

Abstract
We have examined the folding and unfolding of the caspase recruitment domain of procaspase-1 (CP1-CARD), a member of the α-helical Greek key protein family. The equilibrium folding/unfolding of CP1-CARD is described by a two-state mechanism, and the results show CP1-CARD is marginally stable with a ΔGH2O of 1.1 ± 0.2 kcal/mole and an m-value of 0.65 ± 0.06 kcal/mole/M (10 mM Tris-HCl at pH 8.0, 1 mM DTT, 25°C). Consistent with the equilibrium folding data, CP1-CARD is a monomer in solution when examined by size exclusion chromatography. Single-mixing stopped-flow refolding and unfolding studies show that CP1-CARD folds and unfolds rapidly, with no detectable slow phases, and the reactions appear to reach equilibrium within 10 msec. However, double jump kinetic experiments demonstrate the presence of an unfolded-like intermediate during unfolding. The intermediate converts to the fully unfolded conformation with a half-time of 10 sec. Interrupted refolding studies demonstrate the presence of one or more nativelike intermediates during refolding, which convert to the native conformation with a half-time of about 60 sec. Overall, the data show that both unfolding and refolding processes are slow, and the pathways contain kinetically trapped species.
Keywords: caspase recruitment domain, α-helical Greek key, kinetic trap, double jump, interrupted re-folding, equilibrium folding
CARDs are members of the death domain superfamily, which contains the death domain (DD), the death effecter domain (DED), the pyrin domain, and the caspase recruitment domain (CARD; Hofmann 1999; Fairbrother et al. 2001; Weber and Vincenz 2001). Biologically, CARDs are important because they interact through homo- or heterooligomerization to convey cellular signals to downstream reactions (Hofmann 1999; Weber and Vincenz 2001; Shi 2002; Shiozaki et al. 2002). The family members play specific roles in apoptosis, the inflammatory response or transcriptional activation. Although the amino acid sequence identity is quite low within the family, generally less than 20% (Druilhe et al. 2001; Lee et al. 2001; Weber and Vincenz 2001), CARDs demonstrate the structural homology within the death domain superfamily (Chou et al. 1998; Eberstadt et al. 1998; Day et al. 1999; Jeong et al. 1999; Vaughn et al. 1999; Xiao et al. 1999; Zhou et al. 1999; Humke et al. 2000; Weber and Vincenz 2001). These protein domains are comprised of six antiparallel α-helices that tightly surround a hydrophobic core, with many charged residues on the surface (see Fig. 1 ▶). In contrast to most helical bundles in which sequential helices pack adjacently in antiparallel orientations, the arrangement of the helices in CARDs results in a Greek key topology (Vaughn et al. 1999).
Figure 1.

(A) Sequence alignment of ICEBERG, CARD of Procaspase-1 (CASP-1), and CARD of RICK. The helical regions of ICEBERG are indicated above the alignment. Regions of similarity are highlighted in gray. Proline residues are underlined and in bold. (B) The structure of ICEBERG (right panel) and the homology model of CP1-CARD (left panel). ICEBERG (PDB code 1DGN) is shown in ribbon diagram with the proline residues in black and the aromatic residues, two phenylalanines, in gray. For CP1-CARD, the single proline is colored in black, and the aromatic residues, two tyrosines in helices 5 and 6, and one phenylalanine in helix 1, are in gray. Structures were drawn with Pymol (DeLano Scientific, CA).
Recently, several groups have examined the folding of homologous proteins, with the ultimate goal of determining the extent to which a folding pathway is conserved (Gunasekaran et al. 2001). This issue addresses the role of amino acid sequence identity/similarity in determining the folding mechanism of homologous proteins. Of particular interest is whether the folding transition state is conserved within the structural family and to what extent this conservation is due to evolutionary pressures to maintain the transition state (Mirny and Shakhnovich 2001) or the structural topology (Plaxco et al. 1998, 2000). CARDs appear to be ideally suited for addressing these issues. They are relatively small proteins, generally about 95 amino acids. With the exception of residues in the turns, they consist entirely of α-helical secondary structure. Yet, they fold into a complex topology, the Greek key. They are amenable to examination by a number of biochemical and biophysical techniques (Chen and Clark 2003) as well as NMR (Chou et al. 1998; Day et al. 1999; Zhou et al. 1999) and crystallographic studies (Vaughn et al. 1999). Therefore, the issues that are examined with other small proteins can be extended to a more complex folding topology without a large increase in protein size.
So far, the only α-Greek key protein studied has been the CARD of RICK (RICK-CARD; Chen and Clark 2003). RICK (CARDIAK, RIP2) is a pro-inflammatory serine/ threonine kinase (Thome et al. 1998). The studies showed that RICK-CARD follows a simple two-state equilibrium folding mechanism, but the folding kinetics are quite complex. Several phases are observed during both refolding and unfolding. The protein exhibits burst phase kinetics as well as slow reactions with half-times of ~80 sec (folding) or ~50 sec (unfolding). The data suggest the presence of kinetically trapped, or misfolded, species that are on-pathway both in refolding and in unfolding. These complex, and slow, folding kinetics are not predicted by algorithms that relate topology to folding rates (Plaxco et al. 1998; Gromiha and Selvaraj 2001). Generally, it is observed in these algorithms that α-helical proteins are dominated by short-range interactions within the helix (i, i + 3/4). As a consequence, the average sequence distance between interacting residues, that is, the contact order (Plaxco et al. 1998), is small. In general, small helical proteins fold rapidly. In the case of RICK-CARD, the predicted folding rate is not consistent with the experimental folding rate, 30 sec−1. It is not yet clear why the protein folds slowly, and the nature of the folding intermediates is unknown. In addition, the extent to which the folding mechanism is conserved in other CARD proteins has not been determined.
Because of these considerations, we examined the equilibrium and kinetic folding of the CARD of procaspase-1 (CP1-CARD). The CP1-CARD is located within the pro-domain of the caspase and comprises residues 1 (Met) to 92 (Asp). We show that CP1-CARD folds by an apparent two-state equilibrium process like RICK-CARD, but the conformational free energy is much lower. In contrast to RICK-CARD, the time courses of refolding and of unfolding examined by single-mixing stopped-flow techniques show that the reactions appear to reach equilibrium within the dead time for mixing (~10 msec). However, sequential mixing stopped-flow studies demonstrate the formation of unfolded-like intermediate(s) during unfolding and of native-like intermediate(s) during refolding. We compare the results for the folding of CP1-CARD to our previous data for the folding of RICK-CARD.
Results
General properties
CP1-CARD contains two tyrosinyl, a single phenylalanyl, and no tryptophanyl residues. The two tyrosines are located in helices 5 and 6, whereas the phenylalanine is located in helix 1 (Fig. 1 ▶). There are three cysteinyl residues located in helix 5, and CP1-CARD contains one proline in turn four connecting helices 4 and 5. Because the structure of CP1-CARD has not been determined, we generated a homology model using the solution structure of ICEBERG (Humke et al. 2000) as the template because of the relatively high sequence identity (52%) with CP1-CARD (Fig. 1A ▶). The model (Fig. 1B ▶, left panel) is compared to the structure of ICEBERG (Fig. 1B ▶, right panel) and shows a six-helix bundle with a packed hydrophobic core and charged residues on the surface.
Fluorescence emission spectra of CP1-CARD demonstrate a maximum at 330 nm for the native protein and at 340 nm when the protein is incubated in 6 M urea-containing buffer, demonstrating that the aromatic residues are exposed upon unfolding (Fig. 2A ▶). In general, the fluorescence emission signal is low because of the lack of tryptophanyl residues. The spectra shown in Figure 2A ▶ are unusual because tyrosine fluorescence emission usually is observed at 303 nm. There were two possibilities to explain the anomalous fluorescence emission of CP1-CARD. Either there were tryptophan-containing contaminants in our sample, or one of the tyrosines is present as tyrosinate, which exhibits a fluorescence emission maximum at ~340 nm. Following the purification protocol described in Materials and Methods, the protein purity was determined to be >95% pure by SDS-PAGE using Coomassie stain. However, Ruby stain, which has a sensitivity comparable to silver stain, showed that minor contaminants were present. The protein was purified further using Q-sepharose ion exchange and size exclusion chromatography columns. The minor contaminants were mostly removed with this procedure (not shown), and the sample was ~98% pure as assessed by SDS-PAGE and Ruby staining. The fluorescence emission and circular dichroism spectra were measured following each column, and importantly, there were no significant changes from the spectra shown in Figure 2 ▶ (not shown), indicating that tryptophan-containing contaminants were not responsible for the anomalous fluorescence emission spectra. Alternatively, tyrosinate fluorescence emission can be observed at pH > ~10.0. The fluorescence emission of CP1-CARD was examined at pH 12.0 and exhibited a single peak centered on 340 nm (not shown). The shift in emission maximum is due to the ionization of both tyrosine side chains. In addition, the spectrum was nearly superimposable with a control spectrum of tyrosine at pH 12.0, which demonstrated a single peak at 338 nm (not shown). Although these data do not rule out the possible contribution of tryptophan-containing contaminants, they suggest that at least one of the two tyrosines may be present as tyrosinate at pH 8.0. If this is true, then we suggest that tyrosine 75, on helix 5, may have an anomalous Ka. In the model of CP1-CARD (Fig. 1B ▶), the Y75 side chain is predicted to be in close proximity to E78 and E79 (both on helix 5) as well as E28, on helix 2. Hydrogen bonds between tyrosine and glutamate side chains have been shown to result in red-shifted fluorescence emission spectra consistent with tyrosinate (O’Neil and Hofmann 1987).
Figure 2.

(A) Fluorescence emission spectra of CP1-CARD. The protein (3 μM) in buffer (circles) or in 5.8 M urea-containing buffer (squares) was excited at 280 nm. The fluorescence emission was collected from 300 to 400 nm. (B) Far-UV and (C) near-UV CD spectra of CP1-CARD. The spectra of native protein in buffer (solid line) and protein unfolded in 6 M urea-containing buffer (long dashed line) or 6 M guanidine-containing buffer (short dashed line) are shown.
We also examined the secondary and tertiary structures of CP1-CARD using near- and far-UV circular dichroism spectroscopy. The far-UV CD spectrum (Fig. 2B ▶) of native CP1-CARD shows double minima at 203 nm and 220 nm. This suggests a mixture of α-helices and random coil structure. As described below, the results are consistent with a low conformational stability and a low cooperativity of unfolding. Furthermore, the signal is about eightfold lower than that of RICK-CARD (Chen and Clark 2003) or the full-length prodomain of procaspase-1 (Fairbrother et al. 2001). These results are not expected for a well-packed six-helix bundle. The near-UV CD spectrum (Fig. 2C ▶) shows a minimum at 277 nm, demonstrating that the aromatic amino acids are packed in an asymmetric environment. The near-UV CD signal decreases when the protein is incubated in 6 M urea-containing buffer or in 6 M guanidine-containing buffer. Overall, the data are consistent with the loss of secondary and tertiary structure upon incubation in urea- or guanidine-containing buffers, although residual tertiary structure may remain in urea.
Equilibrium folding of CP1-CARD
The equilibrium folding/unfolding of CP1-CARD was examined by monitoring the changes in the fluorescence average emission wavelength (AEW) and the far-UV CD signal at 220 nm upon titration with urea-containing buffer (Fig. 3A ▶). The results show that the two spectroscopic probes are superimposable, demonstrating that the loss of secondary structure is concomitant with the loss of tertiary structure. To examine the effect of protein concentration, if any, on unfolding, the experiments were performed with protein concentrations ranging from 3 to 40 μM. Again, the data were superimposable (not shown), demonstrating that CP1-CARD is a monomer under the conditions of these experiments.
Figure 3.
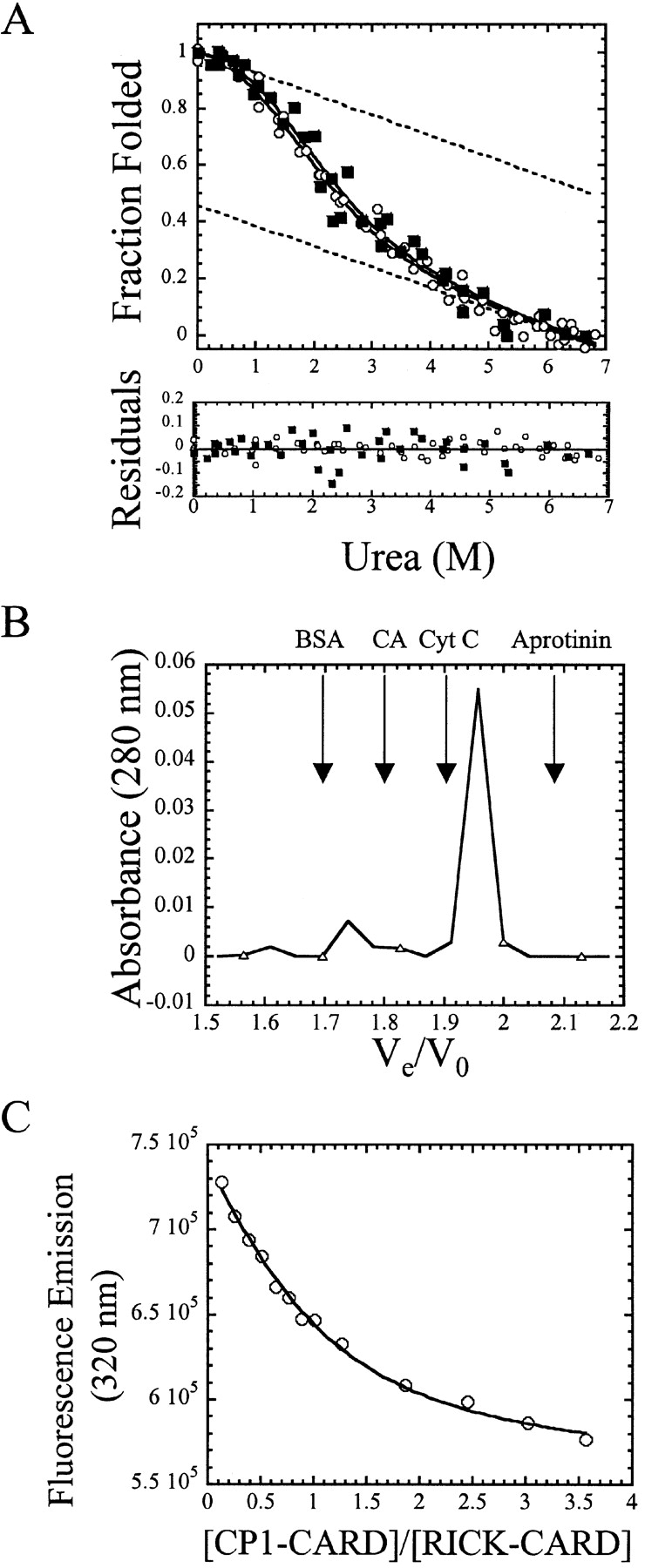
(A) Equilibrium folding/unfolding of CP1-CARD. The normalized CD (filled squares) and fluorescence emission data (open circles) are plotted vs. urea concentration. The protein concentration was 6 μM. The solid lines represent global fits of the data to a two-state equilibrium folding mechanism, as described in the text. The ΔGH2O and m-value obtained from the global fits are 1.1 kcal/mole and 0.65 kcal/mole/M, respectively. The residuals of the fits are shown in the lower panel. The dashed lines represent the native and unfolded baselines. (B) Analytical size exclusion chromatography of CP1-CARD. The absorbance of CP1-CARD (open triangles) is plotted vs. the elution volume. The arrows indicate the elution volumes of the standards, including bovine serum albumin (BSA), carbonic anhydrase (CA), cytochrome c (Cyt C), and aprotinin. (C) Binding of CP1-CARD with RICK-CARD. Samples were excited at 295 nm, and the fluorescence emission at 330 nm (open circles) is plotted vs. the ratio of CP1-CARD to RICK-CARD. The solid line represents a fit of the data to equation 3, as described in Materials and Methods.
To further confirm the results, we examined the protein by analytical size-exclusion chromatography (Fig. 3B ▶). We observed that the majority of the protein (~90%) eluted in the range of 10 kDa, consistent with the molecular weight of the monomer. At the higher protein concentrations of these experiments (85 μM), there is a small population of protein (~10%) that elutes in the range of a tetrameric species. Therefore, we suggest that CP1-CARD is a monomer under the experimental conditions used in our folding studies (10 mM Tris-HCl at pH 8.0, 1 mM DTT, 25°C, 3–40 μM protein).
CP1-CARD contains three cysteinyl residues in helix 5, and two residues (C69 and C72) are separated by one turn of the helix. To examine the possibility that a disulfide bond is present in the protein, we repeated the equilibrium unfolding studies in the absence of reducing agent. The data (not shown) were superimposable with those shown in Figure 3A ▶, demonstrating that disulfide bonds do not form.
The data shown in Figure 3A ▶ were fit globally to a two-state equilibrium folding model (Santoro and Bolen 1988), representing the native and unfolded ensembles of the protein. From the global fit, we determined the conformational free energy, ΔGH2O, to be 1.1 ± 0.2 kcal/mole and the corresponding m-value to be 0.65 ± 0.06 kcal/mole/M. This corresponds to a urea½ of 1.7 M urea. The residuals from the fit are shown in the lower panel of Figure 3A ▶. Both native and unfolded baselines exhibited significant slopes, as shown by the dashed lines in Figure 3A ▶. These lines were obtained from the global fit of the data. From these extrapolations, the midpoint of the unfolding curve is predicted to be ~1.8 M urea, which is within experimental error of that calculated from the free energy and m-value, 1.7 M urea.
The m-value correlates to the change in exposed surface area of the protein upon unfolding (Schellman 1978; Alonso and Dill 1991). Scholtz and coworkers (Myers et al. 1995) have described a qualitative correlation between the m-value, the change in the solvent exposed surface area (ΔASA), and the number of amino acid residues included in the transition. Previously (Chen and Clark 2003), we showed that the ΔASA of RICK-CARD (8200 Å2) agreed well with the total number of amino acids in the protein, 95. For CP1-CARD, we calculate the ΔASA to be 2780 Å2, corresponding to 40 residues. This does not agree with the number of amino acids of CP1-CARD, 92. Overall, the data show that CP1-CARD is marginally stable and suggest that the protein is partially folded. The latter point is consistent with the low far-UV CD signal (Fig. 2B ▶) and the apparent random coil structure observed by far-UV CD.
Binding studies with RICK-CARD
It has been shown that RICK interacts with procaspase-1 via CARD–CARD interactions (Thome et al. 1998). To determine whether CP1-CARD is functional, we examined the binding to RICK-CARD using fluorescence titration studies (Fig. 3C ▶). RICK-CARD contains a single tryptophanyl residue in helix 1 (Fig. 1A ▶) that is quenched upon complex formation. The data were fit to a simple binding model (equation 3, below) assuming a 1:1 complex, as described in Materials and Methods (solid line in Fig. 3C ▶). Overall, the results correlate well with the assumption that the two proteins interact with a stoichiometry of 1:1. The dissociation constant, Kd, obtained from the fit is 1.4 μM and is in good agreement with that obtained previously for the interaction of Pelle and Tube, two DD proteins, of 0.5 μM (Schiffmann et al. 1999). Based on the structure of Apaf-1-CARD complexed with procaspase-9 CARD (Qin et al. 1999), it is inferred that the binding interactions occur via helices 1 and 4 of CP1-CARD with helices 2 and 3 of RICK-CARD (see Fig. 1B ▶). Overall, the results of the binding studies show that CP1-CARD is functional and suggest that the helices of CP1-CARD required for interactions with RICK-CARD, presumably helices 1 and 4, are in the native conformation. An alternative explanation for these results is that the binding induces helical structure in CP1-CARD. However, the far-UV CD signal of the complex was not significantly different from the sum of the two individual spectra (data not shown), indicating that there is not a large change in helical structure upon complex formation.
Folding kinetics of CP1-CARD
The refolding and unfolding kinetics of CP1-CARD were examined initially in single-mixing stopped-flow experiments by monitoring changes in fluorescence emission (Fig. 4A ▶). In both experiments, the data suggest that the reactions are complete within the dead time for mixing. Within 10 msec, for example, the refolding traces overlay with the native protein signal, and the unfolding traces overlay with the unfolded protein signal. There was no further change in signal when monitored to 500 sec (the first 10 sec are shown in Fig. 4A ▶). Thus, aside from the burst phase, there are no detectable kinetic phases in refolding or in unfolding when monitored by single-mixing stopped-flow techniques.
Figure 4.
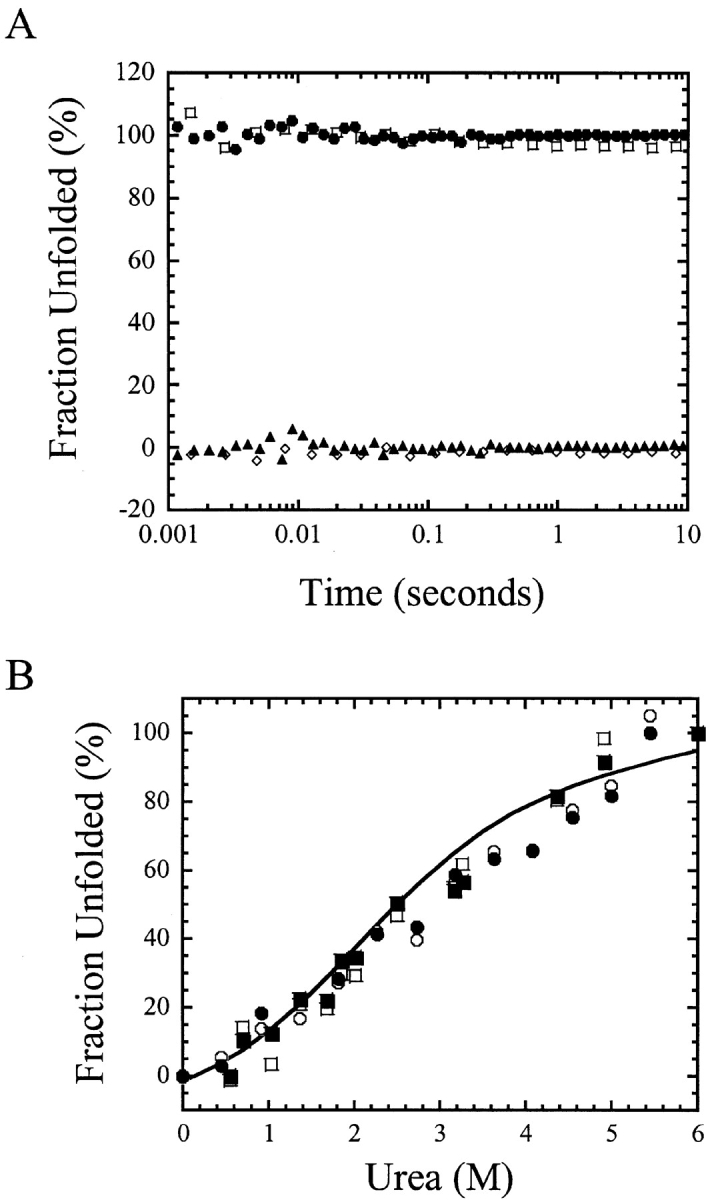
(A) Unfolding (filled circles) and refolding (filled triangles) kinetic traces of CP1-CARD. The signals are normalized to the fraction of unfolded protein. The native (open diamond) and unfolded (open square) protein signals are indicated. (B) The signals of unfolding and of refolding vs. urea concentrations. The initial (open circles, 10 msec) and final (filled circles, 10 sec) signals of unfolding are shown. The initial (open squares, 10 msec) and final (filled squares, 10 sec) signals of refolding are shown as well. The solid line represents the fit of the equilibrium data shown in Figure 3A ▶.
We monitored the burst phase amplitudes as a function of final urea concentration from 0 to 6 M. In all cases (not shown), the traces were linear, that is, without observable exponential phases. The initial signals (10 msec) and final signals (10 sec) are plotted as the fraction of unfolded protein (Fig. 4B ▶) and are compared to the data obtained from equilibrium folding/unfolding experiments (Fig. 3A ▶). The results show that the signals obtained from the kinetic studies are similar to those of the equilibrium folding data. The low cooperativity, which is reflected in the low m-value, results in a very broad folding/unfolding transition. The kinetic data suggest that the reactions have reached equilibrium within 10 msec; however, as we show later, the reactions reach a true equilibrium after about 50 sec. In this experiment the data were collected for a maximum of 10 sec, which may explain why the kinetic amplitudes do not overlay with the equilibrium data at intermediate urea concentrations (3–4 M). These results are very different from those obtained previously for RICK-CARD (Chen and Clark 2003), where we showed that RICK-CARD contains a burst phase intermediate in unfolding followed by three slower unfolding phases. In refolding, RICK-CARD also demonstrates burst phase kinetics, but the amplitudes are linear with final urea concentration, suggesting that the burst phase does not correlate with the formation of an intermediate. In contrast, a fast refolding phase (k = 30 sec−1, t½ ~ 0.02 sec) appears to be the major refolding phase of RICK-CARD.
To further examine the kinetics of folding and of unfolding for CP1-CARD, we employed sequential-mixing stopped-flow techniques. Initially, the protein was examined in double jump experiments, which provide details on the unfolding pathway (Brandts et al. 1975). In these experiments, native CP1-CARD was incubated in urea-containing buffer for various amount of time (delay time). Following a specified delay time, the urea was diluted and the protein was refolded. The second process, that is, refolding, is monitored experimentally. These experiments are used typically to examine slow isomerization reactions that occur from the unfolded state, such as cis-trans prolyl isomerization (Brandts et al. 1975; Nall et al. 1978; Kiefhaber et al. 1990). In this sequential mechanism (N ⇆ Uf ⇆ Us), where Uf and Us are fast- and slow-folding species, respectively, of the unfolded ensemble and N represents the native ensemble. The short delay times of the double jump result in a large population of native protein, which decreases at longer delay times as the slower isomerization reaction occurs in the unfolded state.
In the first jump, native CP1-CARD was mixed with urea-containing buffer so that the final urea concentration was 5 M. As shown in Figure 3 ▶, these conditions are sufficient to unfold the protein. During the first jump, the time in which the protein was incubated in urea (delay time) was varied from 0.1 to 200 sec. In the second jump, the protein was returned to native conditions, and refolding was monitored. In the experiments for CP1-CARD, there were no detectable kinetic phases aside from the burst phase amplitude. This is reflected in the same initial (10 msec) and final (10 sec) fluorescence emission signals. Surprisingly, the results for CP1-CARD show that no native protein is produced following short delay times of unfolding (<1 sec; Fig. 5A ▶), as it is observed that the signal remains similar to that of the unfolded protein. A transition occurs between 1 and 30 sec that corresponds to native protein formation. At delay times greater than 30 sec, one recovers 100% of the signal of the folded protein during the refolding phase of the experiment. The data shown in Figure 5A ▶ were fit to a single exponential equation, and the observed rate constant obtained from the fit is 0.07 sec−1 (t½ ~10 sec). A similar result was observed previously for RICK-CARD (Chen and Clark 2003), although two phases were detected in the double jump experiment. The first phase was a lag phase, with a half-time of about 15 sec. Native RICK-CARD was produced from the second phase, with a half-time of about 50 sec. Overall, the results shown here suggest that an intermediate forms during the unfolding of CP1-CARD and that the fluorescence properties of the intermediate are similar to those of the unfolded protein. In addition, the intermediate does not refold to the native conformation upon dilution of the denaturant, on the time scale that this experiment was carried out. The protein must be unfolded for delay times greater than 30 sec in order to refold to the native state. The latter point is in common with RICK-CARD (Chen and Clark 2003).
Figure 5.
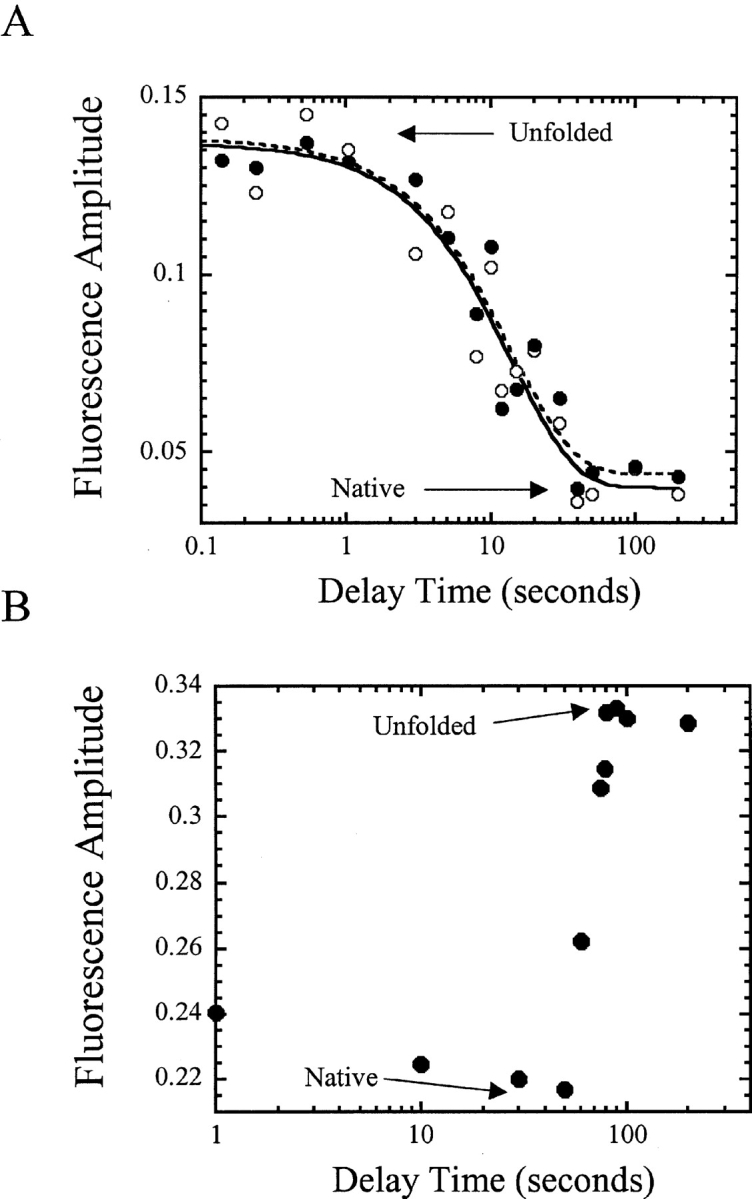
(A) Fluorescence amplitude vs. delay time in double jump experiments. The initial (open circles) and final (filled circles) signals of the traces are plotted vs. delay time. The lines represent fits of the initial (solid line) and final (dashed line) signals to a single exponential equation, as described in the text. (B) Fluorescence amplitude vs. delay time in interrupted refolding experiments. For both A and B, the signals of unfolded and of native protein are indicated.
The second sequential-mixing technique used to examine the protein was that of interrupted refolding. This technique provides details on the refolding pathway (Pappenberger et al. 2001). In these experiments, unfolded CP1-CARD was incubated in buffer for various amount of time to allow the protein to begin to refold. Following a specified time of refolding (delay time), the protein was then unfolded by mixing with urea-containing buffer. The second process, that is unfolding, is monitored experimentally. In this technique, it is assumed that the native protein represents the thermodynamically most stable state; thus intermediates (if any) that form will be unstable under the conditions of the experiment and will unfold faster than the population of native protein that has formed. Therefore, the signal traces will resemble those of the single-mixing experiments only when the native conformation is formed, and the amplitude should reflect the population of native protein. We showed in Figure 4A ▶ that during the unfolding of CP1-CARD, the signal overlays with that of the unfolded protein control within 10 msec because unfolding appears to be very rapid. Thus, if an intermediate were to form during refolding of CP1-CARD, then it may not be observed in this experiment. In other words, in the absence of kinetic phases, it may not be possible to distinguish between the amplitudes of the native protein and of the intermediate states.
This was not the case for CP1-CARD, however. The data showed that with relatively short delay times (between 1 and 50 sec), the signal was close to that of the native protein (Fig. 5B ▶). Approximately 80% of the native signal was obtained with the shortest delay time (1 sec), and this increased to 100% with a delay time of 50 sec. Surprisingly, for those delay times (1–50 sec) the signal did not return to that of the unfolded state when the refolding protein was returned to 5 M urea-containing buffer during the second jump. This demonstrated the formation of a nativelike fluorescent species that was not the native protein. The native protein formed only after longer delay times, that is, the second phase shown in Figure 5B ▶. Overall, the data show a lag of about 50 sec during which a nativelike species is formed (Fig. 5B ▶), and native CP1-CARD is formed from the second phase, with a half-time of about 65 sec. Similar results were observed for RICK-CARD (Chen and Clark 2003), where we showed that there are two phases in the interrupted refolding studies, a lag (t½ ~ 15 sec) followed by a second phase (t½ ~ 100 sec) from which the native state formed.
As described previously by Kiefhaber (1995), the lag phase observed in the interrupted refolding experiment can be explained only if the protein folds by a sequential folding mechanism because the native conformation is not directly accessible from the unfolded state. However, the results shown in Figure 5 ▶ are difficult to reconcile with a simple sequential folding model. The data suggest that the unfolded state may be partitioned between two major species and that folding occurs from both species. The native conformation is not directly accessible from either unfolded state because there is not an early formation of native protein. Over the course of 50 sec, the folding intermediates appear to convert to a conformation with nativelike fluorescence emission, from which the native conformation is accessible. Thus, the refolding pathway may contain early parallel folding events and a late sequential folding event. A mechanism of this type has been described recently for the formation of a kinetic trap in the folding of single-chain FV fragments (Hoyer et al. 2002).
Discussion
We have characterized the equilibrium and kinetic folding of CP1-CARD, a member of the α-helical Greek key protein family. We have shown that the folding of CP1-CARD is described by a two-state equilibrium mechanism representing the native and unfolded ensembles. The protein is marginally stable, with a ΔGH2O of 1.1 ± 0.2 kcal/mole and an m-value of 0.65 ± 0.06 kcal/mole/M (10 mM Tris-HCl at pH 8.0, 1 mM DTT, 25°C). When compared to the homologous protein, RICK-CARD, CP1-CARD is less stable by about 2 kcal/mole. The native CP1-CARD is fully functional for binding to RICK-CARD.
Single-mixing stopped-flow studies showed that both re-folding and unfolding appeared to be complete within the dead time for mixing (10 msec). As a result, both the equilibrium and kinetic properties of the protein would appear to be relatively simple. However, double-jump experiments demonstrated the presence of an unfolded-like intermediate during unfolding. The intermediate converted to the fully unfolded conformation with an apparent rate constant of 0.07 sec−1. As was shown previously for RICK-CARD (Chen and Clark 2003), CP1-CARD must be fully unfolded in order to refold to the native conformation. The interrupted refolding studies showed a lag of about 50 sec followed by a second phase from which native protein formed. During the lag phase, a highly fluorescent species formed that did not unfold when returned to denaturing conditions. Although at first glance the folding/unfolding kinetics may appear straightforward, the sequential-mixing studies show that the reactions are quite complex. Overall, the data suggest that the intermediates that form both in unfolding and in refolding represent kinetically trapped species. Currently, the nature of the intermediates remains unknown.
Although the determinants of folding are still ambiguous, Baker and coworkers (Plaxco et al. 2000) have suggested that the structural topology is a critical determinant of protein folding rates. The contact order (CO) represents the average sequence separation between pairs of contacting residues in the native structure and represents a means to describe the complexity of a folding topology. Based on the CO of CP1-CARD and RICK-CARD (7% and 8%, respectively) and the correlation between CO and refolding rates of two-state proteins (Plaxco et al. 2000), the folding rates of CP1-CARD and RICK-CARD are predicted to be 2.2e4 sec−1 and 1.5e4 sec−1, respectively. Another algorithm, LRO, calculates long-range interactions in the native protein (Gromiha and Selvaraj 2001) and is defined as contacts between two residues that are close in space (≤8 Å) but far apart in the sequence (>12 residues). Using this model and the calculated LRO of CP1-CARD (1.00) and RICK-CARD (1.41), the predicted folding rate is 6900 sec−1 (CP1-CARD) or 2700 sec−1 (RICK-CARD). As described previously (Chen and Clark 2003), the fast phase of RICK-CARD folding occurs with a rate constant of 30 sec−1, several orders of magnitude slower than predicted for a small helical protein. In the case of CP1-CARD, single-mixing stopped-flow studies suggest that refolding is rapid, possibly on the order predicted by CO or LRO. However, the interrupted refolding studies demonstrated that the burst phase kinetics correspond to rapid formation of a kinetically trapped intermediate with nativelike fluorescence properties.
The role of intermediates in the refolding of proteins remains a topic of much debate (Creighton 1994; Baldwin 1996) as the presence of intermediates slows the folding process. In particular, discussions focus on whether the intermediates are obligatory or off-pathway and whether they are misfolded conformations. Kinetically trapped intermediates, those with high activation barriers, have been described for several proteins and have been shown to be due to cistrans prolyl isomerization (Nall et al. 1978; Schmid 1986; Kiefhaber et al. 1990; Kiefhaber 1995; Bhuyan and Udgaonkar 1999; Reader et al. 2001), slow formation of disulfide isomers (Weissman and Kim 1991; Chang et al. 2001; Hua et al. 2002), heme misligation (Rumbley et al. 2002), or inappropriate domain (Hoyer et al. 2002) or oligomer (Ziegler et al. 1993) interactions. In some cases (Capaldi et al. 2001, 2002) the evidence strongly suggests that the intermediates are on-pathway and likely represent an ensemble of conformations representative of the energy landscape (Bhuyan and Udgaonkar 1999). The nature of the intermediates of the CARD domains is unknown at present, but two principles emerge from current studies of α-Greek key proteins. First, both unfolding and refolding processes are slow. Second, the pathways of refolding and of unfolding contain kinetically trapped species.
Two nucleation models have been proposed for the folding of β-Greek key proteins, the β-hairpin model and the β-zipper model. In the first case, the protein folds initially through a long β-hairpin, and then the protein folds up two strands at a time (Richardson 1977). In the second model, the hydrophobic core forms via two adjacent strands connected by a short loop (Hazes and Hol 1992). The relevance of these models to the folding of α-Greek key proteins is currently unknown, but it is interesting to note that Lim (1978) suggested that the β-structures form first through highly helical folding intermediates. Still, the question remains as to why the CARD domains (and the death domain superfamily) fold into the Greek key motif as opposed to antiparallel helical bundles. As described by DeGrado and coworkers (Hill et al. 2000), helical propensity and hydrophobicity are important parameters for defining the folding of a protein. These factors, however, are not sufficient to define conformational specificity as they are geometrically nonspecific. More specific interactions are required to obtain a uniquely folded structure. Working with designed helical peptides that form dimeric four-helix bundles, De-Grado’s group (Hill et al. 2000) has shown that two nearly identical peptides (32 of 35 residues) adopt different folds. In one case the peptide adopted an antiparallel helical arrangement, whereas the second peptide adopted a crossover fold (the bisecting U motif) found in the death domains. As a result of the amino acid changes, new helix interfaces were formed. This process likely occurs on a larger scale with the α-Greek key CARD domains. That is, the helical interfaces cannot be satisfied in an antiparallel arrangement, so the conformational specificity is restricted to the Greek key topology.
Materials and methods
Reagents
Ultrapure urea was purchased from Nacalai Tesque Inc. Dithiothreitol (DTT) was from Fisher. Tris(2-carboxyethyl)-phosphine hydrochloride (TCEP) was from Pierce. Trizma-base (Tris), sodium chloride (NaCl), DEAE sepharose, and molecular weight markers were from Sigma. Guanidium hydrochloride (GdnHCl) was obtained from ICN. All buffers were filtered through either 0.45 or 0.22 μm filter membranes. The urea-containing buffers were prepared as described previously (Pace et al. 1989).
Plasmid construction
The cDNA encoding the CARD domain of procaspase-1 was obtained from PCR amplification using the template cDNA of full-length procaspase-1 (D. Danzeiser and A.C. Clark, unpubl.). The forward and reverse primers used for PCR amplification were 5′-GTCGCGGATCCCATATGGCCGACAAGG-3′ and 5′-GG TAATTTCCTCGAGTTTAATCTGCTG-3′, respectively. The PCR product was subcloned into pET21a (Novagen) using the restriction enzymes NdeI and XhoI. The resulting plasmid, named pCARD-procasp1, was sequenced (both strands) to confirm the sequence. The resulting protein, CARD of procaspase-1 (CP1-CARD), is composed of 92 amino acids from methionine 1 to aspartate 92 of procaspase-1. The calculated molecular weight of CP1-CARD is 10,262 Da.
Protein purification
CP1-CARD was purified using protocols described previously for RICK-CARD (Chen and Clark 2003) with the following modifications. CP1-CARD was loaded onto a DEAE-sepharose column (2.6 × 25 cm; Sigma) equilibrated with buffer A (30 mM Tris at pH 8.0, 1 mM DTT) at 4°C. The column was then washed with 200 mL of buffer A, and a two-step gradient procedure was used to elute the protein. First, a linear gradient of buffer A containing 150 to 400 mM NaCl, with a total volume of 800 mL, was used. This was followed by a linear gradient of buffer A containing 400 to 500 mM NaCl, with a total volume of 400 mL. The flow rate was 4 mL/min. CP1-CARD eluted between 300 and 350 mM NaCl. The fractions were analyzed by 4%–25% SDS-PAGE. The pure protein fractions were pooled, concentrated, and dialyzed against buffer A. The extinction coefficient at 280 nm of CP1-CARD was calculated (Edelhoch 1967) to be 2,650 M−1cm−1. Tryptic digests followed by mass spectral sequencing confirmed the CP1-CARD protein. Two fragments were identified that unambiguously establish the protein identity: SMGEGTINGLLDELLQTRVLN KEEMEK and ALIDSVIPK. RICK-CARD was purified as described (Chen and Clark 2003).
Molecular modeling
The homology model of CP1-CARD was generated using the homology module of Insight II (Molecular Simulation Inc.). The sequence alignment used to generate the homology model is based on the conserved residues, hydrophobic core residues, and the secondary structural elements between CP1-CARD and the template. The alignment is shown in Figure 1A ▶ and the structural model is shown in Figure 1B ▶. The NMR structure of ICEBERG, another CARD-containing protein (Humke et al. 2000), was used as the modeling template (from Protein Data Bank, accession no. 1DGN) because of its higher pairwise sequence identity, 52%, with CP1-CARD.
Fluorescence and circular dichroism (CD) spectroscopy
Fluorescence emission spectra were obtained by using a PTI C-61 spectrofluorometer (Photon Technology International). CP1-CARD (6 μM) was excited at 280 nm, and the emission spectra were measured from 300 to 400 nm in buffer B (10 mM Tris at pH 8.0, 1 mM DTT). CD spectra were obtained with a J600A spec-tropolarimeter (Jasco Inc.). The protein concentrations for far-UV CD in buffer B and 6 M urea-containing buffer B were 92 and 35 μM, respectively. The protein concentrations for near-UV CD in buffer B and 6 M urea-containing buffer B were 225 μM and 117 μM, respectively. The path length was either 0.1 cm (far-UV) or 1 cm (near-UV). All spectra were corrected for buffer signals. The experiments were maintained at a constant temperature of 25°C using a circulating water bath.
Equilibrium folding/unfolding
Equilibrium folding/unfolding experiments were done by manual preparation or by titration. For the manual preparation, each sample was prepared to contain the desired urea concentrations as indicated in Figure 3A ▶ and a desired protein concentration. The experiments were repeated with different protein concentrations ranging from 3 to 40 μM in buffer B. The samples were incubated overnight at 25°C, then the fluorescence and CD spectra were examined. Alternatively, automated titrations were performed as described (Chen and Clark 2003). Samples were excited at 280 nm, and fluorescence emission was monitored from 300 to 400 nm. For the CD experiments, titrations were performed manually by mixing unfolded protein with the native protein, with an incubation time of 2,400 sec. A quartz cuvette with 1 cm path length was used in these experiments. The signal at 220 nm was measured every 0.5 sec for 120 sec, and the data were averaged and corrected for background signals.
The fluorescence emission spectrum at each urea concentration was analyzed as described previously (Royer et al. 1993) to determine the average emission wavelength. The normalized average emission wavelength and the normalized CD data were fit globally to a two-state equilibrium process (N ⇆ U) described by Santoro and Bolen (1988), where N and U are the native and unfolded ensembles, respectively. The program IgorPro (Wavemetrics Inc.) was used for the global fits.
Size-exclusion chromatography
A Superose 6HR 10/30 FPLC column (10 × 300 mm; Amersham Pharmacia) was used to characterize the molecular weight of CP1-CARD. Molecular weight markers (100 μL; Sigma) were loaded onto the column individually in buffer C (30 mM Tris at pH 8.0, 250 mM NaCl). The concentrations of the markers were the following: dextran blue (2000 kDa, 2 mg/mL), bovine serum albumin (66.2 kDa, 5 mg/mL), carbonic anhydrase (29 kDa, 2 mg/mL), cytochrome c (12.4 kDa, 2 mg/mL), and aprotinin (6.5 kDa, 3 mg/mL). The flow rate was 0.1 mL/min, and fractions were collected with 10 drops per fraction (~0.47 mL). CP1-CARD (85 μM, 150 μL) was loaded onto the column in buffer C containing 0.5 mM TCEP. The molecular weight of CP1-CARD obtained from the standard curve is 10,600 Da.
Fluorescence titration studies for CARD-CARD interaction
CP1-CARD, which does not contain a tryptophanyl residue, was added to RICK-CARD (3 μM) so that the final ratio of CP1-CARD to RICK-CARD was 0 to 4. The sample was excited at 295 nm, and the signals were corrected for dilution and background signal. The fluorescence emission of RICK-CARD at 330 nm was plotted versus the ratio of CP1-CARD:RICK-CARD. The results were fit to equation 3, below. Assuming a simple protein-to-ligand binding reaction (P + ↔ L), the fraction of bound ligand, γ (=[L]/[PT]), can be related to the equilibrium dissociation constant, Kd, the total concentration of protein (RICK-CARD), PT, and the total concentration of ligand (CP1-CARD), LT, as shown in equation 1:
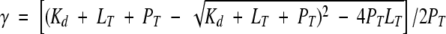 |
(1) |
The fluorescence emission, Fobs, is related to the fraction of bound ligand as shown in equation 2, where F0 is the initial fluorescence signal and F1 is the final fluorescence signal:
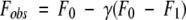 |
(2) |
Combining equation 1 and equation 2 yields equation 3:
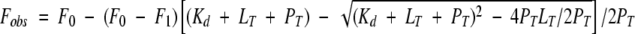 |
(3) |
which describes the observed fluorescence emission as a function of the Kd.
Single-mixing stopped-flow fluorescence studies
The kinetic folding/unfolding experiments were performed using a stopped-flow spectrofluorometer (SX.18MV, Applied Photophysics). The temperature was controlled at 25°C using a circulating water bath. The samples were excited at 280 nm, and the fluorescence emission was measured using a cutoff filter of 305 nm. The final protein concentration was 3 μM, and a mixing ratio of 1:10 was used. For refolding experiments, stock protein (33 μM) in 6 M urea-containing buffer was mixed with buffer B containing urea between 0 and 6 M, as shown in the figures. For unfolding experiments, stock protein (33 μM) in buffer B was mixed with buffer B containing urea between 0 and 6 M. The final protein concentration was 3 μM, and a mixing ratio of 1:10 was used. The final urea concentrations are shown in the figures.
Double jump stopped-flow experiments
The experiments were performed at 25°C as described (Schmid 1986; Chen and Clark 2003). Native CP1-CARD (36 μM) in buffer B was mixed 1:1 with buffer B containing 9.6 M urea for the first jump. After a specified delay time, the protein solution was mixed with buffer B using a mixing ratio of 1:5 for the second jump. The signal trace was monitored for more than 10 sec. The final protein concentration was 3 μM, and the final urea concentration was 0.8 M. The native protein signal was determined from the protein signal in 0.8 M urea-containing buffer B. The unfolded protein signal was obtained from the protein signal in 4.8 M urea-containing buffer B.
Interrupted refolding experiments
The interrupted refolding experiments for CP1-CARD were performed as described previously (Chen and Clark 2003), with the following exceptions. The first jump was performed with a 1:10 mixing ratio, and the second jump was performed with a 1:5 mixing ratio. Initially, CP1-CARD was unfolded in buffer B containing 5 M urea for approximately 1 h to allow for equilibration. Unfolded CP1-CARD (198 μM) in buffer B containing 5 M urea was mixed with buffer B for the first jump. After various delay times, as shown in the figures, the protein solution was mixed with buffer B containing 6 M urea. After the first jump, the urea concentration was 0.45 M. After the second jump, the urea concentration was 5 M, and the final protein concentration was 3 μM. The signal for the unfolded protein was obtained from the final signal of the experiment that used a 100-sec delay time. To obtain the signal of the native protein, the third solution was changed to buffer B.
Acknowledgments
This work was supported by a grant from the NIH (GM065970). We thank Dr. Stefan Franzen for assistance with the homology modeling, Dr. Yun-Chorng Chang for assistance with calculations of long-range order, and Dr. Michael Goshe for mass spectral analysis.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
CARD, caspase recruitment domain
CP1-CARD, CARD of procaspase-1
RICK, RIP-like interacting CLARP kinase
DD, death domain
DED, death effector domain
APAF-1, apoptotic protease activating factor
IPTG, isopropyl β-D-1-thiogalactopyranoside
TCEP, Tris(2-carboxyethyl)-phosphine hydrochloride
CO, relative contact order
LRO, long range order
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03521504.
References
- Alonso, D.O. and Dill, K.A. 1991. Solvent denaturation and stabilization of globular proteins. Biochemistry 30 5974–5985. [DOI] [PubMed] [Google Scholar]
- Baldwin, R.L. 1996. On-pathway versus off-pathway folding intermediates. Fold. Des. 1 R1–R8. [DOI] [PubMed] [Google Scholar]
- Bhuyan, A.K. and Udgaonkar, J.B. 1999. Observation of multistate kinetics during the slow folding and unfolding of barstar. Biochemistry 38 9158–9168. [DOI] [PubMed] [Google Scholar]
- Brandts, J.F., Halvorson, H.R., and Brennan, M. 1975. Consideration of the possibility that the slow step in protein denaturation reactions is due to cistrans isomerism of proline residues. Biochemistry 14 4953–4963. [DOI] [PubMed] [Google Scholar]
- Capaldi, A.P., Shastry, M.C., Kleanthous, C., Roder, H., and Radford, S.E. 2001. Ultrarapid mixing experiments reveal that Im7 folds via an on-pathway intermediate. Nat. Struct. Biol. 8 68–72. [DOI] [PubMed] [Google Scholar]
- Capaldi, A.P., Kleanthous, C., and Radford, S.E. 2002. Im7 folding mechanism: Misfolding on a path to the native state. Nat. Struct. Biol. 9 209–216. [DOI] [PubMed] [Google Scholar]
- Chang, J.Y., Li, L., and Lai, P.H. 2001. A major kinetic trap for the oxidative folding of human epidermal growth factor. J. Biol. Chem. 276 4845–4852. [DOI] [PubMed] [Google Scholar]
- Chen, Y.R. and Clark, A.C. 2003. Equilibrium and kinetic folding of a α-helical Greek key protein domain: Caspase recruitment domain (CARD) of RICK. Biochemistry 42 6310–6320. [DOI] [PubMed] [Google Scholar]
- Chou, J.J., Matsuo, H., Duan, H., and Wagner, G. 1998. Solution structure of the RAIDD CARD and model for CARD/CARD interaction in caspase-2 and caspase-9 recruitment. Cell 94 171–180. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. 1994. The energetic ups and downs of protein folding. Nat. Struct. Biol. 1 135–138. [DOI] [PubMed] [Google Scholar]
- Day, C.L., Dupont, C., Lackmann, M., Vaux, D.L., and Hinds, M.G. 1999. Solution structure and mutagenesis of the caspase recruitment domain (CARD) from Apaf-1. Cell Death Differ. 6 1125–1132. [DOI] [PubMed] [Google Scholar]
- Druilhe, A., Srinivasula, S.M., Razmara, M., Ahmad, M., and Alnemri, E.S. 2001. Regulation of IL-1β generation by pseudo-ICE and ICEBERG, two dominant negative caspase recruitment domain proteins. Cell Death Differ. 8 649–657. [DOI] [PubMed] [Google Scholar]
- Eberstadt, M., Huang, B., Chen, Z., Meadows, R.P., Ng, S.C., Zheng, L., Le-nardo, M.J., and Fesik, S.W. 1998. NMR structure and mutagenesis of the FADD (Mort1) death-effector domain. Nature 392 941–945. [DOI] [PubMed] [Google Scholar]
- Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6 1948–1954. [DOI] [PubMed] [Google Scholar]
- Fairbrother, W.J., Gordon, N.C., Humke, E.W., O’Rourke, K.M., Starovasnik, M.A., Yin, J.P., and Dixit, V.M. 2001. The PYRIN domain: A member of the death domain-fold superfamily. Protein Sci. 10 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromiha, M.M. and Selvaraj, S. 2001. Comparison between long-range interactions and contact order in determining the folding rate of two-state proteins: Application of long-range order to folding rate prediction. J. Mol. Biol. 310 27–32. [DOI] [PubMed] [Google Scholar]
- Gunasekaran, K., Eyles, S.J., Hagler, A.T., and Gierasch, L.M. 2001. Keeping it in the family: Folding studies of related proteins. Curr. Opin. Struct. Biol. 11 83–93. [DOI] [PubMed] [Google Scholar]
- Hazes, B. and Hol, W.G. 1992. Comparison of the hemocyanin β-barrel with other Greek key β-barrels: Possible importance of the “β-zipper” in protein structure and folding. Proteins 12 278–298. [DOI] [PubMed] [Google Scholar]
- Hill, R.B., Raleigh, D.P., Lombardi, A., and DeGrado, W.F. 2000. De novo design of helical bundles as models for understanding protein folding and function. Acc. Chem. Res. 33 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann, K. 1999. The modular nature of apoptotic signaling proteins. Cell. Mol. Life Sci. 55 1113–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer, W., Ramm, K., and Pluckthun, A. 2002. A kinetic trap is an intrinsic feature in the folding pathway of single-chain Fv fragments. Biophys. Chem. 96 273–284. [DOI] [PubMed] [Google Scholar]
- Hua, Q.X., Jia, W., Frank, B.H., Phillips, N.F., and Weiss, M.A. 2002. A protein caught in a kinetic trap: Structures and stabilities of insulin disulfide isomers. Biochemistry 41 14700–14715. [DOI] [PubMed] [Google Scholar]
- Humke, E.W., Shriver, S.K., Starovasnik, M.A., Fairbrother, W.J., and Dixit, V.M. 2000. ICEBERG: A novel inhibitor of interleukin-1β generation. Cell 103 99–111. [DOI] [PubMed] [Google Scholar]
- Jeong, E.J., Bang, S., Lee, T.H., Park, Y.I., Sim, W.S., and Kim, K.S. 1999. The solution structure of FADD death domain. Structural basis of death domain interactions of Fas and FADD. J. Biol. Chem. 274 16337–16342. [DOI] [PubMed] [Google Scholar]
- Kiefhaber, T. 1995. Kinetic traps in lysozyme folding. Proc. Natl. Acad. Sci. 92 9029–9033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefhaber, T., Quaas, R., Hahn, U., and Schmid, F.X. 1990. Folding of ribo-nuclease T1. 1. Existence of multiple unfolded states created by proline isomerization. Biochemistry 29 3053–3061. [DOI] [PubMed] [Google Scholar]
- Lee, S.H., Stehlik, C., and Reed, J.C. 2001. Cop, a caspase recruitment domain-containing protein and inhibitor of caspase-1 activation processing. J. Biol. Chem. 276 34495–34500. [DOI] [PubMed] [Google Scholar]
- Lim, V.I. 1978. Polypeptide chain folding through a highly helical intermediate as a general principle of globular protein structure formation. FEBS Lett. 89 10–14. [DOI] [PubMed] [Google Scholar]
- Mirny, L. and Shakhnovich, E. 2001. Evolutionary conservation of the folding nucleus. J. Mol. Biol. 308 123–129. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nall, B.T., Garel, J.R., and Baldwin, R.L. 1978. Test of the extended two-state model for the kinetic intermediates observed in the folding transition of ribonuclease A. J. Mol. Biol. 118 317–330. [DOI] [PubMed] [Google Scholar]
- O’Neil, J.D. and Hofmann, T. 1987. Tyrosine and tyrosinate fluorescence of pig intestinal Ca2+-binding protein. Biochem. J. 243 611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C.N., Shirley, B., and Thompson, J. 1989. Measuring the conformational stability of a protein. In rotein structure, a practical approach (ed. T.E. Creighton), pp. 311–330. IRL Press, New York.
- Pappenberger, G., Aygun, H., Engels, J.W., Reimer, U., Fischer, G., and Kiefhaber, T. 2001. Nonprolyl cis peptide bonds in unfolded proteins cause complex folding kinetics. Nat. Struct. Biol. 8 452–458. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., and Baker, D. 1998. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 277 985–994. [DOI] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., Ruczinski, I., and Baker, D. 2000. Topology, stability, sequence, and length: Defining the determinants of two-state protein folding kinetics. Biochemistry 39 11177–11183. [DOI] [PubMed] [Google Scholar]
- Qin, H., Srinivasula, S.M., Wu, G., Fernandes-Alnemri, T., Alnemri, E.S., and Shi, Y. 1999. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399 549–557. [DOI] [PubMed] [Google Scholar]
- Reader, J.S., Van Nuland, N.A., Thompson, G.S., Ferguson, S.J., Dobson, C.M., and Radford, S.E. 2001. A partially folded intermediate species of the β-sheet protein apopseudoazurin is trapped during proline-limited folding. Protein Sci. 10 1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, J.S. 1977. β-sheet topology and the relatedness of proteins. Nature 268 495–500. [DOI] [PubMed] [Google Scholar]
- Royer, C.A., Mann, C.J., and Matthews, C.R. 1993. Resolution of the fluorescence equilibrium unfolding profile of trp aporepressor using single tryp-tophan mutants. Protein Sci. 2 1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbley, J.N., Hoang, L., and Englander, S.W. 2002. Recombinant equine cytochrome c in Escherichia coli: High-level expression, characterization, and folding and assembly mutants. Biochemistry 41 13894–13901. [DOI] [PubMed] [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethane-sulfonyl α-chymotrypsin using different denaturants. Biochemistry 27 8063–8068. [DOI] [PubMed] [Google Scholar]
- Schellman, J.A. 1978. Solvent denaturation. Biopolymers 17 1305–1322. [DOI] [PubMed] [Google Scholar]
- Schiffmann, D.A., White, J.H., Cooper, A., Nutley, M.A., Harding, S.E., Jumel, K., Solari, R., Ray, K.P., and Gay, N.J. 1999. Formation and biochemical characterization of tube/pelle death domain complexes: Critical regulators of postreceptor signaling by the Drosophila toll receptor. Biochemistry 38 11722–11733. [DOI] [PubMed] [Google Scholar]
- Schmid, F.X. 1986. Fast-folding and slow-folding forms of unfolded proteins. Methods Enzymol. 131 70–82. [DOI] [PubMed] [Google Scholar]
- Shi, Y. 2002. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 9 459–470. [DOI] [PubMed] [Google Scholar]
- Shiozaki, E.N., Chai, J., and Shi, Y. 2002. Oligomerization and activation of caspase-9, induced by Apaf-1 CARD. Proc. Natl. Acad. Sci. 99 4197–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome, M., Hofmann, K., Burns, K., Martinon, F., Bodmer, J.L., Mattmann, C., and Tschopp, J. 1998. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr. Biol. 8 885–888. [DOI] [PubMed] [Google Scholar]
- Vaughn, D.E., Rodriguez, J., Lazebnik, Y., and Joshua-Tor, L. 1999. Crystal structure of Apaf-1 caspase recruitment domain: An α-helical Greek key fold for apoptotic signaling. J. Mol. Biol. 293 439–447. [DOI] [PubMed] [Google Scholar]
- Weber, C.H. and Vincenz, C. 2001. The death domain superfamily: A tale of two interfaces? Trends Biochem. Sci. 26 475–481. [DOI] [PubMed] [Google Scholar]
- Weissman, J.S. and Kim, P.S. 1991. Reexamination of the folding of BPTI: Predominance of native intermediates. Science 253 1386–1393. [DOI] [PubMed] [Google Scholar]
- Xiao, T., Towb, P., Wasserman, S.A., and Sprang, S.R. 1999. Three-dimensional structure of a complex between the death domains of Pelle and Tube. Cell 99 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P., Chou, J., Olea, R.S., Yuan, J., and Wagner, G. 1999. Solution structure of Apaf-1 CARD and its interaction with caspase-9 CARD: A structural basis for specific adaptor/caspase interaction. Proc. Natl. Acad. Sci. 96 11265–11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler, M.M., Goldberg, M.E., Chaffotte, A.F., and Baldwin, T.O. 1993. Re-folding of luciferase subunits from urea and assembly of the active heterodimer. Evidence for folding intermediates that precede and follow the dimerization step on the pathway to the active form of the enzyme. J. Biol. Chem. 268 10760–10765. [PubMed] [Google Scholar]