

Abstract
Signaling by Hedgehog (Hh) proteins shapes most tissues and organs in both vertebrates and invertebrates, and its misregulation has been implicated in many human diseases. Although components of the signaling pathway have been identified, key aspects of the signaling mechanism and downstream targets remain to be elucidated. We performed an enhancer/suppressor screen in Drosophila to identify novel components of the pathway and identified 26 autosomal regions that modify a phenotypic readout of Hh signaling. Three of the regions include genes that contribute constituents to the pathway—patched, engrailed, and hh. One of the other regions includes the gene microtubule star (mts) that encodes a subunit of protein phosphatase 2A. We show that mts is necessary for full activation of Hh signaling. A second region includes the gene second mitotic wave missing (swm). swm is recessive lethal and is predicted to encode an evolutionarily conserved protein with RNA binding and Zn+ finger domains. Characterization of newly isolated alleles indicates that swm is a negative regulator of Hh signaling and is essential for cell polarity.
HH signaling is essential to the development of many tissues and organs in both vertebrates and invertebrates, and it has important medical implications. hh was first identified as a gene that is required for segmentation of the Drosophila embryo (Nusslein-Volhard and Wieschaus 1980); subsequent studies have established roles at all developmental stages in a variety of cell types. When signaling is reduced in human, sheep, fish, and mouse embryos, severe holoprosencephaly and cyclopia result (Belloni et al. 1996; Chiang et al. 1996; Roessler et al. 1996; Schier et al. 1997). Increased signaling in human adults can lead to cancers of the skin, cerebellum, muscle, digestive tract, pancreas, and prostate (reviewed in Pasca Di Magliano and Hebrok 2003). Hh signaling is complex, and a complete understanding of Hh signaling will encompass the mechanism and regulation of active Hh protein production, its release from producing cells, its transit to target cells, the mechanism that senses its presence in target cells, and the output of the pathway on patterning and growth.
Synthesis of the mature Hh peptide involves multiple steps, including cleavage of a signal sequence from an inactive precursor, autoproteolysis, N-terminal palmitoylation, and C-terminal cholesteroylation (Porter et al. 1996; Pepinsky et al. 1998). The N-terminal lipid is essential for Hh activity (Chamoun et al. 2001; Lee and Treisman 2001; Micchelli et al. 2002). The C-terminal cholesterol is needed to generate the normal distribution of transported protein (Porter et al. 1996; Gallet et al. 2003; Dawber et al. 2005; Callejo et al. 2006; Gallet et al. 2006). Release of lipid-modified Hh requires the Dispatched protein (Burke et al. 1999), but how Dispatched interacts with and affects Hh awaits clarification. Movement of Hh away from producing cells may involve interactions with proteoglycans (reviewed in Eaton 2006) and may require assembly into a multimer form (Zeng et al. 2001; Chen et al. 2004; Feng et al. 2004; Gallet et al. 2006;).
In target cells, several membrane proteins contribute to the recognition of Hh— Patched (Ptc), Interference Hedgehog (Ihog), Brother of Ihog, and Smoothened (Smo) (Hooper 1994; Chen and Struhl 1996; Quirk et al. 1997; Yao et al. 2006). In the absence of Hh, Ptc inhibits Smo-dependent signal transduction. In the presence of Hh, Smo accumulates at the cell surface (Zhang et al. 2001, 2004; Apionishev et al. 2005; Lu et al. 2006) and binds through its intracellular C-terminal tail to a Hh signaling complex. Other constituents of this complex include the kinase Fused, (Fu), the kinesin-related motor protein Costal2 (Cos2), Suppressor of Fused (SuFu), and the Zn+ finger transcription factor Cubitus interruptus (Ci). It is thought that Hh binding to Ptc alters the composition and localization of this and derivative protein complexes in ways that are sensitive to Hh concentration (reviewed in Hooper and Scott 2005; Ogden et al. 2006). Although we lack a complete understanding of how the presence of Hh at the cell surface leads to changes in expression of target genes, we know that the responses of Smo, Cos2, and Ci to Hh are mediated, in part, by phosphorylation. Smo requires several phosphorylation sites in its C-terminal tail for activity (Chen et al. 1998; Price and Kalderon 2002; Jia et al. 2004; Collins and Cohen 2005), phosphorylation of Cos2 by Fu promotes cell surface accumulation and stabilization of Smo (Nybakken et al. 2002; Lu et al. 2006), and Ci phosphorylation is required for conversion to either its repressor or activator forms (Chen et al. 1998; Price and Kalderon 2002).
Hh signaling is arguably best understood in the context of its roles in the imaginal discs where it governs patterning and cell growth. In the eye imaginal discs, for instance, Hh signals regulate cell proliferation directly in the receiving cells by inducing Cyclin D and Cyclin E (Duman-Scheel et al. 2002). In the wing disc, hh is positively regulated by Engrailed (En), and both hh and en are expressed by all P-compartment cells (Tabata et al. 1992). Hh protein moves across the A/P compartment border (Tabata and Kornberg 1994) to activate such genes as ptc and decapentaplegic (dpp) in A cells, creating a developmental organizer among the Anterior Hh-responding cells adjacent to the compartment border (Tabata et al. 1995; Zecca et al. 1995). Hh signals regulate cell proliferation in the receiving cells by inducing Dpp, the predominant mitogen for this tissue. In addition, Hh itself has a mitogenic role independent of Dpp in the wing cells between veins 3 and 4 (Mullor et al. 1997; Strigini and Cohen 1997). The sensitivity of these cells to Hh levels is manifested by an expansion of the intervein region when signaling increases and a corresponding reduction when it decreases. We have tapped this sensitivity to identify novel components of the Hh signaling pathway with a genetic screen.
Several types of screens for Hh pathway components have been carried out previously. Published genetic screens were based on the wing phenotypes of known pathway mutants (Vegh and Basler 2003), on suppression of a gain of function hh mutant (Haines and van den Heuvel 2000), or on enhancement or suppression of a partial loss-of-function phenotype generated by a dominant negative form of Smo (Collins and Cohen 2005). These screens identified a new class of ptc mutants (Vegh and Basler 2003), mutations that affect Hh stability (Haines and van den Heuvel 2000), and implicated the translation factors eRF1 and elF1A and the kinesin-like protein Pavarotti in the Hh response (Collins and Cohen 2005). Genomewide RNA interference (RNAi) assays in cultured cells identified many functions that contribute to Hh responses (Lum et al. 2003; Nybakken et al. 2005). These include components of the ribosome, the vesicular transport pathway, the RNA splicing and transport pathways, and several protein kinases (Nybakken et al. 2005), as well as Hh-binding proteins such as Ihog (Lum et al. 2003; Yao et al. 2006).
In this report, we describe a deficiency screen for genomic regions that enhance or suppress a smo partial loss-of-function wing vein phenotype. Twenty-six regions were identified that scored in this screen; twenty-two of these do not contain genes that encode known Hh pathway components. We report on the characterization two genes located in these regions: microtubule star (mts; Snaith et al. 1996), which positively regulates Hh signaling and whose loss of function enhances the smo knockdown phenotype; and second mitotic wave missing (swm; Gay and Contamine 1993), which negatively regulates Hh signaling and whose loss of function suppresses the smo knockdown phenotype.
MATERIALS AND METHODS
Drosophila stocks and culture:
Deficiency kit collections of stocks carrying deletions for chromosomes 2 and 3 were obtained from the Bloomington Drosophila Stock Center (Bloomington, IN). These deletions are estimated to cumulatively delete 92–95% of the euchromatin (flystocks.bio.indiana.edu). Stocks carrying mutations in candidate genes were obtained from individual labs and from the Bloomington and Szeged Stock Centers. Stocks expressing RNAi's of candidate genes were generously provided by R. Ueda and K. Fujitani (National Institute of Genetics, Mishima, Japan). Flies were cultured on standard cornmeal/molasses medium at 25°; crosses with smo RNAi were carried out at 29°.
Molecular cloning and sequence analysis:
PCR products were generated using Vent DNA Polymerase (New England Biolabs, Beverly, MA), subcloned into pCR2.1-TOPO (Invitrogen, Carlsbad, CA), and sequenced. A smo hairpin RNAi in “tail to tail” orientation was cloned into the GAL4-inducible RNAi vector pWIZ (Lee and Carthew 2003) to create pWIZ-smo (Ogden et al. 2006). To generate pUAS-swm, a swm cDNA was amplified (primer pair: TTTCTCGAGATGATTCTGGAGAATTCGGACAAGC and TTTTCTAGATCAACGACGCCAGGAGCGATCCTCG) and the product was subcloned into pUAST (Brand and Perrimon 1993). To generate GFP-Swm, GFPS65T was amplified (primer pair: TTTCTCGAGATGATTCTGGAGAATTCGGACAAGC and TTTCTCGAGTTTGTATAGTTCATCCATGCC), a swm (CG10084) cDNA was amplified (primer pair: TTTCTCGAGATGATTCTGGAGAATTCGGACAAGC and CGAGGATCGCTCCTGGCGTCGTTGATCTAGAAAA), and the products were ligated together after XhoI digestion. The resulting fusion protein was subcloned into pUAST after XbaI digestion.
Protein sequence was analyzed for motifs with InterProScan (http://www.ebi.ac.uk/InterProScan; Apweiler et al. 2001) and PredictNLS (http://cubic.bioc.columbia.edu/predictNLS; Cokol et al. 2000).
Cell and tissue staining:
Drosophila S2 cells were cultured in Shields and Sang M3 media (Sigma, St. Louis) supplemented with 10% heat-inactivated fetal bovine serum. To determine the subcellular localization of GFP-Swm, S2 cells were cotransfected with pA5c-GAL4 (Ramirez-Weber et al. 2000) and pUAS-GFP-swm using Effectene (QIAGEN, Valencia, CA). After 48 hr, cells were transferred to Lab-Tek Permanox chamber slides (Nagle Nunc International, Rochester, NY) coated with concanavalin A (Sigma-Aldrich, St. Louis) for 20 min prior to examination using a 100× objective. Imaginal discs from wandering third instar larvae were fixed in PBS containing 4% formaldehyde for 20 min, and stained overnight in PBS containing 5% normal donkey serum and 0.3% Triton X-100 with affinity-purified monoclonal mouse anti-Ptc antisera (1:50; Capdevila et al. 1994), or rat monoclonal anti-Ci antisera (1:2000; Motzny and Holmgren 1995), followed by detection with Alexaflour-488 or -555 conjugated secondary antibodies (Invitrogen, Carlsbad, CA). Wings were mounted in Euparol. To estimate cell size in the wing blade, the intervein region between veins 4 and 5 of 11 wings of each genotype was photographed, and the number of wing hairs from equal-sized regions was counted.
smo interaction screen:
A y1 w*; ptcGAL4 P{WIZ-smo}2B chromosome which causes a partial fusion of longitudinal wing veins 3 and 4 was screened for enhancement or suppression when crossed to deficiency kits for chromosomes 2 and 3 (Bloomington Drosophila Stock Center). Chromosomes harboring deletions from deficiency kits were initially outcrossed into a y− background and placed over a corresponding autosome marked with y+: males carrying chromosome 2 deletions were crossed with stock B-1816 females (y1 w1118; P{Car20y}25F FRT40A), and males with chromosome 3 deletions were crossed with B-16761 females (y1 w67c23; P{EPgy2}CG17370EY06831). Males with third chromosome deletions balanced over TM2 (emc2 Ubx130 es) were initially outcrossed to B-20124 females (y1 w67c23; P{EPgy2}emcEY01657/TM3, Sb1 Ser1). Phenotypically y+ male progeny from these crosses carrying the deficiency over y+ were then mated with y1 w*; ptcGAL4 P{WIZ-smo}2B females at 29°. (For deficiencies balanced over TM2, the y+ emc+ male progeny that carry deficiencies over the y+ marked third chromosome were used.) Among the progeny of this second cross, y+ control male flies were compared with their y− siblings carrying the deletion chromosomes. In this way, each of the deficiency stocks was backcrossed once before testing its effects on the wing smo RNAi phenotype, and all of the crosses scored for enhancement/suppression had an internal sibling control. An example of this strategy for chromosome 2 is diagrammed below. Any deficiency chromosome which carried a y+ marker was backcrossed initially to y1, w* females, male progeny were then crossed to y1 w*; ptcGAL4 P{WIZ-smo}2B females at 29°, and y+ male progeny carry the deficiency were compared to their y− control siblings. Deficiencies that scored as enhancers or suppressors were retested. Crossing scheme for enhancer/suppressor screen is as follows:
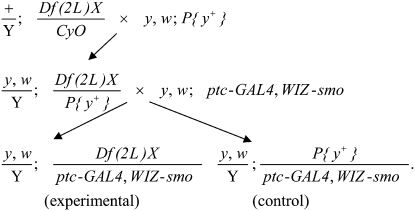 |
Candidate smo modifier genes in regions that scored in the screen were tested with stocks carrying smaller deletions, UAS-RNAi lines, or extant alleles. RNAi lines were screened using both ptcGAL4 P{WIZ-smo} (in which smo activity is reduced in the Hh-receiving cells) and y1 w*; C765GAL4 UAS-smo5A (in which a dominant negative allele of smo is expressed in both anterior and posterior compartment cells, causing a reduction in wing size (Collins and Cohen 2005). Any mutant alleles, RNAi lines, or minimal deficiencies found to enhance or suppress both smo RNAi and smo5a were then tested for nonspecific effects in a cross to y1 w*; P{GAL4-vg.M}2 pUAS-cutRNAi1 (generously provided by Wesley Gruber and Yuh-Nun Jan), which produces a jagged wing-blade margin.
Isolation of swm alleles:
Male flies (B-1646) w1118; P{white-un1}30C FRT40A were desiccated and starved at room temperature for 6 hr, followed by 24 hr with 7.5 mm 1,3-butadiene diepoxide (Sigma-Aldrich) in 1% sucrose (Reardon et al. 1987). As diagrammed below, treated flies were mated with y1 w67c23; In(2LR)Gla wgGla-1/SM6a females en masse for 3 days, after which the males were removed. Progeny carrying mutagenized second chromosomes balanced with SM6a were crossed to swm37Dh-1, pr1 (Gay and Contamine 1993) balanced with CyO-WeeP, a GFP-tagged CyO balancer (Clyne et al. 2003). Noncomplementing w+, pr+, and GFP+ progeny were recovered and tested for complementation with Df(2L) Exel8041, which removes swm among ∼40 genes in 37D7–37F2. To identify lesions in the swm gene from swm mutant stocks, the swm locus was amplified in two segments from genomic DNA essentially as described (Casso et al. 2005) using the following primer pairs: AAATCGCAGGAGCACAGTCG and TGCTGTCGTTATGCCAGAAGACCT; and AACGAAACGAAAGGTGCCG and GGTAACACCGATAAAAAGTGGCG. Genomic DNA from GFP− larval progeny of swm/CyO-GFP stocks was isolated and three independent PCR products were subcloned into pCR2.1-TOPO and sequenced. Crossing scheme for swm allele isolation is as follows:
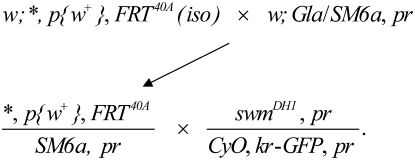 |
Wing analysis:
Interactions with hhMrt were scored by classifying the severity of wing malformations with a system similar to that described by Felsenfeld and Kennison (1995). Class I, wild type; II, margin defects (e.g., thickened margin), without any distortion of wing shape; III, anterior overgrowths, but without duplications or blistering; and IV, severe overgrowths which are rounded, have duplications, or show blistering. Approximately 100 wings from male flies were analyzed from each cross. The results of representative hhMrt crosses are presented in the text as percentages for each class.
To calculate the relative sizes of the regions between veins 3 and 4 in swmDH1/swmF15 escaper and wild-type wings, wings were mounted in Euparol and photographed. Prints were cut between the alula and costa to remove the hinge, then along veins 3 and 4. The size of each region (anterior margin to vein 3, from vein 3 to vein 4, and from vein 4 to posterior margin) was measured by weighing the respective piece and normalizing it to the total size of the wing. In all cases of quantitation, errors indicate standard deviation.
Clonal analysis:
Clones were induced with either a negative marking system (hsflp, Ubi-GFP(S65T)nls2L FRT40A), or the positive MARCM system (Lee et al. 2000) using y* w* hsFLP; tubP-GAL80LL10 FRT40A/CyO ActGFPJMR1; tubP-GAL4LL7, UAS-mCD8∷GFPLL6/TM6 Tb1 (Bello et al. 2006). M+ clones of swm in a M/+ background were generated with w*; M(2)24F1 P{piM}36F FRT40A. Germline clones were generated by the FLP-DFS technique with the stock P{ovoD1-18}2La P{ovoD1-18}2Lb FRT40A/CyO (Chou and Perrimon 1992).
RESULTS
A screen for enhancers and suppressors of smo wing phenotype:
The screen we carried out is based on comparisons of euploid and partially haploid flies that have reduced smo function due to expression of smo RNAi in the Hh-responsive intervein region of the wing. In developing this screen, we first established that knocking down components of the Hh signal transduction pathway by RNAi provides a reliable measure of Hh signaling. Injection of RNAi directed against components of the Hh signaling pathway strongly inhibits embryonic segmentation, and affected embryos recapitulate full loss-of-function phenotypes observed in null mutants (Ramirez-Weber et al. 2000). To assess the efficacy of RNAi knockdown in the adult wing, we designed strains that target smo with RNAi.
smo RNAi expressed under the control of ptc produced partial proximal fusions of longitudinal veins 3 and 4 near the anterior cross vein (Figure 1, A and B; Ogden et al. 2006). These fusions are similar to the defects that are observed with weak loss-of-function alleles of fused (fu), a gene that encodes a putative protein kinase that is required downstream of smo for Hh pathway activation. Although viable loss-of-function smo alleles have not been described, the fused vein phenotype is consistent with reduced hh signaling. This phenotype was obtained with either T80GAL4 (ubiquitous expression in the wing disc; not shown) or ptcGAL4, but not when expression was restricted to the posterior compartment (with either hhGAL4 or enGAL4). Wings with these latter genotypes were indistinguishable from wild type (not shown), indicating that the vein-fusion phenotype was dependent on expression of the RNAi in the anterior compartment cells that require Hh signaling.
Figure 1.—

smo RNAi in the adult wing. (A) wild type, (B) ptcGAL4 WIZ-smo/+, (C) ptcGAL4 WIZ-smo/UAS-smo, (D) ptcGAL4 WIZ-smo/smo3, (E) ptcGAL4 WIZ-smo/+, hh10/+, (F) ptcGAL4 WIZ-smo/enE, (G) ptcGAL4 WIZ-smo/ptc10, (H) ptcGAL4 WIZ-smo/Df(2L)BSC41, (I) ptcGAL4 WIZ-smo/mtsXE-2258, (J) ptcGAL4 WIZ-smo/Df(2L)Exel8041, (K) ptcGAL4 WIZ-smo/swmDh-1, (L) ptcGAL4 WIZ-smo/swmF11, (M) ptcGAL4/10084R1 (swm RNAi), and (N) ptcGAL4 WIZ-smo/10084R1. Enhancement and suppression of the smo RNAi phenotype was scored as the distance between veins 3 and 4 in the proximal part of the wing near the anterior crossvein (see arrow in B). Ectopic vein tissue between veins 3 and 4 was seen in many genetic interactions with smo RNAi (e.g., D, G, and J), but it did not correlate with enhancement or suppression.
Since the ptcGAL4 smo RNAi phenotype was both consistent and highly penetrant, its sensitivity to levels of Hh pathway components could be tested. The smo RNAi phenotype was completely suppressed by coexpression of a smo cDNA (Figure 1C), indicating that the wing phenotype was most likely due to RNAi-mediated reduction in smo expression. In contrast, reducing the number of functional smo genes by introducing an amorphic allele (either the deletion, smoD, or the truncation generated by a stop codon in the second intracellular loop, smo3) increased the extent of the 3–4 vein fusion. This is indicative of an enhanced effect (Figure 1D and data not shown; Chen et al. 1998; Alcedo et al. 2000). Reducing hh function (with either the hhAC deletion or hh10 amorphic allele) enhanced vein fusion in smo RNAi-expressing flies, although to a lesser extent than the enhancement observed with smo3 or smoD (Figure 1E and data not shown). We also reduced hh expression by introducing the mutant enE (Gustavson et al. 1996), which is deleted for the positive regulators of hh expression, en and invected (inv). Hemizygosity for en/inv enhanced the smo RNAi phenotype; although the enhancement was less than with either the smo or hh mutants, it was reproducible (Figure 1F). Suppression of the smo RNAi phenotype was observed when ptc function was reduced in ptc10 heterozygotes (Figure 1G). ptc10 is an EMS-induced null allele (Ramirez-Weber et al. 2000). Since Ptc is a negative regulator of Hh signaling, we conclude that smo RNAi assay is a reliable indicator for the pathway.
Identification of deficiencies that modify smo RNAi:
We challenged the smo RNAi phenotype with 216 lines from the Bloomington Drosophila Stock Center deficiency collection for chromosomes 2 and 3. In aggregate, these lines are estimated to delete >90% of the autosomal euchromatin. Since their diverse genetic backgrounds are likely to affect the smo RNAi phenotype, each line was crossed into a common background prior to testing. Wing phenotype was monitored among F2 progeny, and since flies carrying the deficiency could be distinguished from nondeficiency bearing flies, each cross was internally controlled for subtle background effects. Crosses were scored by comparing the vein morphology of a minimum of 25 flies of each genotype. Every deficiency observed to enhance or suppress the smo RNAi phenotype was retested, and regions that gave consistent effects were characterized further. If available, additional genomic deletions in the region were tested, and any available lines harboring mutations in genes uncovered by the smallest deletion were tested for enhancement or suppression. This analysis entailed >2500 crosses.
The deficiencies that modify the smo RNAi phenotype are listed in Tables 1 and 2. Figure 2 depicts the cytological positions of deficiencies and mutations that either enhance (red) or suppress (blue) the 3–4 vein-fusion phenotype. Among the deficiencies identified in the screen are ones that removed the known Hh pathway components en, ptc, and hh. (There are currently no multigenic deficiencies available for the smo region.) The en, ptc, and hh deficiencies produced phenotypes that mimicked the mutant alleles of these genes. In addition to these regions, there were nine that suppressed and thirteen that enhanced the vein-fusion phenotype. We have identified protein-encoding genes that affect Hh signaling in two of these regions.
TABLE 1.
Deficiencies on chromosomes 2 and 3 that enhance smo RNAi
| Deficiency | Start | End | Interaction | Candidate gene |
|---|---|---|---|---|
| Df(2L)BSC37 | 22D2–3 | 22F1–2 | Lethal | |
| Df(2L)GpdhA | 25D7–E1 | 26A8–9 | Enhancer | |
| Df(2L)cl7 | 25E1–2 | 26A7 | Enhancer | |
| Df(2L)ED334 | 25F2 | 26B2 | Weak enhancer | |
| Df(2L)E110 | 25F3–26A1 | 26D3–11 | Enhancer | |
| In(2LR)DTD116[L]DTD24[R] | 26A4–6 | 26C1–2 | Weak enhancer | |
| Df(2L)BSC5 | 26B1–2 | 26D1–2 | Weak enhancer | |
| Df(2L)ED353 | 26B2 | 26B5 | Enhancer | |
| Df(2L)XE-3801 | 27E2 | 28D1 | Enhancer | mts |
| Df(2L)RF | 27E3–F | 28B3–4 | Weak enhancer | |
| Df(2L)BSC41 | 28A4–B1 | 28D3–9 | Enhancer | mts |
| Df(2L)N22-14 | 29C1–2 | 30C8–9 | Weak enhancer | |
| Df(2L)N22-5 | 29D1–2 | 30C4–D1 | Weak enhancer | |
| Df(2L)30A-C | 29F7–30A1 | 30C2–5 | Weak enhancer | |
| Df(2L)N22-3 | 30A1–2 | 30D1–2 | Enhancer | |
| Df(2L)gamma7 | 30A9–B1 | 30D2–F4 | Weak enhancer | |
| Df(2L)BSC17 | 30C3–5 | 30F1 | Weak enhancer | |
| Df(2L)J2 | 31B | 32A | Enhancer | |
| Df(2L)J3 | 31D | 31F | Enhancer | |
| Df(2L)J106 | 31D | 31F2 | Weak enhancer | |
| In(2LR)PL | 31F | 51C | Weak enhancer | |
| Df(2L)BSC32 | 32A1–2 | 32C5–D1 | Weak enhancer | |
| Df(2L)BSC30 | 34A3 | 34B7–9 | Enhancer | |
| Df(2R)E3363 | 47A | 47F | Enhanced, small wing | |
| Df(2R)en-B | 47E3 | 48A4 | Enhancer | en |
| Df(2R)en-SFX31 | 48A | 48B | Enhancer | en |
| Df(2R)RM2-1 | 54F2 | 56A1 | Enhancer | |
| Df(2R)PC4 | 55A | 55F | Weak enhancer | |
| Df(3L)M21 | 62F | 63D | Enhancer | |
| Df(3L)HR119 | 63C2 | 63F7 | Enhancer | |
| Df(3L)vin6 | 68C8–11 | 69A4–5 | Enhancer | |
| Df(3L)Exel6116 | 68F2 | 69A2 | Enhancer | |
| Df(3L)vin5 | 69A2–3 | 69A1–3 | Enhancer | |
| Df(3R)M-Kx1 | 86C1 | 87B1–5 | Weak enhancer | |
| Df(3R)Exel6162 | 87A1 | 87B5 | Lethal | |
| Df(3R)ry615 | 87B11–13 | 87E8–11 | Weak enhancer | |
| Df(3R)Exel8157 | 87D8 | 87D10 | Weak enhancer | |
| Df(3R)Exel6193 | 94D3 | 94E4 | Enhancer | hh |
| Df(3R)BSC56 | 94E1–2 | 94F1–2 | Enhancer | hh |
| Df(3R)96B | 96A21 | 96B8–10 | Enhancer | |
| Df(3R)Espl3 | 96F1 | 97B1 | Enhancer |
Note that there are no multigenic deficiencies that span the smo locus.
TABLE 2.
Deficiencies on chromosomes 2 and 3 that suppress smo RNAi
| Deficiency | Start | End | Interaction | Candidate gene |
|---|---|---|---|---|
| Df(2L)cact-255rv64 | 35F–36A | 36D | Suppressor | |
| Df(2L)TW50 | 36E4–F1 | 38A6–A7 | Suppressor | swm |
| Df(2L)pr-A16 | 37B2–12 | 38D2–D5 | Suppressor | swm |
| Df(2L)TW158 | 37B2–B8 | 37E2–F1 | Suppressor | swm |
| Df(2L)VA17 | 37C1 | 37F5 | Suppressor | swm |
| Df(2L)VA12 | 37C2–5 | 38B2–C1 | Suppressor | swm |
| Df(2L)Sd77 | 37D1–D2 | 38C1–C2 | Suppressor | swm |
| Df(2L)Sd37 | 37D2–D5 | 38A6–B2 | Suppressor | swm |
| Df(2L)pr-A14 | 37D2–D7 | 39A4–A7 | Suppressor | swm |
| Df(2L)E55 | 37D2–E1 | 37F5–38A1 | Suppressor | swm |
| Df(2L)Exel8041 | 37D7 | 37F2 | Suppressor | swm |
| Df(2R)H3C1 | 43F | 44D3–D8 | Suppressor | |
| Df(2R)44CE | 44C4 | 44E4 | Suppressor | ptc |
| Df(2R)H3E1 | 44D1–D4 | 44F12 | Suppressor | ptc |
| Df(2R)H3D3 | 44D1–4 | 44F4–5 | Suppressor | ptc |
| Df(2R)Exel8047 | 44D4 | 44D5 | Suppressor | ptc |
| Df(2R)Exel7098 | 44D5 | 44E3 | Suppressor | ptc |
| Df(2R)CX1 | 49C1–4 | 50C23–D1 | Weak suppressor | |
| Df(2R)X58-8 | 58B3 | 59A1 | Suppressor | |
| Df(2R)59AD | 59A1–A3 | 59D1–D4 | Suppressor | |
| Df(3L)CH39 | 64 | 65B5–C1 | Weak suppressor | |
| Df(3L)ED210 | 64B9 | 64C13 | Weak suppressor | |
| Df(3L)CH18 | 64B–C | 65B5–C1 | Weak suppressor | |
| Df(3L)ZN47 | 64C | 65C | Suppressor | |
| Df(3L)CH20 | 64D1–2 | 65C3 | Suppressor | |
| Df(3L)Exel6107 | 64E5 | 64F5 | Suppressor | |
| Df(3L)vin2 | 67F2–F3 | 68D6 | Weak suppressor | |
| Df(3L)ED4470 | 68A6 | 68E1 | Weak suppressor | |
| Df(3L)D-5rv12 | 70C2 | 72A1 | Suppressor | |
| Df(3R)D605 | 97E3 | 98A5 | Suppressor | |
| Df(3R)R38.3 | 97E3–11 | 98A | Suppressor |
Figure 2.—

Results of the smo RNAi enhancer-suppressor screen. Chromosomes 2 and 3 are depicted graphically as black lines. Below, deletions and mutant chromosomes that enhance (red) or suppress (blue) this phenotype are indicated with bars. Cytological locations are marked, and candidate enhancer and suppressor genes are indicated.
microtubule star:
The screen mapped a mild enhancement function to the 28A–D interval using deficiencies Df(2L)XE-3801 and Df(2L)BSC41 (Table 1, Figure 1H, and data not shown). This region contains microtubule star (mts; CG7109), and we confirmed that these deficiencies fail to complement mts (Wassarman et al. 1996). We tested five mts alleles in the smo RNAi assay. One, mtsXE-2258, recapitulated the effects of the deficiencies by weakly enhancing the phenotype (Figure 1I); it has a 16-bp deletion that includes the translation start (Wassarman et al. 1996). None of the four P-element insertions (P{lacW}mtsk12502, P{PZ}mts02496, P{PZ}mts05559, and P{lacW}mtss5286) yielded significant enhancement.
To test mtsXE-2258 for effects on Hh signaling with an assay that is independent of smo RNAi and the GAL4 system, crosses were made to the hh allele, hhMrt. hhMrt is a dominant gain-of-function allele that causes ectopic expression of hh along the dorsal/ventral border in the anterior compartment of wing discs; it causes overgrowth of anterior distal wing structures (Felsenfeld and Kennison 1995). Reducing the dosage of mts by introducing mtsXE-2258 or either of the two mts deficiencies strongly suppressed the hhMrt wing overgrowth, resulting in wings that approached wild type. In control crosses, over two-thirds of the hhMrt/+ wings showed overgrowths (class I, 0%; II, 13%; III, 71%; and IV, 16%). When mts function was reduced in mtsXE-2258/+; hhMrt/+, the percentage of wings with overgrowths was reduced by >50% (class I, 32%; II, 36%; III, 28%; and IV, 4%). At the same time, the percentage of wings with normal overall shape (classes I and II) increased from 13 to 68%. mts encodes the catalytic subunit of protein phosphatase type 2A (PP2A); its name derives from the unusual appearance of centrosomal microtubules in mutant embryos (Snaith et al. 1996). Consistent with our in vivo data, in vitro experiments from an RNAi-based screen of Drosophila CL8 cultured cells also suggest that mts is required for maximal Hh pathway activation (Nybakken et al. 2005).
second mitotic wave missing:
The screen identified a strong suppression function in the 37D7–F1 interval. Nine overlapping deletions were identified in this region that suppressed the smo RNAi phenotype (Table 2); these define a region that is predicted to include 32 genes. Among 13 RNAi lines and 12 point mutants in this region that we tested, four suppressed smo RNAi in the manner of the smallest deletion, Df(2L)Exel8041 (37D7–37F2; Figure 1J and data not shown). These four are 10084R1 and 10084R2 (further examination revealed that these two GAL4-driven RNAi lines are isogenic), swm37Dh-1 and swm37Dh-9 (Figure 1, K, M, and N, and data not shown). swm37Dh-1 and 10084R2 suppressed expression of dominant negative smo5A, but not cut RNAi expression (data not shown).
The swm gene (CG10084; 37E4) was first identified genetically as a recessive lethal complementation group in a screen for new alleles of ref(2)P (Gay and Contamine 1993). ref(2)P determines susceptibility to Sigma virus. Nine ethyl methanesulfonate-induced swm alleles, including swm37Dh-1 and swm37Dh-9, were isolated. More recently, swm alleles [S(rux)2B1 and S(rux)2B2, aka swm1 and swm2] were identified in a screen for suppressors of the CDK inhibitor gene roughex (rux; Dong et al. 1997; Foley and Sprenger 2001). S(rux)2B is a recessive lethal that partially rescues a rux roughened eye phenotype (B. Thomas, personal communication; Dong et al. 1997). We confirmed that swmDh-1 increases the size of the eye and partially suppresses the rux1 rough eye phenotype (data not shown).
swm is predicted to produce two transcripts that have identical open reading frames and are distinguished by alternative transcriptional start sites. These transcripts putatively encode a 1062 amino acid protein. Their coding regions distribute over 5 exons that are preceded by an alternatively-spliced 5′ noncoding exon; their five introns are small, totaling <600 nucleotides. The Swm protein is predicted to have four sequence motifs. A CCCH-type Zn+ finger domain (residues 364–398) might function in nucleic acid binding or in protein–protein interactions (Figure 3A, yellow). Residues 574–632 might represent an RNA-binding RNA recognition motif (RRM; Figure 3A, blue), although it might function in nucleotide binding or as a structural domain. Two nuclear localization sequences are predicted (residues 242–250 and 748–769). Expression of a N-terminally tagged GFP–Swm fusion protein in transfected S2 cells generated strong nuclear fluorescence (Figure 6, A–C), consistent with nuclear localization of Swm. Expression in salivary glands was also predominantly nuclear (Figure 6D).
Figure 3.—

The swm locus and sequence. (A) The swm locus has six exons in its swm-RA transcript (thick bars) and five introns (thin lines). The swm-RB transcript is predicted (FlyBase) to have only five exons and an alternative transcriptional start site within intron one (not shown). Both transcripts have the same predicted protein coding region (1062 amino acids, green). Noncoding regions of exons one, two, and six are in black. Two putative functional domains are shown: a CCCH-type Zn+ finger (yellow) and an RNA recognition motif (blue). Mutations in five swm alleles are indicated. (B) Swm protein aligned with human RBM-26 (GenBank, EAW80593) and RBM-27 (GenBank, Q9P2N5). Identity (red), strong similarity (orange), weak similarity (green), no similarity (black), Zn+ finger domain (yellow box), RRM (blue box). Mutations in swm alleles, in boldface type: swmF4 (after D175 GT to AT, the last residues before the splice site); swmDh-1, 157-bp deletion at S249; swmF14, Q314 to stop (CAA to TAA); swmF11, W418 to stop (TAG to TGG); swmF15, D923N.
Figure 6.—

Swm localization and function. (A–C) S2 cells expressing GFP–Swm localize GFP fluorescence to nuclei (A, phase contrast; B, fluorescence; C, merge). (D) GFP–Swm localized to nuclei in third instar salivary glands. (E and F) Wing from a swmF15/Df(2L)Exel8041 escaper (F) has intervein regions 3–4 expanded and misoriented hairs (H), in contrast to wild-type control (E and G).
BLAST searches (Altschul et al. 1990) identified several potential Swm homologs in animal genomes, as well as more distantly related proteins in plants and fungi. The mouse and human genomes each have two putative homologs. The two human gene products are RBM26 (also known as cutaneous T-cell lymphoma tumor antigen se70-2) and RBM27 (also known as peri-implantation stem cell protein 1 for mouse, and KIAA1311 and POU domain, class IV, transcription factor 3 for human; Kavanagh et al. 2005); both have the same predicted domain structure as Drosophila Swm. Their strongest sequence similarities are in the amino terminus, zinc finger, RRM domain, and acidic carboxy terminus (Figure 3B). The mouse and human RBM26 proteins are almost identical to each other, as are the corresponding RBM27 proteins. Multiple splice variants of these vertebrate genes have been identified.
We monitored expression of Drosophila swm by in situ hybridization and detected transcripts at all stages that were examined (Figure 4). Expression was strong in cellularized embryos and was enhanced along the ventral midline. Segmental stripes were evident at germ-band retraction. Late in embryogenesis, swm expression appeared to be ubiquitous. In third instar larval tissues, strong swm expression was observed in imaginal discs, salivary gland, optic lobe, fat body, and in the wreath cells and gastric caecae of the gut.
Figure 4.—

swm expression in embryos and larva. In situ hybridization reveals swm expression in (A) precellular embryo, (B) germ-band extension embryo, (C) germ-band retraction embryo, (D) third instar wing disc (posterior is on the right), (E) eye/antennal disc (mf, morphogenetic furrow; st, stalk), (F) salivary gland (s, secretory cells; d, duct cells), (G) gut (gc, gastric caecae; wc, wreath cells), (H) larval brain (o, optic lobe), and (I) larval fatbody.
Isolation and analysis of swm alleles:
To isolate additional swm mutants, we carried out an F2 screen for noncomplementing alleles of swm37Dh-1. We used an isogenized, viable second chromosome carrying FRT40A that was also marked at 30A with a viable w+ insertion. We found five noncomplementing chromosomes among ∼4000 progeny that were screened.
To determine if the noncomplementing chromosomes have mutant swm alleles, swm coding sequences were determined from the new lines as well as from both the isogenized starting strain, swm37Dh-1 and from a swm cDNA obtained from the Berkeley Drosophila Genome Project collection. The wild-type isogenized chromosome differed from both the swm cDNA and GenBank swm sequences at two positions. Both are likely to be polymophisms; one is silent in codon 996 (ACA to ACT) and the other (TCC to GCC) changes serine 402 to alanine. The swm37Dh-1 allele has a 157-bp deletion in exon 4 that shifts the reading frame at Serine 249 (Figure 3, A and B, mutations are in gray). Mutations were found within the swm transcription unit of four mutagenized chromosomes. The swmF11 and swmF14 alleles have stop codons at residues 418 and 344, respectively, and are predicted to produce truncated proteins. The swmF4 allele has a G-to-A mutation in the first base of the 5′ splice site of intron 3. These three new swm alleles, all of which are predicted to produce proteins deleted of the RRM domain, are strong suppressors of smo RNAi (Figure 1, J–L and data not shown). They are presumed to be nulls.
Heteroallelic combinations of the null swm alleles (swmF4, swmF11, swmF14, swm37Dh-1, swm37Dh-9, and Df(2L)Exel8086) died in L3. Mutant larvae were severely developmentally delayed, and most had wing discs that were <25% the size of wild type.
A missense mutation was identified in swmF15 (D923N; Figure 3B). D923 is conserved in both human Rbm26 (D873) and Rbm27 (D972); it may be in a casein kinase I phosphorylation site. Although swmF15 is recessive lethal, rare transheterozygous escapers with swm37Dh-1, swmF14, and Df(2L)Exel8041 were recovered. In addition, swmF15 is not a dominant suppressor of smo RNAi. We therefore conclude that swm15 retains some function and is hypomorphic.
swmF15 adult escapers (e.g., swmF15/swmDh-1, swmF15/swmF14, and swmF15/Df(2L)Exel8041) were normal size, but were abnormal in several respects. Ocelli were reduced or absent; cephalic macrochaetae were frequently missing; the base of the aristae were frequently enlarged, resembling a weak aristapedia phenotype; eyes were rough; and wings, though normal in size, had ectopic vein tissue and an increased distance between veins 3 and 4 (Figure 6, G and I and data not shown). In swmDH1/swmF15 wings, the region between veins 3 and 4 was 12% larger than the same region in wild-type wings. The 3–4 intervein region comprised 23 ± 1%, (n = 23) of a swm− wing vs. 21 ± 0.3% of a wild-type wing (n = 11). These two populations are different by Student's t-test, P < 3.6 × 10−10. Wings also had a weak wing hair polarity phenotype that is described below.
To investigate how cells that lack swm function develop in the wing disc, three methods were used to induce clones of swm mutant alleles. First, negatively marked clones of two swm amorphic alleles (swmF4 and swmF14) were induced such that the clones lacked GFP expression and twin spots had increased fluorescence relative to nonrecombined cells. Despite an abundance of large twin spots, no swm clones were found. Second, the MARCM technique (Lee et al. 2000) was used to positively mark clones of four alleles (swmF4, swmF11, swmF14, and swmF15). swmF4, swmF11, and swmF14 clones were identified, but they were small (1–2 cells) and few in number. swmF15 clones were slightly larger (up to 8 cells) and were slightly more frequent. Third, M+ mutant clones of three alleles (swmF4, swmF11, and swmF14) were induced in trans with M(2)24F1 to endow swm mutant cells with a growth advantage. Neither the number nor size of clones increased. We conclude that loss of swm function is cell lethal in swm/+ discs. Although mutant cells cannot survive in a background of swm/+ cells, the survival of swmF15 transheterozygotes indicates that swm insufficiency is not generally cell lethal. We do not understand the basis for this behavior. We investigated whether swm function is required in the germline. Although germline clones of the hypomorph swmF15 generated fertile eggs and swmF15/+ adults from such eggs were phenotypically normal, no swmF11 or swmF14 mutant eggs were recovered. In crosses to swmDh-1/CyO-WeeP, swmF15/swmDh-1 progeny of swmF15 female germline clones were larval lethal like other swm transheterozygotes.
swm is a negative regulator of Hh signaling:
Genetic and molecular analyses provided evidence indicating that swm negatively regulates Hh signaling. In the sensitized smo RNAi background, hemizygosity for swm strongly and consistently suppressed the wing vein phenotypes characteristic of reduced Hh signaling (Figure 1, J–L). swm RNAi generated more extreme phenotypes both alone and in combination with smo RNAi (Figure 1, M and N), with a reduction of the distance between veins 2 and 3 reminiscent of ectopic expression of Hh or a dominant negative form of Ptc (Tabata and Kornberg 1994; Porter et al. 1996; Ramirez-Weber et al. 2000; Lu et al. 2006;). When driven by ptcGAL4 at 29°, the swm RNAi phenotype was completely penetrant, and when coexpressed with smo RNAi the fused wing phenotype was suppressed in every fly. In control crosses, swm RNAi did not suppress the wing margin phenotype caused by cut RNAi expression (data not shown), showing that the effects of swm RNAi are not targeted generally and nonspecifically to the RNAi pathway.
Expression of a dominant negative Smo protein, Smo5A (Collins and Cohen 2005), causes wing vein fusions that are similar to smo RNAi. This mutant protein has alanines substituted for serines and threonines in the C-terminal cytoplasmic tail that are phosphorylated upon Hh activation, and its expression in the wing partially blocks Hh signaling (Collins and Cohen 2005). Reducing swm function (e.g., swm37Dh-1/+ or swm RNAi) partially suppressed the vein-fusion phenotype of smo5A-expressing flies (not shown); the capacity for swm37Dh-1 hemizygosity to counteract the effects of reduced Hh signaling is not specific to smo RNAi.
We also examined the swm insufficiency phenotype in a Hh gain-of-function background. As noted above, in the hhMrt mutant, Hh is ectopically expressed along the dorsal ventral border of the wing disc and the majority of wings develop with anterior overgrowths (class I, 0%; II, 13%; III, 71%; and IV, 16%; see Figure 5, A and B). swm37Dh-1/+; hhMrt/+ wings had more severe overgrowths (class I, 0%; II, 5%; III, 30%; and IV, 65%; see Figure 5, C and D). While severely distorted class IV wings were rare in hhMrt/+ flies, the majority of swm37Dh-1/+; hhMrt/+ wings were in this class. Antibody staining of the Hh targets Ptc and Ci revealed patterns that were consistent with elevated Hh signaling. Levels of Ptc and Ci are normally elevated at the A/P compartment border, and both were also present along the D/V border in the anterior compartment where Hh is ectopically expressed in hhMrt discs (Figure 5, E, F, H, and I). In swm37Dh-1/+; hhMrt/+ discs, expression of both proteins was greatly enhanced along the D/V border (Figure 5, G and J).
Figure 5.—

hhMrt is enhanced by swm. Wings: (A) wild type, (B) hhMrt/+, (C) and (D) swmDh-1/+; hhMrt/+. Third instar wing imaginal discs were stained for Ptc (E, F, and G) or Ci (H, I, and J) protein. Genotypes are as follows: (E and H) wild type; (F and I) hhMrt/+; and swmDh-1/+; (G and J) hhMrt/+. The arrows indicate ectopic expression of Ptc and Ci in the anterior compartment along the dorsal ventral compartment boundary.
Cell polarity and cell-size defects in swm mutants:
In addition to the abnormalities in the wings noted above, wings of the three heteroallelic swm genotypes that produced escapers had subtle wing hair polarity defects. Whereas the orientation of hairs in wild-type wings is highly regular, with hairs of neighboring cells having almost identical proximodistal orientations, most wings of swm mutants (swmF15/swmDh-1, 70%; swmF15/swmF14, 76%; swmF15/Df(2L)Exel8041, 100%) had patches that deviated 30–40% from the normal orientation. A mild form of this phenotype was also evident in posterior wing compartments in which swm function was reduced with enGAL4 swm RNAi (data not shown). These patches did not generate “whorls” characteristic of mutants in planar cell polarity (PCP) genes, but had hairs with orientations that were noticeably askew (Figure 6, G and H) and had occasional cells with multiple wing hairs (not shown). We also observed patches of misoriented hairs in wings that had not been mounted under a coverslip, establishing that the phenotype is not an artifact of preparation. We did not find an effect on bristle orientation or on the orientation of hairs in the abdomen.
swm and wild-type wings were approximately equal size, but the density of wing hairs in swm wings was less than wild type. As each cell normally produces a single distal wing hair, the number of hairs in a given area provides a measure of the number and the size of cells. In equivalent areas between veins 4 and 5 that we chose for purposes of comparison, swm wings had ∼20–28% fewer cells (swmF14/swmF15, 76 ± 5 cells; swmF15/swmDh-1, 68 ± 8 cells; wild type, 95 ± 3 cells; n = 11 wings), indicating that reduction of swm causes an increase in wing cell size.
DISCUSSION
The screen for effectors of the Hh pathway:
This screen identified twenty-six autosomal regions that modified a smo hypomorphic phenotype in a dosage-sensitive manner. Two aspects of its design were key to its success. First, its two-generation crossing scheme eliminated background effects by homogenizing the genetic backgrounds of both experimental and control flies. It also generated reasonably large numbers of both classes of progeny so that a good estimate of an average phenotype could be obtained. These features allowed us to monitor subtle variations in wing vein morphology, despite the significant strain differences among the many lines we tested. Second, its high scoring threshold rendered it relatively insensitive to changes in Hh signaling strength, thereby helping to submerge weak influences. Key to this property was the ptcGAL4 driver that was used to express smo RNAi; it functioned in part as a “genetic buffer.” Since ptcGAL4 is itself responsive to Hh, a modifier that increased Hh signaling would also be predicted to increase the expression of ptcGAL4 and smo RNAi, while a modifier that decreased Hh signaling might be expected to decrease the expression of the ptcGAL4 and smo RNAi. ptcGAL4 therefore buffered against changes in signaling strength and decreased the effects of genetic factors that enhance or suppress signaling; as a consequence, only highly penetrant and consistent phenotypes were scored.
Our screen netted many of the known core components of the Hh signaling pathway, including smo, ptc, hh, and en. We report on mts and swm, two genes whose haplo-insufficiency phenotypes were sufficiently strong to score above the threshold set by our genetic tests. Many other known regulators of Hh signaling were not identified in our screen. There are perhaps multiple reasons, including the high scoring threshold of the smo RNAi screen, or the possibility that not all pathway regulators have haplo-insufficiency phenotypes. skinny hedgehog or suppressor of fused were not included among those identified in the screen, despite the fact that deficiencies that removed them interacted with smo RNAi. The reason is that mutant alleles of these genes that were tested did not yield similar interaction phenotypes. Since we observed many examples of interaction between null alleles of Hh pathway regulators and smo RNAi but consistent failure of hypomorphic alleles to interact, we do not view that lack of interaction as evidence against a gene being a smo RNAi enhancer/suppressor. The possibility that stronger alleles might interact cannot be discounted. We were surprised that hemizygosity of cos2 did not show an interaction with smo RNAi. This could be because it is not haplo-insufficient in our particular assay or because of the complex positive and negative roles cos2 plays in Hh signaling. Finally, there was no apparent overlap between the regions we identified and the mutant lines that were identified in previous screens for modifiers of Hh phenotypes (Haines and van den Heuvel 2000; Collins and Cohen 2005); the smo RNAi assay may be less sensitive but more specific.
microtubule star:
mts lies within 1 of 16 regions that enhanced the smo RNAi phenotype, suggesting that its wild-type function augments the Hh response. mts encodes the catalytic (C) subunit of PP2A, a heterotrimeric phosphatase that has two regulatory subunits, B and B′. It was previously identified as a Hh pathway regulator in CL8 cells (Nybakken et al. 2005); our work now provides in vivo evidence for a role in Hh signaling during development. Three proteins in Hh signal transduction have been shown to be functionally phosphorylated. Phosphorylation of the Smo C terminus is induced by Hh and is required for surface accumulation of Smo and normal activation of the pathway (Zhang et al. 2001, 2004; Apionishev et al. 2005). Thus, reduction of PP2A activity and increased phosphorylation of Smo would not be expected to decrease Hh signaling and enhance the smo RNAi phenotype. Other possible targets of Mts are Ci and Cos2. Phosphorylation of Ci by PKA, casein kinase 1α, and GSK3β is required to convert Ci from its full-length form to its transcriptional repressor form, Ci-75 (Chen et al. 1998; Jia et al. 2002; Price and Kalderon 2002). Hh signaling blocks this proteolytic transformation and also promotes conversion of Ci to an activator form (Aza-Blanc et al. 1997). A decrease in phosphatase activity might increase levels of phosphorylated Ci to effect enhanced conversion to Ci-75 and reduced levels of Ci activator. Levels of Hh signaling would be predicted to decrease. Alternatively, Mts might control phosphorylation of Cos2 by Fu. Phosphorylation of Cos2 prevents its binding to Smo and release of Smo from Cos2 increases the cell surface accumulation of Smo that is necessary for pathway activation (Denef et al. 2000; Liu et al. 2007). Therefore, a reduction of Smo on the plasma membrane due to loss of PP2A activity might attenuate Hh pathway activation.
While the catalytic subunit of PP2A carries enzymatic phosphatase activity, the substrate specificity of PP2A is directed by its regulatory subunits. The phenotypes of mutants in genes that encode the B and B′ regulatory subunits of PP2A, twins and widerborst (wdb), respectively, are interesting to consider in the context of Hh signaling. Wing discs in the twinsP mutant have mirror symmetrical posterior compartment duplications that are associated with ectopic compartment borders (Uemura et al. 1993). Symmetric wing duplications have also been observed after ectopic expression of Hh or Dpp (Zecca et al. 1995 and reviewed in Tabata and Takei 2004), or after loss of en/inv induces an ectopic compartment border (Tabata et al. 1995). Since loss of PP2A function should reduce Hh signaling, it is not obvious how loss of the B twins regulatory subunit leads to an ectopic signaling center. Understanding this interesting aspect of the twins phenotype warrants further investigation.
Misexpression of PP2A can cause cell planar polarity defects in the wing. Misexpression of mts, wdb, or mutant alleles of these genes disrupted wing hair polarity (Hannus et al. 2002). Like mts, reducing wdb expression with RNAi reduced Hh signaling in CL8 cells (Nybakken et al. 2005). This evidence, as well as the wing hair polarity phenotype of swm mutants, raises the possibility that PP2A links Hh signaling with cell polarity. The PCP and Hh pathways may be parallel and independent if PP2A activity is simply common to both, but evidence that Hh is required to establish PCP in the Drosophila embryonic and adult epidermis has recently been described (Colosimo and Tolwinski 2006; Lawrence et al. 2007). As discussed below, the phenotype of swm mutants provides additional evidence for an association of Hh signaling with cell polarity.
second mitotic wave missing:
swm was first identified as l(2)37Dh by Gay and Contamine (1993) in a screen for recessive lethal alleles within Df(2L)E55 (37D2–38A1). It was shown to exhibit synthetic lethality as an enhancer of Minutes. Among the mutant chromosomes from our screen that failed to complement swm, one had a Minute-like phenotype. No changes in the swm coding sequence were found in this mutant; rare escapers that eclosed as heterozygotes with our verified swm alleles had a variety of phenotypes including loss of ocelli, thin macrochetae, and deformed legs. In contrast to swm mutant escapers, however, both their eyes and wings were phenotypically normal (not shown).
More recently, swm was identified as a suppressor of the roughex eye phenotype (Dong et al. 1997). Alleles of ptc were also isolated in this screen (B. Thomas, personal communication). We confirmed these interactions between rux, ptc, and swm (data not shown). Since Ptc is a negative regulator of the Hh pathway and ptc mutations are therefore likely to elevate Hh signaling, and since Hh plays a key role in eye morphogenesis, the rux phenotype is apparently sensitive to Hh levels. We therefore interpret the identification of both ptc and swm mutants as rux suppressors as a consequence of the same mechanism—an increase in Hh signaling caused by a decrease in the level of a negative regulator.
Our results provide several additional lines of evidence that swm negatively regulates Hh signaling. swm mutants dominantly suppress smo hypomorphic phenotypes (smo RNAi and smo5A; Figure 1 and data not shown), enhance a Hh gain-of-function phenotype (hhMrt; Figure 5), and increase targets of Hh signaling such as Ptc and Ci (Figures 5 and 6). These effects on Hh signaling seem to occur through swm activity in the anterior compartment since swm RNAi expressed in these cells is sufficient to suppress smo RNAi. Although these interactions implicate Swm, we have not determined how and where Swm impacts signal transduction or what its molecular function might be. Swm protein has features suggestive of a function in nucleic acid metabolism—it has a putative RRM RNA binding domain and a CCCH Zn+ finger (Figure 3), and a GFP–Swm fusion we examined localized to nuclei in cultured cells (Figure 6). Presumably, Swm affects expression, production, or presentation of proteins involved in Hh signaling or signal transduction. However, swm function is not specific to Hh signaling, since many aspects of the phenotype (e.g., ectopic venation, wing hair polarity, cell size, and interaction with Minutes) are not attributable to defects in Hh signaling.
swm is expressed broadly in both embryos and larvae, and in wing discs, it appears to be required in all cells. Null alleles, which are cell lethal in a swm/+ background, share some, but not all characteristics of Minute ribosomal protein mutants. Although swm mutants do not have thin bristles as is characteristic of Minutes, they are recessive lethal and developmentally delayed, and they interact genetically with Minutes and Minute-like loci (Gay and Contamine 1993). The wings of the Minute locus RpL38 have defects which are similar to swm wings—extra venation, expanded distance between veins 3 and 4, wing hair polarity abnormalities, and increased cell size (Marygold et al. 2005). Although RpL3845-72, Df(2R)M41A10, and M41A4 suppressed hhMrt, they did not interact with smo RNAi or smo5A (data not shown). We can speculate only on the basis of aspects of the Minute phenotype that are shared with Hh signaling.
While the interactions between swm and Minutes, as well as the similar phenotypes of swm and the RpL38 genes, might indicate a direct role in ribosome function, both Drosophila Swm and one of its two vertebrate homologs (RBM-27) are nuclear (Kavanagh et al. 2005). The presence of RRM sequences in Swm and its homologs might suggest a role in RNA binding or metabolism, and the RRM of RBM-27 binds RNA (Birney et al. 1993; Burd and Dreyfuss 1994; Kavanagh et al. 2005). However, RRMs can have a structural role in protein–protein interactions independent of RNA binding (Fribourg et al. 2003), so we cannot determine the molecular function of Swm and its homologs by genetic methods alone. We are intrigued by the fact that the other vertebrate homolog, RBM-26, was identified as se70-2, an autoantigen that is recognized by sera of cutaneous T-cell lymphoma patients and has been used as a diagnostic marker for this tumor (Eichmuller et al. 2001; Dummer et al. 2004). In addition, the mouse RBM-26/se70-2 locus was identified as one of four genes deleted in a region required for normal murine skeletal, cartilage, and craniofacial development (Peterson et al. 2002). Perhaps the roles of Hh that extend beyond pattern formation to cell cycle regulation, growth control, and cell polarity signify that Hh signal transduction integrates inputs from all three pathways. The pleiotropy of swm and mts may reflect these multiple inputs.
Acknowledgments
We thank Susan Younger, Barbara Thomas, Brenda Ng, Li Lin, Frank Hsuing, Gretchen Ehrenkauffer, Brian Biehs, Arjun Guha, Xingwu Lu, Shiho Kawamura, Sougata Roy, Joan Hooper, Chen-Ming Fan, Jeremy Reiter, Chris Wilson, and Pao-Tien Chuang for helpful discussions. For contributing stocks and reagents, we thank the Bloomington Drosophila Stock Center, Ryu Ueda and Kazuko Fujitani (National Institute of Genetics, Mishima, Japan), the Szeged Drosophila Stock Center, Joan Hooper, Wesley Gruber, Yuh-Nun Jan, Suzanne Eaton, Marcel van den Heuvel, Alan Shearn, Michelle Beaucher, Barbara Thomas, Tulle Hazelrigg, Steven Cohen, Fabrice Roegiers, and Bruno Bello. This work was supported by grants from the National Institutes of Health to T.B.K.
References
- Alcedo, J., Y. Zou and M. Noll, 2000. Posttranscriptional regulation of smoothened is part of a self-correcting mechanism in the Hedgehog signaling system. Mol. Cell 6 457–465. [DOI] [PubMed] [Google Scholar]
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers and D. J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol. 215 403–410. [DOI] [PubMed] [Google Scholar]
- Apionishev, S., N. M. Katanayeva, S. A. Marks, D. Kalderon and A. Tomlinson, 2005. Drosophila Smoothened phosphorylation sites essential for Hedgehog signal transduction. Nat. Cell Biol. 7 86–92. [DOI] [PubMed] [Google Scholar]
- Apweiler, R., T. K. Attwood, A. Bairoch, A. Bateman, E. Birney et al., 2001. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 29 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aza-Blanc, P., F. A. Ramirez-Weber, M. P. Laget, C. Schwartz and T. B. Kornberg, 1997. Proteolysis that is inhibited by hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell 89 1043–1053. [DOI] [PubMed] [Google Scholar]
- Bello, B., H. Reichert and F. Hirth, 2006. The brain tumor gene negatively regulates neural progenitor cell proliferation in the larval central brain of Drosophila. Development 133 2639–2648. [DOI] [PubMed] [Google Scholar]
- Belloni, E., M. Muenke, E. Roessler, G. Traverso, J. Siegel-Bartelt et al., 1996. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 14 353–356. [DOI] [PubMed] [Google Scholar]
- Birney, E., S. Kumar and A. R. Krainer, 1993. Analysis of the RNA-recognition motif and RS and RGG domains: conservation in metazoan pre-mRNA splicing factors. Nucleic Acids Res. 21 5803–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand, A. H., and N. Perrimon, 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118 401–415. [DOI] [PubMed] [Google Scholar]
- Burd, C. G., and G. Dreyfuss, 1994. Conserved structures and diversity of functions of RNA-binding proteins. Science 265 615–621. [DOI] [PubMed] [Google Scholar]
- Burke, R., D. Nellen, M. Bellotto, E. Hafen, K. A. Senti et al., 1999. Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell 99 803–815. [DOI] [PubMed] [Google Scholar]
- Callejo, A., C. Torroja, L. Quijada and I. Guerrero, 2006. Hedgehog lipid modifications are required for Hedgehog stabilization in the extracellular matrix. Development 133 471–483. [DOI] [PubMed] [Google Scholar]
- Capdevila, J., F. Pariente, J. Sampedro, J. L. Alonso and I. Guerrero, 1994. Subcellular localization of the segment polarity protein patched suggests an interaction with the wingless reception complex in Drosophila embryos. Development 120 987–998. [DOI] [PubMed] [Google Scholar]
- Casso, D. J., S. Tanda, B. Biehs, B. Martoglio and T. B. Kornberg, 2005. Drosophila signal peptide peptidase is an essential protease for larval development. Genetics 170 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoun, Z., R. K. Mann, D. Nellen, D. P. von Kessler, M. Bellotto et al., 2001. Skinny hedgehog, an acyltransferase required for palmitoylation and activity of the hedgehog signal. Science 293 2080–2084. [DOI] [PubMed] [Google Scholar]
- Chen, Y., and G. Struhl, 1996. Dual roles for patched in sequestering and transducing Hedgehog. Cell 87 553–563. [DOI] [PubMed] [Google Scholar]
- Chen, Y., N. Gallaher, R. H. Goodman and S. M. Smolik, 1998. Protein kinase A directly regulates the activity and proteolysis of cubitus interruptus. Proc. Natl. Acad. Sci. USA 95 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M. H., Y. J. Li, T. Kawakami, S. M. Xu and P. T. Chuang, 2004. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 18 641–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, C., Y. Litingtung, E. Lee, K. E. Young, J. L. Corden et al., 1996. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383 407–413. [DOI] [PubMed] [Google Scholar]
- Chou, T. B., and N. Perrimon, 1992. Use of a yeast site-specific recombinase to produce female germline chimeras in Drosophila. Genetics 131 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clyne, P. J., J. S. Brotman, S. T. Sweeney and G. Davis, 2003. Green fluorescent protein tagging Drosophila proteins at their native genomic loci with small P elements. Genetics 165 1433–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokol, M., R. Nair and B. Rost, 2000. Finding nuclear localization signals. EMBO Rep. 1 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, R. T., and S. M. Cohen, 2005. A genetic screen in Drosophila for identifying novel components of the hedgehog signaling pathway. Genetics 170 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo, P. F., and N. S. Tolwinski, 2006. Wnt, Hedgehog and junctional Armadillo/beta-catenin establish planar polarity in the Drosophila embryo. PLoS ONE 1 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawber, R. J., S. Hebbes, B. Herpers, F. Docquier and M. van den Heuvel, 2005. Differential range and activity of various forms of the Hedgehog protein. BMC Dev. Biol. 5 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denef, N., D. Neubuser, L. Perez and S. M. Cohen, 2000. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 102 521–531. [DOI] [PubMed] [Google Scholar]
- Dong, X., K. H. Zavitz, B. J. Thomas, M. Lin, S. Campbell et al., 1997. Control of G1 in the developing Drosophila eye: rca1 regulates Cyclin A. Genes Dev. 11 94–105. [DOI] [PubMed] [Google Scholar]
- Duman-Scheel, M., L. Weng, S. Xin and W. Du, 2002. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 417 299–304. [DOI] [PubMed] [Google Scholar]
- Dummer, R., J. C. Hassel, F. Fellenberg, S. Eichmuller, T. Maier et al., 2004. Adenovirus-mediated intralesional interferon-gamma gene transfer induces tumor regressions in cutaneous lymphomas. Blood 104 1631–1638. [DOI] [PubMed] [Google Scholar]
- Eaton, S., 2006. Release and trafficking of lipid-linked morphogens. Curr. Opin. Genet. Dev. 16 17–22. [DOI] [PubMed] [Google Scholar]
- Eichmuller, S., D. Usener, R. Dummer, A. Stein, D. Thiel et al., 2001. Serological detection of cutaneous T-cell lymphoma-associated antigens. Proc. Natl. Acad. Sci. USA 98 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenfeld, A. L., and J. A. Kennison, 1995. Positional signaling by hedgehog in Drosophila imaginal disc development. Development 121 1–10. [DOI] [PubMed] [Google Scholar]
- Feng, J., B. White, O. V. Tyurina, B. Guner, T. Larson et al., 2004. Synergistic and antagonistic roles of the Sonic hedgehog N- and C-terminal lipids. Development 131 4357–4370. [DOI] [PubMed] [Google Scholar]
- Foley, E., and F. Sprenger, 2001. The cyclin-dependent kinase inhibitor Roughex is involved in mitotic exit in Drosophila. Curr. Biol. 11 151–160. [DOI] [PubMed] [Google Scholar]
- Fribourg, S., D. Gatfield, E. Izaurralde and E. Conti, 2003. A novel mode of RBD-protein recognition in the Y14-Mago complex. Nat. Struct. Biol. 10 433–439. [DOI] [PubMed] [Google Scholar]
- Gallet, A., R. Rodriguez, L. Ruel and P. P. Therond, 2003. Cholesterol modification of hedgehog is required for trafficking and movement, revealing an asymmetric cellular response to hedgehog. Dev. Cell 4 191–204. [DOI] [PubMed] [Google Scholar]
- Gallet, A., L. Ruel, L. Staccini-Lavenant and P. P. Therond, 2006. Cholesterol modification is necessary for controlled planar long-range activity of Hedgehog in Drosophila epithelia. Development 133 407–418. [DOI] [PubMed] [Google Scholar]
- Gay, P., and D. Contamine, 1993. Study of the ref(2)P locus of Drosophila melanogaster. II. Genetic studies of the 37DF region. Mol. Gen. Genet. 239 361–370. [DOI] [PubMed] [Google Scholar]
- Gustavson, E., A. S. Goldsborough, Z. Ali and T. B. Kornberg, 1996. The Drosophila engrailed and invected genes: partners in regulation, expression, and function. Genetics 142 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines, N., and M. van den Heuvel, 2000. A directed mutagenesis screen in Drosophila melanogaster reveals new mutants that influence hedgehog signaling. Genetics 156 1777–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannus, M., F. Feiguin, C. P. Heisenberg and S. Eaton, 2002. Planar cell polarization requires Widerborst, a B′ regulatory subunit of protein phosphatase 2A. Development 129 3493–3503. [DOI] [PubMed] [Google Scholar]
- Hooper, J. E., 1994. Distinct pathways for autocrine and paracrine Wingless signalling in Drosophila embryos. Nature 372 461–464. [DOI] [PubMed] [Google Scholar]
- Hooper, J. E., and M. P. Scott, 2005. Communicating with Hedgehogs. Nat. Rev. Mol. Cell. Biol. 6 306–317. [DOI] [PubMed] [Google Scholar]
- Jia, J., K. Amanai, G. Wang, J. Tang, B. Wang et al., 2002. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature 416 548–552. [DOI] [PubMed] [Google Scholar]
- Jia, J., C. Tong, B. Wang, L. Luo and J. Jiang, 2004. Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432 1045–1050. [DOI] [PubMed] [Google Scholar]
- Kavanagh, S. J., T. C. Schulz, P. Davey, C. Claudianos, C. Russell et al., 2005. A family of RS domain proteins with novel subcellular localization and trafficking. Nucleic Acids Res. 33 1309–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, P. A., G. Struhl and J. Casal, 2007. Planar cell polarity: One or two pathways? Nat. Rev. Genet. 8 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. D., and J. E. Treisman, 2001. Sightless has homology to transmembrane acyltransferases and is required to generate active Hedgehog protein. Curr. Biol. 11 1147–1152. [DOI] [PubMed] [Google Scholar]
- Lee, T., C. Winter, S. S. Marticke, A. Lee and L. Luo, 2000. Essential roles of Drosophila RhoA in the regulation of neuroblast proliferation and dendritic but not axonal morphogenesis. Neuron 25 307–316. [DOI] [PubMed] [Google Scholar]
- Lee, Y. S., and R. W. Carthew, 2003. Making a better RNAi vector for Drosophila: use of intron spacers. Methods 30 322–329. [DOI] [PubMed] [Google Scholar]
- Liu, Y., X. Cao, J. Jiang and J. Jia, 2007. Fused Costal2 protein complex regulates Hedgehog-induced Smo phosphorylation and cell-surface accumulation. Genes Dev. 21 1949–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, X., S. Liu and T. B. Kornberg, 2006. The C-terminal tail of the Hedgehog receptor Patched regulates both localization and turnover. Genes Dev. 20 2539–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum, L., S. Yao, B. Mozer, A. Rovescalli, D. Von Kessler et al., 2003. Identification of Hedgehog pathway components by RNAi in Drosophila cultured cells. Science 299 2039–2045. [DOI] [PubMed] [Google Scholar]
- Marygold, S. J., C. M. Coelho and S. J. Leevers, 2005. Genetic analysis of RpL38 and RpL5, two minute genes located in the centric heterochromatin of chromosome 2 of Drosophila melanogaster. Genetics 169 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micchelli, C. A., I. The, E. Selva, V. Mogila and N. Perrimon, 2002. Rasp, a putative transmembrane acyltransferase, is required for Hedgehog signaling. Development 129 843–851. [DOI] [PubMed] [Google Scholar]
- Motzny, C. K., and R. Holmgren, 1995. The Drosophila Cubitus interruptus protein and its role in the wingless and hedgehog signal transduction pathways. Mech. Dev. 52 137–150. [DOI] [PubMed] [Google Scholar]
- Mullor, J. L., M. Calleja, J. Capdevila and I. Guerrero, 1997. Hedgehog activity, independent of decapentaplegic, participates in wing disc patterning. Development 124 1227–1237. [DOI] [PubMed] [Google Scholar]
- Nusslein-Volhard, C., and E. Wieschaus, 1980. Mutations affecting segment number and polarity in Drosophila. Nature 287 795–801. [DOI] [PubMed] [Google Scholar]
- Nybakken, K., S. A. Vokes, T. Y. Lin, A. P. McMahon and N. Perrimon, 2005. A genome-wide RNA interference screen in Drosophila melanogaster cells for new components of the Hh signaling pathway. Nat. Genet. 37 1323–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nybakken, K. E., C. W. Turck, D. J. Robbins and J. M. Bishop, 2002. Hedgehog-stimulated phosphorylation of the kinesin-related protein Costal2 is mediated by the serine/threonine kinase fused. J. Biol. Chem. 277 24638–24647. [DOI] [PubMed] [Google Scholar]
- Ogden, S. K., D. J. Casso, M. Ascano, Jr., M. M. Yore, T. B. Kornberg et al., 2006. Smoothened regulates activator and repressor functions of Hedgehog signaling via two distinct mechanisms. J. Biol. Chem. 281 7237–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca di Magliano, M., and M. Hebrok, 2003. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 3 903–911. [DOI] [PubMed] [Google Scholar]
- Pepinsky, R. B., C. Zeng, D. Wen, P. Rayhorn, D. P. Baker et al., 1998. Identification of a palmitic acid-modified form of human Sonic hedgehog. J. Biol. Chem. 273 14037–14045. [DOI] [PubMed] [Google Scholar]
- Peterson, K. A., B. L. King, A. Hagge-Greenberg, J. J. Roix, C. J. Bult et al., 2002. Functional and comparative genomic analysis of the piebald deletion region of mouse chromosome 14. Genomics 80 172–184. [DOI] [PubMed] [Google Scholar]
- Porter, J. A., K. E. Young and P. A. Beachy, 1996. Cholesterol modification of hedgehog signaling proteins in animal development. Science 274 255–259. [DOI] [PubMed] [Google Scholar]
- Price, M. A., and D. Kalderon, 2002. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 108 823–835. [DOI] [PubMed] [Google Scholar]
- Quirk, J., M. van den Heuvel, D. Henrique, V. Marigo, T. A. Jones et al., 1997. The smoothened gene and hedgehog signal transduction in Drosophila and vertebrate development. Cold Spring Harbor Symp. Quant. Biol. 62 217–226. [PubMed] [Google Scholar]
- Ramirez-Weber, F. A., D. J. Casso, P. Aza-Blanc, T. Tabata and T. B. Kornberg, 2000. Hedgehog signal transduction in the posterior compartment of the Drosophila wing imaginal disc. Mol. Cell 6 479–485. [DOI] [PubMed] [Google Scholar]
- Reardon, J. T., C. A. Liljestrand-Golden, R. L. Dusenbery and P. D. Smith, 1987. Molecular analysis of diepoxybutane-induced mutations at the rosy locus of Drosophila melanogaster. Genetics 115 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler, E., E. Belloni, K. Gaudenz, P. Jay, P. Berta et al., 1996. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 14 357–360. [DOI] [PubMed] [Google Scholar]
- Schier, A. F., S. C. Neuhauss, K. A. Helde, W. S. Talbot and W. Driever, 1997. The one-eyed pinhead gene functions in mesoderm and endoderm formation in zebrafish and interacts with no tail. Development 124 327–342. [DOI] [PubMed] [Google Scholar]
- Snaith, H. A., C. G. Armstrong, Y. Guo, K. Kaiser and P. T. Cohen, 1996. Deficiency of protein phosphatase 2A uncouples the nuclear and centrosome cycles and prevents attachment of microtubules to the kinetochore in Drosophila microtubule star (mts) embryos. J. Cell Sci. 109(Pt 13): 3001–3012. [DOI] [PubMed] [Google Scholar]
- Strigini, M., and S. M. Cohen, 1997. A Hedgehog activity gradient contributes to AP axial patterning of the Drosophila wing. Development 124 4697–4705. [DOI] [PubMed] [Google Scholar]
- Tabata, T., and T. B. Kornberg, 1994. Hedgehog is a signaling protein with a key role in patterning Drosophila imaginal discs. Cell 76 89–102. [DOI] [PubMed] [Google Scholar]
- Tabata, T., and Y. Takei, 2004. Morphogens, their identification and regulation. Development 131 703–712. [DOI] [PubMed] [Google Scholar]
- Tabata, T., S. E. Eaton and T. B. Kornberg, 1992. The Drosophila hedgehog gene is expressed specifically in posterior compartment cells and is a target of engrailed regulation. Genes Dev. 6 2635–2645. [DOI] [PubMed] [Google Scholar]
- Tabata, T., C. Schwartz, E. Gustavson, Z. Ali and T. B. Kornberg, 1995. Creating a Drosophila wing de novo, the role of engrailed, and the compartment border hypothesis. Development 121 3359–3369. [DOI] [PubMed] [Google Scholar]
- Uemura, T., K. Shiomi, S. Togashi and M. Takeichi, 1993. Mutation of twins encoding a regulator of protein phosphatase 2A leads to pattern duplication in Drosophila imaginal discs. Genes Dev. 7 429–440. [DOI] [PubMed] [Google Scholar]
- Vegh, M., and K. Basler, 2003. A genetic screen for hedgehog targets involved in the maintenance of the Drosophila anteroposterior compartment boundary. Genetics 163 1427–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman, D. A., N. M. Solomon, H. C. Chang, F. D. Karim, M. Therrien et al., 1996. Protein phosphatase 2A positively and negatively regulates Ras1-mediated photoreceptor development in Drosophila. Genes Dev. 10 272–278. [DOI] [PubMed] [Google Scholar]
- Yao, S., L. Lum and P. Beachy, 2006. The ihog cell-surface proteins bind Hedgehog and mediate pathway activation. Cell 125 343–357. [DOI] [PubMed] [Google Scholar]
- Zecca, M., K. Basler and G. Struhl, 1995. Sequential organizing activities of engrailed, hedgehog and decapentaplegic in the Drosophila wing. Development 8 2265–2278. [DOI] [PubMed] [Google Scholar]
- Zeng, X., J. A. Goetz, L. M. Suber, W. J. Scott, Jr., C. M. Schreiner et al., 2001. A freely diffusible form of Sonic hedgehog mediates long-range signalling. Nature 411 716–720. [DOI] [PubMed] [Google Scholar]
- Zhang, C., E. H. Williams, Y. Guo, L. Lum and P. A. Beachy, 2004. Extensive phosphorylation of Smoothened in Hedgehog pathway activation. Proc. Natl. Acad. Sci. USA 101 17900–17907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., X. G. Jiang and J. Yao, 2001. Lowering of sodium deoxycholate-induced nasal ciliotoxicity with cyclodextrins. Acta Pharmacol. Sin. 22 1045–1050. [PubMed] [Google Scholar]