either vehicle (a–b), 100 nM CP 55,940 alone (c–f), or pretreated with 1 μM SR1 followed by application of 100 nM CP 55,940 in the presence of 1μM SR1 (g–h). Phospho-ERK1/2 was detected immunoctyochemically (see Methods). Both wild type and S426A/S430A CB1 increased phospho-ERK1/2 immunoreactivity at 7 min. However, with S426A/S430A CB1 substantial phospho-ERK1/2 immunoreactivity was still evident at 20 min. In contrast, with wild type CB1 phospho-ERK1/2 immunoreactivity was transient, returning to control levels after 20 min. Pre-treatment with SR1 blocked ERK1/2 activation by both receptors. Scale bar, 20 μm. (C) Prolonged activation of ERK1/2 by the S426A/S430A CB1 receptor was rapidly (t1/2= 2.97 min) reversed by addition of 1μM SR1 after 35 min of 10 nM CP 55,940 stimulation (time of SR1 addition indicated by arrow in A and C). Data are mean ± SEM with n = 6 from three experiments performed in duplicate.
To determine if the prolonged phosphorylation of ERK1/2 reflects sustained S426A/S430A CB1 receptor activation, cells were first treated with 10 nM CP 55,940 for 35 min. Then 1μM of the CB1 receptor antagonist, SR141716A was added at 2.5 min intervals in the continued presence of 10 nM CP 55,940 until 45 min had elapsed (Fig. 1C). Phosphorylation of ERK1/2 by S426A/S430A CB1 was rapidly (t1/2= 2.97 min) reversed by addition of SR141716A, suggesting sustained ERK1/2 phosphorylation was due to the continued signaling by the mutant S426A/S430A CB1 receptor.
3.2 CB1 receptor internalization is not necessary for ERK1/2 activation and does not contribute to the decay of ERK1/2 phosphorylation
Agonist-induced internalization of G-protein coupled receptors is a widely observed phenomenon. Receptor internalization has been suggested to be necessary for the activation of ERK1/2, as well as for receptor resensitization (Daaka et al., 1998). In both heterologous and endogenous expression systems, wild-type CB1 receptors are internalized following agonist treatment (Coutts et al., 2001; Hsieh et al., 1999; Rinaldi-Carmona et al., 1998). Previously, we have shown using confocal microscopy that serines 426 and 430 are not required for rapid (< 30 min), agonist-induced CB1 receptor internalization in AtT20 cells (Jin et al., 1999). In the present study we extended this observation to CB1 receptors expressed in HEK293 cells in a more quantitative fashion. Consistent with previous findings, after 15 minutes of 100 nM CP 55,940 treatment (a time window that includes the period of peak ERK1/2 activation) S426A/S430A CB1 and wild-type CB1 receptors are internalized to the same extent (78 ± 2% versus 78 ± 0.6% of receptors on the cell surface, respectively) (Fig. 2A). However, for agonist treatments >15 min, the extent of internalization for the mutant CB1 receptor is significantly attenuated compared to wild-type receptors (for example, 77 ± 2% versus 58 ± 0.4% of receptors remain at the cell surface after 120 minutes of agonist treatment) (Fig. 2A).
Fig. 2. S426A/S430A CB1 receptor internalization and pharmacological blockade of CB1 receptor internalization with Concanavalin A.
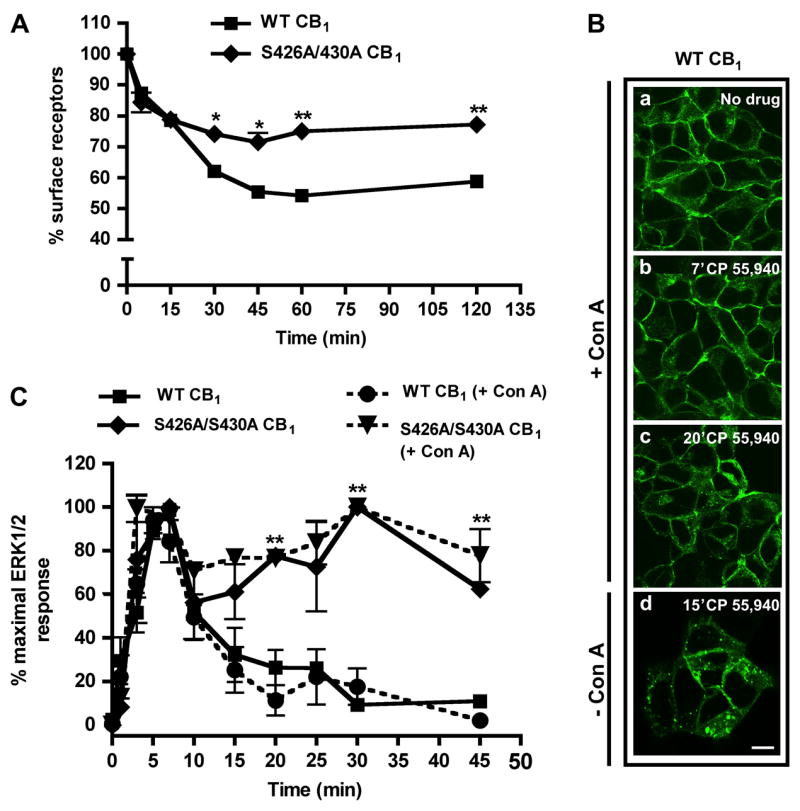
(A) Agonist-induced internalization of wild-type and S426A/S430A CB1. Cells were treated with 100 nM CP 55,940 for the indicated times at 37°C. Loss of cell surface receptors was quantified as described in Methods. Rapid (≤ 15 min) receptor endocytosis was unaffected by the S426A/S430A mutation. However, the extent of S426A/S430A CB1 receptor internalization was significantly attenuated following prolonged drug treatment (≥ 30 min). Data are mean ± SEM; n = 15–20 from three to five experiments performed in quadruplicate. **p<0.01 and *p<0.05 compared with wild-type CB1 receptor internalization by unpaired t-test. (B) Immunostaining of wild-type CB1 receptors following Con A treatment. Cells were pretreated with HBS containing 100 μg/ml Con A (a–c) or HBS alone (d) for 30 min, followed by stimulation with 100 nM CP 55,940 for the indicated times. Con A prevented significant CB1 receptor internalization at both time points evaluated. In the absence of Con A, a significant fraction of CB1 receptors were internalized after 15 min (d). Scale bar, 20 μm. (C) Time course of ERK1/2 activation in the presence or absence of Con A. Cells were pretreated with either HBS alone or 100 μg/ml Con A for 30 min (+Con A groups). Following pretreatment, the cells were stimulated with 10 nM CP 55,940 alone or in the presence of Con A (+Con A groups). Inhibition of CB1 receptor internalization by Con A (wild-type or S426A/S430A) does not affect the kinetics of ERK1/2 activation. Data are mean ± SEM; n = 5–15 from two to three experiments performed in duplicate. **p<0.01 compared with wild-type CB1, no Con A by unpaired t-test.
To determine if CB1 receptor internalization is required for ERK1/2 activation and/or the decay of phosphorylation in the continued presence of agonist, we examined the time course under conditions where internalization was blocked. We used a pharmacological inhibitor, concanavalin A (Con A), to prevent clathrin-dependent endocytosis following agonist treatment, a strategy utilized in previous studies (Arttamangkul et al., 2006). HEK293 cells stably expressing wild-type CB1 receptors were treated with Con A (100 μg/ml) and stimulated with 10 nM CP 55,940 (Fig. 2B). Con A completely inhibited CB1 receptor internalization during peak ERK1/2 activation (7 min) and greatly inhibited endocytosis following prolonged drug treatment (Fig. 2Bb, c). In the absence of Con A, a significant fraction of CB1 receptors were endocytosed (Fig. 2Bd). To examine whether CB1 receptor internalization was required for the transient activation of ERK1/2, we stimulated HEK293 cells expressing wild-type CB1 receptors in the presence of Con A (Fig. 2C). Blockade of endocytosis by Con A did not significantly alter the time course of receptor-mediated ERK1/2 phosphorylation. Additionally, 10 nM CP 55,940-induced ERK1/2 phosphorylation by S426A/S430A CB1 receptors was unaffected by Con A treatment (Fig. 2C).
A complementary approach was taken to further determine if CB1 receptor internalization contributes to the decay of ERK1/2 phosphorylation. We evaluated the ability of a previously characterized mutant CB1 receptor, D164N CB1 (aspartate to asparagine at residue 164), (Roche et al., 1999) to phosphorylate ERK1/2. CB1 receptors with a D164N modification do not undergo WIN 55,212-2-induced internalization in AtT20 cells but remain partially functional – in that they can inhibit Ca2+ channels and adenylyl cylase, and activate MAP kinase, yet do not activate GIRK channels (Roche et al., 1999). We first verified the internalization-deficient finding in a quantitative manner by assessing agonist-induced internalization of D164N CB1 receptors expressed in HEK293 cells by CP 55,940 (100 nM). Consistent with previous work, no significant internalization of D164N CB1 receptors was observed (101 ± 3% surface receptors at 120 min) relative to wild-type CB1 receptors (58 ± 0.4% surface receptors at 120 min) following treatment with 100 nM CP 55,940 at time points ≥ 30 min (Fig. 3A). To preclude the possibility of agonist-specific trafficking, the extent of internalization of D164N CB1 receptors was evaluated using a different CB1 agonist, WIN 55,212-2 (1 μM). In agreement with the CP 55,940 results, no internalization of D164N CB1 receptors was observed with WIN 55,212-2 stimulation (99 ± 0.8% surface receptors at 120 min for D164N versus 50 ± 0.1% for wild-type; data not shown). We then examined the time course of D164N CB1 receptor-mediated activation of ERK1/2. Both wild-type and D164N CB1 receptors activated ERK1/2 to a similar extent (2-fold over basal; data not shown) and with the same temporal pattern of activation (Fig. 3B). Taken together, these results indicate that CB1 receptor internalization is not required for ERK1/2 phosphorylation nor does it modulate the time course of receptor-mediated activation.
Fig. 3. The kinetics of ERK1/2 activation for an internalization-deficient mutant CB1 receptor (D164N) is temporally similar to that of the wild-type receptor.

(A) 100 nM CP 55,940-induced internalization of wild-type and D164N CB1 receptors. Mutation of aspartate 164 to asparagine prevented receptor internalization in HEK293 cells. Data shown are mean ± SEM; n = 15–20 for three to four experiments. **p<0.01 compared with the extent of wild-type CB1 receptor internalization by unpaired t-test. (B) Activation of ERK1/2 in cells expressing wild-type or D164N CB1 receptors. The time course of D164N CB1 receptor-mediated activation of ERK1/2 was not significantly different from the wild-type receptor. Data shown are mean ± SEM; n = 15–20 for three to four experiments.
3.3 Wild-type and S426A/S430A CB1 receptors activate ERK1/2 independent of Gi/o G-protein signaling
Agonist stimulation of CB1 receptors has been reported to activate ERK1/2 via pertussis toxin (PTX) sensitive G-proteins in heterologous expression systems (Bouaboula et al., 1999; Bouaboula et al., 1995; Kearn et al., 2005). Thus, we expected ERK1/2 activation in HEK293 cells to be PTX-sensitive. However, overnight treatment with 500 ng/ml PTX followed by 100 nM CP 55,940, did not alter the time course of wild-type or S426A/S430A CB1 receptor-mediated phosphorylation of ERK1/2 (Fig. 4). However, the same treatment did inhibit depolarization-induced suppression of excitation in cultured hippocampal neurons (data not shown), attesting to the activity of the toxin. CB1 receptors have been shown to functionally couple to stimulatory Gαs G-proteins under certain conditions (Glass and Felder, 1997; Kearn et al., 2005). However, selective down-regulation of Gαs with overnight cholera toxin treatment (100 ng/ml) did not prevent ERK1/2 activation (data not shown). Collectively these results suggest that CB1 receptors expressed in HEK293 cells can mediate ERK1/2 activation through G-proteins other than those of the Gαi/o and Gαs families.
Fig. 4. ERK1/2 activation is pertussis toxin-insensitive.

Time course of ERK1/2 activation following pertussis toxin treatment. Cells were treated overnight with 500 ng/ml pertussis toxin (+PTX groups) and stimulated the next day with 100 nM CP 55,940 for the indicated times. Inhibition of Gi/o sensitive G-proteins with pertussis toxin did not attenuate agonist-activation of ERK1/2 for wild-type or mutant CB1 receptors. Data shown are mean ± SEM; n = 16–20 for three to four experiments. **p<0.01 and *p<0.05 compared with wild-type CB1 receptor without pertussis toxin treatment by unpaired t-test.
3.4 Wild-type and S426A/S430A CB1 receptors recruit βarrestin-2-RFP to the plasma membrane following agonist stimulation
Phosphorylation of serine and threonine residues in the carboxy-terminus of many GPCRs by GRKs and subsequent βarrestin binding is thought to be a common mechanism for both receptor internalization and desensitization of GPCRs (Gainetdinov et al., 2004). βarrestin-2 has been implicated as a key regulator of desensitization of CB1 receptor-mediated coupling to GIRK currents and excitatory glutamatergic transmission in Xenopus oocytes and cultured hippocampal neurons, respectively (Jin et al., 1999; Kouznetsova et al., 2002). These studies determined the necessity of βarrestin-2 for GPCR desensitization using dominant negative arrestin constructs or direct protein over-expression. If βarrestin binding is necessary for receptor desensitization, this work indirectly suggests that βarrestin-2 is recruited to CB1 receptors on the cell surface following agonist activation. To test this hypothesis, we evaluated βarrestin-2 translocation in HEK293 cells transiently expressing RFP-tagged βarrestin-2 and wild-type CB1 receptors by confocal imaging (Fig. 5A). In the absence of receptor stimulation, βarrestin-2-RFP is uniformly expressed within the cytosol (Fig. 5Ac-d). Agonist activation of wild-type CB1 receptors with 100 nM CP 55,940 causes a rapid redistribution of βarrestin-2-RFP (Fig. 5Ag). Clusters of this protein are found at the surface in close proximity to CB1 receptors following CP 55,940 treatment (highlighted by arrows) (Fig. 5Ag). Quantitative analysis of βarrestin-2-RFP recruitment to wild-type CB1 receptors shows a 2-fold increase in the membrane/cytosol fluorescence ratio within 2 min (0.8 ± 0.05 at time 0 versus 1.6 ± 0.1 at 2 min) indicative of an increase in membrane associated βarrestin-2 (Fig. 5B). Interestingly, the membrane/cytosol ratio remains constant (1.5 ± 0.1 at 1 min versus 1.5 ± 0.2 at 15 min) indicating a sustained association of βarrestin-2-RFP with the plasma membrane and perhaps with activated CB1 receptors.
Fig. 5. βarrestin-2-RFP is rapidly recruited following CB1 receptor activation.

(A) Confocal images of HEK293 cells transiently expressing βarrestin-2-RFP and either wild-type or S426A/S430A CB1 receptors. CB1 receptors were detected immunocytochemically, while βarrestin-2-RFP was detected by its red fluorescence. Transiently expressed CB1 receptors were found both at the surface and within the cytosol (a–b). In unstimulated cells, βarrestin-2-RFP was diffusely present throughout the cytosol (c–d). CB1 receptor activation with 100 nM CP 55,940 for 2 min caused a dramatic translocation of βarrestin-2-RFP to the surface in cells expressing both wild type and S426A/S430A CB1 (g–h). Arrows indicate the regions of βarrestin-2-RFP enrichment at the plasma membrane. Scale bar, 5 μm. (B) Quantitation of βarrestin-2-RFP recruitment. Cells were treated with 100 nM CP 55,940 for the indicated times. βarrestin2-RFP translocation was quantified as described in Methods and the ratio of membrane averaged intensity values to cytosolic averaged intensity values are plotted. Data shown are mean ± SEM; n = 5–10 cells per time point (40–80 defined regions total).
Since residues 426 and 430 are putative GRK phosphorylation sites and are likely to contribute to a βarrestin-2 binding site, we examined whether βarrestin-2-RFP recruitment was reduced when these sites were mutated (S426A/S430A receptor). Surprisingly, βarrestin-2-RFP was still rapidly recruited to the surface in cells expressing S426A/S430A receptors stimulated with 100 nM CP 55,940. (Fig. 5Ah). The extent (1.6 ± 0.1 at 2 min for wild-type versus 1.4 ± 0.1 at 2 min) of βarrestin-2-RFP translocation in S426A/S430A expressing cells was not significantly different than that seen in wild-type cells (Fig. 5B). Furthermore, the time course of βarrestin-2-RFP translocation in S426A/S430A expressing cells is similar to that of the wild-type receptor. These data suggest that the receptor domain responsible for CB1 receptor desensitization is not required for βarrestin-2 recruitment.
4. Discussion
The principal finding of this study is that CB1 receptor desensitization dictates the kinetics of agonist-mediated ERK1/2 phosphorylation (activity). Using HEK293 cells stably expressing modified CB1 receptors, we have shown that mutation of serines 426 and 430 of CB1 to alanines dramatically prolongs the duration of ERK1/2 phosphorylation during protracted receptor stimulation (Fig. 1A). Importantly, the sustained phosphorylation of ERK1/2 (at time points ≤ 35 min) is a direct result of continued agonist activation of the S426A/S430A CB1 receptor and is not a consequence of a receptor-independent phenomenon (Fig. 1B, C). The results of the present study, coupled with findings from our earlier work (Jin et al., 1999), suggest that the time course of both ERK1/2 de-phosphorylation and GIRK channel inactivation are dynamically regulated at the receptor level by a distinct domain of the CB1 receptor – a domain that mediates G-protein uncoupling, likely by receptor phosphorylation.
In contrast to the desensitization results, mutation of serines 426 and 430 to alanine had no effect on the rate of rapid CB1 receptor internalization (< 15 min), but did significantly attenuate the extent of internalization following prolonged agonist treatment (Fig. 2A). These results contrast observations made previously using this same mutant receptor in AtT20 cells (Jin et al., 1999). In this study, the S426A/S430A mutation did not appear to affect CB1 receptor internalization after a 30 min application of WIN 55,212-2. The apparent discrepancy between the current and the previous findings may be due to the increased sensitivity of the quantitative approach for detecting receptor loss from the surface in the current study or may be due to a cell-type specific difference in trafficking. Whatever the cause, our present results suggest that the desensitization domain of CB1 may have a role in the late phase of receptor endocytosis.
Our results strongly support the notion that internalization of the CB1 receptor following agonist stimulation is not required for ERK1/2 activation nor does it dictate the time course of ERK1/2 inactivation. Pharmacological inhibition of agonist-induced CB1 receptor internalization was without effect on the kinetics of ERK1/2 phosphorylation (Fig. 2C). Consistent with these findings, we have found that a non-internalizing, modified D164N CB1 receptor is still capable of activating ERK1/2 with a similar temporal pattern to that of the wild-type receptor (Fig 3B). These latter data are in agreement with the initial observation in AtT20 cells that found robust D164N CB1 receptor mediated ERK1/2 activation at a single time point (Roche et al., 1999). Taken together, these data indicate that CB1 receptor internalization is not required for the activation of ERK1/2. An early study found that β2-adrenergic receptor-mediated activation of ERK1/2 was blocked by the over-expression of βarrestin-2 and dynamin dominant negative constructs in HEK293 cells (Daaka et al., 1998). This observation has caused some to conclude that receptor internalization is a necessary step in ERK1/2 activation. However, subsequent studies have suggested that agonist-induced internalization of GPCR’s, including the β2-adrenergic receptor, is not required for MAP kinase activation (Huang et al., 2004; Jimenez-Sainz et al., 2003; Kramer and Simon, 2000; Li et al., 1999). These results, coupled with our present findings, suggest that internalization is not a general requirement for ERK1/2 activation. Furthermore, at least one study (Huang et al., 2004) suggests that the requirement for receptor internalization in ERK1/2 activation may also be cell-type dependent, adding an additional layer of complexity.
CB1 receptor-mediated activation of ERK1/2 did not require active G protein αi/o or αs subunits. Specifically, overnight treatment with PTX and/or CTX did not block agonist-induced ERK1/2 phosphorylation by either wild-type or S426A/S430A CB1 receptors (Fig. 4). This is surprising considering that the majority of CB1 mediated signal transduction pathways involve Gαi/o or Gαs G-proteins. Possible interpretations of these results are the involvement of Gz or another G protein alpha subunit, or the involvement of a G protein-independent (potentially βarrestin-2 mediated) signaling pathway. This possibility will need to be addressed by additional experiments beyond the scope of the present study.
GPCRs are specifically phosphorylated by GRKs following agonist activation. The phosphorylated receptor then interacts with βarrestin. The physical association of βarrestin with the activated receptor has been shown to uncouple many GPCR’s from their cognate G-proteins, inducing homologous desensitization (Gainetdinov et al., 2004). In Xenopus oocytes and cultured hippocampal neurons, homologous CB1 receptor desensitization is mediated in part by βarrestin-2 (Jin et al., 1999; Kouznetsova et al., 2002). In these studies βarrestin over-expression or dominant negative βarrestin-2 mutants were used to reveal the necessity of this protein for the desensitization of CB1 receptor-mediated activation of GIRK channels or inhibition of voltage-gated Ca2+ channels. While these results suggest a role for βarrestin-2 in receptor desensitization, βarrestin-2 recruitment to activated CB1 receptors has not been reported. Figure 5 shows that wild-type CB1 receptors rapidly recruit (within 2 min) βarrestin-2 to the plasma membrane during agonist treatment, where it co-localizes with CB1. Since CB1 serines 426 and 430 are likely candidates for mediating βarrestin-2 recruitment, we also examined βarrestin-2 recruitment by the desensitization-deficient CB1 receptor. Surprisingly, S426A/S430A CB1 receptors recruited βarrestin-2 to a similar extent and with the same kinetics as the wild-type receptor (Fig. 5B), indicating that the residues involved in desensitization are not required for βarrestin-2 recruitment. One interpretation of these results is that the gross recruitment of βarrestin-2 seen following CB1 activation reflects the bulk recruitment of βarrestin-2 for its scaffolding functions (Lefkowitz and Shenoy, 2005; Shenoy and Lefkowitz, 2005) and not the much more modest quantities (i.e., equimolar with the CB1 receptor) likely needed for desensitization.
In summary, our results suggest that the rapid inactivation of ERK1/2 during stimulation of CB1 is mediated by phosphorylation of serines 426 and 430 and desensitization of the CB1 receptor. CB1 receptor internalization does not seem to play a role in ERK1/2 activation (or inactivation) as its blockade did not effect on the kinetics of ERK1/2 phosphorylation. Thus, increases in the ERK1/2 cascade following chronic cannabinoid administration in vivo (Derkinderen et al., 2003; Valjent et al., 2001), are likely to be regulated by CB1 receptor desensitization at the cellular level.
Acknowledgments
This work was supported by DA07278 (T.D.), DA11322 (K.M.), and DA00286 (K.M.). We would like to thank Giovanna Cacciola for generating the concentration-response curve for ERK1/2 activation by CP 55,940 and for help with the initial assay development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersson H, D’Antona AM, Kendall DA, Von Heijne G, Chin CN. Membrane assembly of the cannabinoid receptor 1: impact of a long N-terminal tail. Mol Pharmacol. 2003;64:570–577. doi: 10.1124/mol.64.3.570. [DOI] [PubMed] [Google Scholar]
- Arttamangkul S, Torrecilla M, Kobayashi K, Okano H, Williams JT. Separation of mu-opioid receptor desensitization and internalization: endogenous receptors in primary neuronal cultures. J Neurosci. 2006;26:4118–4125. doi: 10.1523/JNEUROSCI.0303-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass CE, Martin BR. Time course for the induction and maintenance of tolerance to Delta(9)-tetrahydrocannabinol in mice. Drug Alcohol Depend. 2000;60:113–119. doi: 10.1016/s0376-8716(99)00150-7. [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Bianchini L, McKenzie FR, Pouyssegur J, Casellas P. Cannabinoid receptor CB1 activates the Na+/H+ exchanger NHE-1 isoform via Gi-mediated mitogen activated protein kinase signaling transduction pathways. FEBS Lett. 1999;449:61–65. doi: 10.1016/s0014-5793(99)00395-6. [DOI] [PubMed] [Google Scholar]
- Bouaboula M, Poinot-Chazel C, Bourrie B, Canat X, Calandra B, Rinaldi-Carmona M, Le Fur G, Casellas P. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995;312(Pt 2):637–641. doi: 10.1042/bj3120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breivogel CS, Scates SM, Beletskaya IO, Lowery OB, Aceto MD, Martin BR. The effects of delta9-tetrahydrocannabinol physical dependence on brain cannabinoid receptors. Eur J Pharmacol. 2003;459:139–150. doi: 10.1016/s0014-2999(02)02854-6. [DOI] [PubMed] [Google Scholar]
- Brown SM, Wager-Miller J, Mackie K. Cloning and molecular characterization of the rat CB2 cannabinoid receptor. Biochim Biophys Acta. 2002;1576:255–264. doi: 10.1016/s0167-4781(02)00341-x. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claing A, Laporte SA, Caron MG, Lefkowitz RJ. Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog Neurobiol. 2002;66:61–79. doi: 10.1016/s0301-0082(01)00023-5. [DOI] [PubMed] [Google Scholar]
- Coutts AA, Anavi-Goffer S, Ross RA, MacEwan DJ, Mackie K, Pertwee RG, Irving AJ. Agonist-induced internalization and trafficking of cannabinoid CB1 receptors in hippocampal neurons. J Neurosci. 2001;21:2425–2433. doi: 10.1523/JNEUROSCI.21-07-02425.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SS, Caron MG, Lefkowitz RJ. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- Derkinderen P, Valjent E, Toutant M, Corvol JC, Enslen H, Ledent C, Trzaskos J, Caboche J, Girault JA. Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci. 2003;23:2371–2382. doi: 10.1523/JNEUROSCI.23-06-02371.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewire SM, Ahn S, Lefkowitz RJ, Shenoy SK. beta-Arrestins and Cell Signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Gonzalez S, Cebeira M, Fernanadez-Ruiz J. Cannabinoid tolerance and dependence: a review of studies in laboratory animals. Pharmacol Biochem Behav. 2005;81:300–318. doi: 10.1016/j.pbb.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AF, Oz M, Caulder T, Lupica CR. Functional tolerance and blockade of long-term depression at synapses in the nucleus accumbens after chronic cannabinoid exposure. J Neurosci. 2003;23:4815–4820. doi: 10.1523/JNEUROSCI.23-12-04815.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Fleming RM. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol Pharmacol. 1984;26:532–538. [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- Huang J, Sun Y, Huang XY. Distinct roles for Src tyrosine kinase in beta2-adrenergic receptor signaling to MAPK and in receptor internalization. J Biol Chem. 2004;279:21637–21642. doi: 10.1074/jbc.M400956200. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sainz MC, Fast B, Mayor F, Jr, Aragay AM. Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal-regulated kinase activation. Mol Pharmacol. 2003;64:773–782. doi: 10.1124/mol.64.3.773. [DOI] [PubMed] [Google Scholar]
- Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, Chavkin C, Mackie K. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J Neurosci. 1999;19:3773–3780. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearn CS, Blake-Palmer K, Daniel E, Mackie K, Glass M. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: a mechanism for receptor cross-talk? Mol Pharmacol. 2005;67:1697–1704. doi: 10.1124/mol.104.006882. [DOI] [PubMed] [Google Scholar]
- Kouznetsova M, Kelley B, Shen M, Thayer SA. Desensitization of cannabinoid-mediated presynaptic inhibition of neurotransmission between rat hippocampal neurons in culture. Mol Pharmacol. 2002;61:477–485. doi: 10.1124/mol.61.3.477. [DOI] [PubMed] [Google Scholar]
- Kramer HK, Simon EJ. mu and delta-opioid receptor agonists induce mitogen-activated protein kinase (MAPK) activation in the absence of receptor internalization. Neuropharmacology. 2000;39:1707–1719. doi: 10.1016/s0028-3908(99)00243-9. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Leterrier C, Bonnard D, Carrel D, Rossier J, Lenkei Z. Constitutive endocytic cycle of the CB1 cannabinoid receptor. J Biol Chem. 2004;279:36013–36021. doi: 10.1074/jbc.M403990200. [DOI] [PubMed] [Google Scholar]
- Li JG, Luo LY, Krupnick JG, Benovic JL, Liu-Chen LY. U50,488H-induced internalization of the human kappa opioid receptor involves a beta-arrestin- and dynamin-dependent mechanism. Kappa receptor internalization is not required for mitogen-activated protein kinase activation. J Biol Chem. 1999;274:12087–12094. doi: 10.1074/jbc.274.17.12087. [DOI] [PubMed] [Google Scholar]
- Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Sim-Selley LJ, Selley DE. Signaling pathways involved in the development of cannabinoid tolerance. Trends Pharmacol Sci. 2004;25:325–330. doi: 10.1016/j.tips.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, Manzoni OJ. A single in vivo exposure to delta 9THC blocks endocannabinoid-mediated synaptic plasticity. Nat Neurosci. 2004;7:585–586. doi: 10.1038/nn1251. [DOI] [PubMed] [Google Scholar]
- McDonald NA, Henstridge CM, Connolly CN, Irving AJ. An Essential Role for Constitutive Endocytosis, but Not Activity, in the Axonal Targeting of the CB1 Cannabinoid Receptor. Mol Pharmacol. 2007;71:976–984. doi: 10.1124/mol.106.029348. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Le Duigou A, Oustric D, Barth F, Bouaboula M, Carayon P, Casellas P, Le Fur G. Modulation of CB1 cannabinoid receptor functions after a long-term exposure to agonist or inverse agonist in the Chinese hamster ovary cell expression system. J Pharmacol Exp Ther. 1998;287:1038–1047. [PubMed] [Google Scholar]
- Roche JP, Bounds S, Brown S, Mackie K. A mutation in the second transmembrane region of the CB1 receptor selectively disrupts G protein signaling and prevents receptor internalization. Mol Pharmacol. 1999;56:611–618. doi: 10.1124/mol.56.3.611. [DOI] [PubMed] [Google Scholar]
- Rubino T, Forlani G, Vigano D, Zippel R, Parolaro D. Modulation of extracellular signal-regulated kinases cascade by chronic delta 9-tetrahydrocannabinol treatment. Mol Cell Neurosci. 2004;25:355–362. doi: 10.1016/j.mcn.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Seven-transmembrane receptor signaling through beta-arrestin. Sci STKE 2005. 2005:cm10. doi: 10.1126/stke.2005/308/cm10. [DOI] [PubMed] [Google Scholar]
- Sim-Selley LJ. Regulation of cannabinoids CB1 receptors in the central nervous system by chronic cannabinoids. Crit Rev Neurobiol. 2003;15:91–119. doi: 10.1615/critrevneurobiol.v15.i2.10. [DOI] [PubMed] [Google Scholar]
- Valjent E, Pages C, Rogard M, Besson MJ, Maldonado R, Caboche J. Delta 9-tetrahydrocannabinol-induced MAPK/ERK and Elk-1 activation in vivo depends on dopaminergic transmission. Eur J Neurosci. 2001;14:342–352. doi: 10.1046/j.0953-816x.2001.01652.x. [DOI] [PubMed] [Google Scholar]