

Abstract
A technique for systematic peptide variation by a combination of rational and evolutionary approaches is presented. The design scheme consists of five consecutive steps: (i) identification of a “seed peptide” with a desired activity, (ii) generation of variants selected from a physicochemical space around the seed peptide, (iii) synthesis and testing of this biased library, (iv) modeling of a quantitative sequence-activity relationship by an artificial neural network, and (v) de novo design by a computer-based evolutionary search in sequence space using the trained neural network as the fitness function. This strategy was successfully applied to the identification of novel peptides that fully prevent the positive chronotropic effect of anti-β1-adrenoreceptor autoantibodies from the serum of patients with dilated cardiomyopathy. The seed peptide, comprising 10 residues, was derived by epitope mapping from an extracellular loop of human β1-adrenoreceptor. A set of 90 peptides was synthesized and tested to provide training data for neural network development. De novo design revealed peptides with desired activities that do not match the seed peptide sequence. These results demonstrate that computer-based evolutionary searches can generate novel peptides with substantial biological activity.
Keywords: β-adrenergic receptor/evolutionary design/genetic algorithm/peptide library/screening
Molecular design aims to identify compounds with a desired activity and to rationally modify molecular structures to yield desired or improved molecular properties. Two principal problems need to be solved: first, a search strategy must be developed for exploiting the potentially huge number of compounds; second, for any rational design approach a model must be available to guide the systematic modification of molecular structures. Rational molecular design can be successful only with profound knowledge of the influence of structural modifications on molecular function. An alternative approach is to use evolutionary strategies in which optimization of molecular properties is achieved by a cyclic variation-selection process; no detailed understanding of the respective structure-activity relationship is required. This process can be performed in vivo, in vitro, or even entirely by computer (“in machina” or “in silico”). Usually, a large number of compounds must be synthesized and tested per evolutionary cycle to avoid trapping into premature convergence, and the optimization process bears the danger of being merely superior to pure random search. If, however, this limitation can be overcome, e.g., by massively parallel screening and smart variation of molecular structures, evolutionary design can be a powerful method for rapid identification of potential lead compounds.
We have developed a design approach combining the advantages of a computer-based evolutionary search with a knowledge-based rational access to reduce the time and effort needed to obtain desirable molecules. The goal is to minimize the number of bench experiments by making extensive use of the information provided by the results of each in vitro or in vivo experiment. Our concept can be divided into successive steps: (i) identification of a single compound with some desired activity, e.g., by expert knowledge, database or random screening, combinatorial libraries, or phage display (1–3); (ii) generation of a focusing library taking the compound obtained in step i as a “seed structure.” A limited set of variants is generated, approximately gaussian-distributed, in some physicochemical space around the seed (4); (iii) synthesis and testing of the new variants for their activity; (iv) training of artificial neural networks providing simple heuristic models of (quantitative) structure–activity relationships (SARs) based on the activities measured in step iii (5); and (v) computer-based evolutionary search for highly active compounds using the network models as the fitness function (6, 7).
Here we describe the successful application of our approach to the design of a peptide that fully prevents the positive chronotropic effect of anti-β1-adrenoreceptor autoantibodies from the serum of patients with idiopathic dilated cardiomyopathy (DCM). Autoantibodies directed against the first and second extracellular loops of human β1-adrenoreceptor were shown to contribute to the harmful chronic cardiac adrenergic drive to which patients with DCM are believed to be exposed (8–12). Short synthetic peptides encompassing the natural epitopes were able to neutralize the chronotropic effect of the autoantibodies (12). Our idea was to test whether it is possible to derive novel artificial epitope sequences that could be used as potential immunotherapeutic agents following the design strategy described above. The peptide–antibody interaction investigated here is thought to provide a model of specific peptide–protein interactions.
EXPERIMENTAL PROTOCOL
Peptide Synthesis.
Polypropylene pin-based peptide synthesis was performed according to the method introduced by Gheysen et al. (13, 14) with the modifications described previously (15, 16). Distribution of the peptides on the pin-plate was randomized to avoid systematic errors, e.g., border effects influenced by the microtiter-plate format of the pin-plate. Soluble peptides were prepared by the solid-phase method (17) in a MilliGen 9050 continuous-flow peptide synthesizer using Fmoc/tBu (18) fast-cycle strategy and TOPPipU (19) as the coupling reagent. After cleavage from the resin using standard protocols (18) the crude peptides were purified to homogeneity by preparative HPLC on a Waters Delta-Pak C18 300-Å column and lyophilized. Characterization was accomplished by analytical HPLC and matrix-assisted laser desorption time-of-flight mass spectroscopy. Multipin noncleavable pin-kits (pin-plates) were purchased from Abimed Analyses-Technik (Langenfeld, Germany), the 9-fluoroenylmethyloxycarbonyl (Fmoc)-protected and the pentafluorophenyl (Pfp)- or 3-hydroxy-2,3-dihydroxy-4-oxobenzotriazoyl (Dhbt)-activated amino acids were obtained from PerSeptive Biosystems (Wiesbaden, Germany); TentaGel S resins were from Rapp Polymere (Tübingen, Germany); 2-[2-oxo-1(2H)-pyridyl]-1,1,3,3-bispentamethylneuronium tetrafluoroborate (TOPPipU) was purchased from Calbiochem-Nova Biochem (Bad Soden/Ts., Germany).
Preparation of the Gamma Globulin Fraction.
Serum was obtained from a patient with idiopathic DCM whose left ventricular ejection fraction averaged 15%. The gamma globulin fraction was isolated by using 40% ammonium sulfate (wt/vol) precipitation performed three times. The samples were dialyzed against 1 liter of 10 mM phosphate-buffered 0.9% NaCl solution (pH 7.2) for 30 hr. The solution was changed five times. The purified Igs were taken up in PBS (pH 7.2).
Neutralization of the Autoantibodies.
Gamma globulin fractions were used for the neutralization experiments. The corresponding peptides (20 μl) were added to 50 μl of the gamma globulin fraction. The mixtures were shaken and placed in a refrigerator for 1 hr. After this procedure the samples (70 μl) were added to neonatal heart-muscle cells which were cultured in 2 ml of Halle SM20-I medium (21). The final dilution of the gamma globulin fraction was 1:40. The beating rate of the cells was measured 5 and 60 min after the addition of the peptide/gamma globulin mixture.
Cardiomyocyte Cell Culture.
Single cells were dissociated from the minced ventricles of 1- to 2-day-old Wistar rats with a 0.2% solution of trypsin and were cultured, as detailed elsewhere (20), in Halle SM20-I medium in the presence of 10% neonatal calf serum and 2 μM fluorodeoxyuridine (21). On the third or fourth day of cultivation the cells were incubated for 2 hr in 2 ml of fresh serum-containing medium. Thereafter the beating frequency of the myocytes was determined on the heated stage of an inverted microscope at 37°C. Seven to ten selected cells or synchronously contracting cell clusters were counted for 15 s. Antibodies and peptides were added cumulatively. This procedure was repeated twice in different cultures to yield results representing a total of 20–30 cells or cell clusters for each sample of a given gamma globulin fraction.
Enzyme Immunoassay (EIA).
Pin-EIA was performed in a 96-well microtiter plate (Nunc) directly with the Pin-peptides (peptides remained pin-bound). Gamma globulins were diluted 1:500 in PBS according to Dulbecco (pH 7.4) (13) with 1% BSA (Sigma) and 0.1% Tween 20 (Sigma). Blocking of nonspecific binding sites was achieved by pin incubation in sample buffer (200 μl/well) for 1 hr at room temperature in a humid chamber prior to addition of Ig. For the test 175 μl/well of diluted gamma-globulin fraction was incubated for 18 hr at 4–7°C in a humid chamber. Then the pins were extensively washed with PBS buffer (see above). For detection of bound Ig the pins were incubated with conjugate [goat anti-human IgG, IgM, and IgA coupled to horseradish peroxidase, and goat anti-human IgG-Fc coupled to horseradish peroxidase as a mixture (Dianova), diluted 1:4000 in PBS] for 1 hr at room temperature. After four washings, the substrate for peroxidase reaction [0.1 mg of 3,3",5,5"-tetramethylbenzidine (Fluka) in 11 ml of phosphate-citrate buffer containing 0.012% sodium perborate (Sigma)] was added. After a 10-min incubation the pins were removed from the wells, and 50 μl of 2 M sulfuric acid was added per well. The absorbance per well (i.e., per pin-peptide) was detected at 450 nm in a microtiter-plate photometer. For statistical purposes the EIA was repeated five times, and for data analysis the background activity was subtracted from the average absorbance per well.
Neural Network Training.
Three-layered feed-forward networks with a single hidden layer containing different numbers of neurons were trained by using an evolutionary algorithm (5, 6, 32). The number of training cycles (generations) was 200 per network, and the population size per generation was 500. Network weights were initialized in [−1, 1]. The weight vector leading to the lowest mean square error in training-data prediction was selected as the “parent” for the next generation. New weight vectors were generated approximately gaussian-distributed around the parent. The learning-step size determining the standard deviation of the gaussian distribution was automatically adapted during the network training process, i.e., it was also subjected to a variation/selection scheme. The initial step-size value was 1. If the step-size value was below 0.001 it was automatically reset to a value of 0.01 to avoid premature convergence of the training process. All networks were implemented by using the programming language ANSI C (22), and training was performed on an SGI R4400 central processing unit.
RESULTS AND DISCUSSION
Identification of a Natural Peptide with Some Desired Activity by Epitope Mapping.
To obtain a seed peptide, parts of the sequence of human β1-adrenoreceptor encompassing loop 2 were analyzed by epitope mapping. The amino acid sequence was chopped into overlapping fragments by using a sliding window of 10 residues and a step size of 2 residues. The ability of the peptide fragments to bind to human anti-β1-adrenoreceptor antibodies was measured by ELISA (Fig. 1). The amino acid sequence ARRCYNDPKC (positions 107–116) was identified as a natural epitope with specific affinity to the antibodies and was therefore selected as the seed peptide. This sequence is in agreement with the epitope identified by Wallukat et al. (12).
Figure 1.

Epitope mapping in the second extracellular loop of human β1-adrenoreceptor (positions 197–222). Overlapping peptides encompassing 10 residues each were synthesized and tested for autoantibody binding by ELISA. Two peptides show a pronounced affinity to the antibodies.
Generation of a Focusing Synthetic Peptide Library.
Using the amino acid sequence ARRCYNDPKC as the seed peptide, a set of 90 variants was generated by a simple algorithm describing each residue by the respective property values for hydrophobicity (23) and volume (24). This led to a 20-dimensional vector representation in terms of the two properties. The Box–Muller formula was used to generate 90 vectors approximately gaussian-distributed around the seed-peptide vector, where g is a gaussian-distributed random number and i and j are random numbers in ]0,1]:
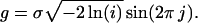 |
A standard deviation of σ = 0.1 was used to obtain many vectors close to the seed peptide vector as well as some distant variant vectors. The rationale behind this scheme was that we expected peptides with an activity similar to the seed peptide to be in close proximity to the seed peptide in sequence space (25). On the other hand, a normal distribution also contains a number of rather dissimilar vectors, which are important for neural network training (see below). The property vectors were translated back into amino acid sequences by selecting the most similar residues at each sequence position according to their physicochemical properties.
Synthesis and Testing of the New Peptide Variants for Their Activity.
The ELISA used in the epitope-mapping procedure was applied to the computer-generated peptides to test for their ability to bind to human anti-β1-adrenoreceptor antibodies [gamma globulin fractions of β1- or antibody-positive sera, or control sera, respectively; the “positive” gamma globulin fractions were also positive in the bioassay (12)]. In Fig. 2 absorbance is plotted versus the euclidian distance between the peptides and the seed peptide in sequence space. The seed peptide is located at the origin. On average, peptide activity decreases with increasing distance from the seed peptide, as expected. Although a gaussian fit could be used to model the relationship between distance in sequence space and activity, a linear or low-order polynomial fit would be equally valid. Two exceptions from the general trend given by the plot are striking: first, in the immediate sequence-space vicinity of the seed peptide several peptides (distance <0.5) were identified that have a higher activity than the seed peptide; and second, a number of peptides located some distance away from the seed peptide in sequence space revealed activities comparable to the seed peptide or even higher. The first observation may result from local hill-climbing in the natural fitness landscape assuming the seed peptide is in a suboptimal location on a practical, rather than a global, optimum. The second observation may either be a consequence of an inaccurate distance measurement in sequence space or of remote active peptides corresponding to different local optima in the fitness landscape. Despite these uncertainties a clear result of the in vitro test is the confirmation of the applicability of our selection scheme to the generation of a small focusing peptide library. Several nonnatural peptides with increased antibody-binding ability compared with the natural epitope were identified (Fig. 2). The error of absorption measurement was determined to be 15% (overall average of experiments repeated five times).
Figure 2.

Activity of 90 peptides constituting a focusing library. Average absorbance as measured by ELISA is shown. The dashed line marks the seed peptide activity. Vertical bars give the minimal and maximal values found in the distance intervals marked on the x-axis.
Training of Neural Networks on Sequence–Activity Relationships.
The next step of our optimization procedure was neural network training. The goal was to establish an artificial fitness function modeling the underlying SAR that can be used for further computer-based optimization of peptide activity. The sequences of the 90 tested peptides plus the seed peptide were described by two property values per residue position, hydrophobicity (23) and volume (24), leading to 91 20-dimensional pattern vectors. The task of the network system was to correctly predict peptide activity on the basis of the 20-dimensional description.
Two-dimensional projections of the data were made to obtain an idea of the distribution of active (absorbance ≥103) and inactive (absorbance <103) peptides in this physicochemical space (Fig. 3). Principal component analysis (PCA) (26) and Sammon mapping (27) were used to generate the projections. PCA leads to a linear projection of high-dimensional data onto a plane spanned by the two axes (the principal components) covering most of the data variance. The nonlinear Sammon projection was obtained by using an optimization procedure where the relative distances between the peptides in the high-dimensional space are conserved in the two-dimensional x–y representation. For a more in-depth treatment of Sammon mapping and PCA, see refs. 28 and 29. The resulting maps consistently reveal two adjacent clusters of active compounds (labeled I and II in Fig. 3); the active peptides are not spread over the whole map. However, because of the relatively broad distribution of active compounds and the apparently nonhomogeneous clusters, SAR modeling may be confronted with a difficult if not ill-posed problem. The surprising existence of two clusters of active peptides may be a consequence of the polyclonal antibody pool used in the assay. Perhaps the two clusters represent supertope motifs of peptides binding to different antibodies. Another possibility could be that different peptides bind to the same antibody in different binding modes (30).
Figure 3.

Projections of the peptide distributions in physicochemical space by principal component analysis (a) and nonlinear mapping (b). Black circles represent active peptides, and open circles represent inactive peptides. Activity was measured by ELISA. Two adjacent clusters of active peptides are labeled I and II.
We selected supervised neural networks as a framework for derivation of an SAR because these systems provide a flexible and adaptive framework for modeling arbitrary nonlinear relationships and have been shown to be relatively noise-tolerant (29, 31). Realistic SARs usually are of a complex nonlinear nature. Other modeling techniques, e.g., polynomial fitting or partial least squares, could also be used for this task. In a number of applications neural networks were shown to lead to more accurate SAR models (29, 31). However, the accuracy of an SAR model mainly depends on the descriptors used for compound encoding, and the choice of a modeling technique is of minor importance. Neural networks are not superior per se to other nonlinear function-estimation approaches.
It is important for successful feature extraction to include both positive examples (active peptides) and negative examples (inactive peptides) in the training data. This is one reason for the selection of a gaussian distribution for the generation of the above described peptide library. The general theory of neural computation and applications of neural networks to sequence analysis and SAR modeling can be found elsewhere (29, 31). The networks employed here were identical to the ProFI (Protein Filter Induction) system, which was successfully applied to similar tasks before (6, 7, 32). Fig. 4 shows the network architecture used, a three-layered feed-forward network with sigmoidal hidden-unit activity and a single linear output unit.
Figure 4.

Scheme of the neural network architecture used for modeling the sequence-activity relationship. Formal neurons are drawn as circles, with lines representing connection weights. The input-layer neurons are “fan-out” units, hidden-layer neurons have sigmoidal activity, and the output neuron is a linear unit.
The overall function represented by the network type shown in Fig. 4 is:
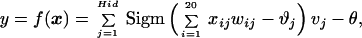 |
where x is a 20-dimensional input vector, w is the weight vector connecting the input units with the single output neuron, and θ is the output neuron’s bias value; Hid is the number of hidden neurons, v is the weight vector connecting the hidden layer with the output neuron, and ϑ is the hidden neurons’ bias values. Sigm(arg) is the common sigmoidal transfer function Sigm(arg) = 1/[1 − exp(−arg)]. Multilayered networks of this type are able to model arbitrary nonlinear input/output relationships. We used an evolutionary algorithm for network training, i.e., determination of the weights and bias values (32). The 90 peptides plus the seed peptide and their measured absorbance values provided the training and test data.
Cross-validation of the data was performed 10 times using random 8+2 splits; in addition, a complete leave-one-out procedure was applied to assess the usefulness of the SAR models. Several networks with different numbers of hidden neurons were trained on the prediction of absorbance values. A network with five hidden-layer neurons was able to reproduce the training data absorbance values with a relative deviation of 15%, a linear correlation coefficient of r = 0.87 (t = 16.4) (Fig. 5a). Independent test data were predicted with a deviation of 17%, r = 0.79 (t = 4.8) (Fig. 5b). Complete cross-validation (leave-one-out) resulted in a test data deviation of 27%, r = 0.59 (t = 6.8). As indicated by the t test values, the null hypothesis of chance correlation can be rejected. We therefore expect the network to represent a useful SAR model for exploiting sequence space. The error of ELISA measurements was determined to be 15%. Networks with more hidden-layer units showed overfitting (lower training error with a drastically increased test error), whereas networks with fewer hidden units led to higher error values (data not shown). Because even the neural network used in the peptide-design step gave a relatively poor correlation and elevated test error in the complete leave-one-out procedure, this SAR model could only be used for semiquantitative predictions (e.g., differentiating among high, medium, and low levels of activity). The problem mainly results from noise in the ELISA measurements, many free variables in the network system, and shortcomings of our peptide-encoding scheme (Fig. 3). As the number of peptides used for network training increases, greater prediction accuracy is gained. A more appropriate residue-encoding may also lead to improved results (29). However, because continuous-property scales were used, the danger of generating arbitrary chance correlation was reduced.
Figure 5.

Neural network predictions of peptide activity expressed as absorbance values. (a) Complete training data (r = 0.87). (b) An example of test data (r = 0.79). Linear regression lines (solid) are shown with 95% confidence bands (dashed lines).
Computer-Based Evolutionary Search for New Peptides.
The neural network was used as a heuristic for searching in sequence space. An evolutionary algorithm was applied with the network as the fitness function. The method is described in detail elsewhere (6, 7). Briefly, in a cyclic process virtual peptide libraries are generated by variation of a “parent sequence,” the activity of each peptide is predicted by the neural network, and the peptide with the highest predicted activity is selected as the parent for the next cycle. This is repeated until no further optimization can be observed.
In total, a series of six peptides with a range of predicted activities was evaluated in a bioassay (Table 1). Two de novo-designed peptides with predicted high binding potentials were obtained by the algorithm, DRFGDKDIAF (peptide 1) and GWFGGADWHA (peptide 2). These sequences have only one residue (Asp-7) in common with the seed peptide. The aspartic acid in position 7 seems to be an invariant part of the binding motif. Indeed, 73% of the potentially strong-binding peptides possess this residue, and 18% contain an asparagine. In addition, a medium-binding sequence, IWGCSGKLIC (peptide 3) (33) sharing Cys-4 and Cys-10 with the seed peptide, and a low-binding peptide, KLDAPTNKWG (peptide 4) lacking any identity to the seed peptide were selected for the bioassay. Peptides 3 and 4 were not obtained by computer-based design, rather they were selected from the pepScan in-house depository by neural network evaluation. Only these 2 peptides, of 45 decapeptides, were predicted to have some activity. As an additional negative control the evolutionary-design strategy was used to generate a completely inactive peptide (peptide 5). Surprisingly, the resulting sequence FVRRTYYPER has two identical residues to the seed peptide (Arg-3 and Pro-8). Nevertheless, it was predicted to be the most inactive by the neural network.
Table 1.
Activity of peptides in a bioassay
| Peptide description | Amino acid sequence | Activity
|
|
|---|---|---|---|
| Predicted | Measured | ||
| Seed peptide | ARRCYNDPKC | High | High |
| Peptide 1 | DRFGDKDIAF | High | High |
| Peptide 2 | GWFGGADWHA | High | Medium |
| Peptide 3 | IWGCSGKLIC | Medium | Medium |
| Peptide 4 | KLDAPTNKWG | Low | Medium |
| Peptide 5 | FVRRTYYPER | Zero | Zero |
Peptides 1, 2, and 5 were designed de novo. Underlined residues are identical to the respective seed peptide residues (natural epitope).
Testing the Computer-Designed Peptides for Their Activity in in Vitro Bioassays.
To test the designed peptides for their in vitro activity the beating rate of rat myocytes was monitored when adding mixtures of the peptides and gamma globulin fractions from patient serum containing human anti-β1-adrenoreceptor. The results are summarized in Fig. 6. As predicted, the natural epitope (seed) peptide prevented the positive chronotropic effect of the autoantibodies, i.e., the beating rate reverted to basal values. A similar effect was observed for all four peptides that were predicted to have at least a marginal activity (peptides 1–4). The anti-designed inactive peptide (peptide 5) led to no effect on the myocyte beating rate, as expected. Two random peptides also showed no activity at a concentration of 100 μg/ml (data not shown). The most striking differences between the peptides were observed at a concentration of 1 μg/ml (Fig. 6, bars C). One designed-active peptide (peptide 1) revealed similar or more significant effects than the natural epitope at all tested concentrations (10 μg/ml, 1 μg/ml, and 0.1 μg/ml). At a peptide concentration of 0.1 μg/ml the natural epitope showed only marginal activity, i.e., 10% of the normal activity of the myocytes was observed, whereas peptide 1 was still able to restore 50% of the normal myocyte beating rate (Fig. 6, bars B). Peptides 2, 3, and 4 revealed medium activity in the bioassay even though peptide 2 was predicted to have high activity and peptide 4 was expected to show only low activity. The predicted medium activity of peptide 3 was substantiated in the bioassay. The fact that two of six predictions were in conflict with the observed activities (peptides 2 and 4) reflects shortcomings of the SAR model which were already revealed by the neural network test.
Figure 6.

In vitro response of the beating rate of rat myocytes to different peptide concentrations plus gamma globulin fractions from patient serum containing human anti-β1-adrenoreceptor antibodies. Four different peptide concentrations were tested: no peptide added (A), 0.1 μg/ml (B), 1 μg/ml (C), and 10 μg/ml (D).
From the results of the bioassay we conclude that our minimalist design approach using neural networks as a guide in sequence space was successful in that we identified an artificial antigen mimicking a natural linear epitope sequence. The designed peptide has an activity comparable to its natural counterpart but has a significantly different residue sequence. We were able to demonstrate the applicability of our approach to the systematic design of both active and inactive peptides, and the most active peptide represents a good starting point for further optimization. This success is even more surprising because instead of using purified autoantibodies in the binding assays we used gamma globulin pools of patient sera. Future applications of the method will include the use of affinity-purified autoantibodies instead of raw gamma-globulin fractions. This should also further improve the SAR model derived from neural network training.
Peptide selection by iterative trial-and-error approaches—including focusing compound libraries and computer-based SAR models—may be regarded as the reversal of natural polyclonal B-cell selection, which leads to the generation of antigen-specific antibodies. With this challenging interpretation in mind we may also start thinking of similar strategies for the future development of immunotherapeutic drugs. A first step may be the design of peptidomimetics using our approach. It is easily possible to include nonnatural residues in the design strategy presented here. This can lead to peptide-like structures that are suitable as leads for drug development.
Acknowledgments
Karin Karczewski, Monika Wegner, Holle Schmidt, and Manuela Dierenfeld are thanked for excellent experimental support. Uwe Hobohm is thanked for inspiring discussions. G.S. was supported by grants from the Fonds der Chemischen Industrie. The project was financially supported by the Freie Universität Berlin (Kommission für Forschung und Wissenschaftlichen Nachwuchs; to P.W. and G.S.).
ABBREVIATIONS
- DCM
dilated cardiomyopathy
- EIA
enzyme immunoassay
- PCA
principal component analysis
- SAR
structure–activity relationship
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1. Mewes H-W, Doelz R, George D C. J Biotechnol. 1994;35:239–256. doi: 10.1016/0168-1656(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 2.Cortese R, editor. Combinatorial Libraries. Berlin: de Gruyter; 1996. [Google Scholar]
- 3.Katz B A. Annu Rev Biophys Biomol Struct. 1997;26:27–45. doi: 10.1146/annurev.biophys.26.1.27. [DOI] [PubMed] [Google Scholar]
- 4.Schneider G, Grunert H-P, Schuchhardt J, Wolf K U, Müller G, Habermehl K-O, Zeichhardt H, Wrede P. Min Invas Med. 1995;6:106–115. [Google Scholar]
- 5.Schneider G, Schuchhardt J, Wrede P. Biol Cybern. 1995;73:245–254. doi: 10.1007/BF00201426. [DOI] [PubMed] [Google Scholar]
- 6.Schneider G, Wrede P. Biophys J. 1994;66:335–344. doi: 10.1016/s0006-3495(94)80782-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneider G, Schuchhardt J, Wrede P. Biophys J. 1995;68:434–447. doi: 10.1016/S0006-3495(95)80205-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Limas C J, Goldenberg I F, Limas C. Circ Res. 1989;64:97–103. doi: 10.1161/01.res.64.1.97. [DOI] [PubMed] [Google Scholar]
- 9.Magnusson Y, Wallukat G, Waagstein F, Hjalmarson Å, Hoebke J. Circulation. 1994;89:2760–2767. doi: 10.1161/01.cir.89.6.2760. [DOI] [PubMed] [Google Scholar]
- 10.Cetta F, Michels V V. Ann Med. 1995;27:169–173. doi: 10.3109/07853899509031954. [DOI] [PubMed] [Google Scholar]
- 11.Wallukat G, Wollenberger A. Biomed Biochim Acta. 1987;78:634–639. [PubMed] [Google Scholar]
- 12.Wallukat G, Wollenberger A, Morwinski R, Pitschner H-F. J Mol Cell Cardiol. 1995;27:397–406. doi: 10.1016/s0022-2828(08)80036-3. [DOI] [PubMed] [Google Scholar]
- 13.Gheysen H M, Moelen R H, Barteling S J. Proc Natl Acad Sci USA. 1984;81:3998–4002. doi: 10.1073/pnas.81.13.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gheysen H M, Rodda S J, Mason T J, Tribbik G, Schoofs P G. J Immunol Methods. 1987;102:259–274. doi: 10.1016/0022-1759(87)90085-8. [DOI] [PubMed] [Google Scholar]
- 15.Schneider T, Hildebrand W, Rönspeck W, Weigelt W, Pauli G. AIDS Res Hum Retroviruses. 1990;6:943–949. doi: 10.1089/aid.1990.6.943. [DOI] [PubMed] [Google Scholar]
- 16.Vinga-Martins C, Schneider T, Werno A, Rönspeck W, Pauli G, Müller-Lansch N. AIDS Res Hum Retroviruses. 1992;8:1301–1309. doi: 10.1089/aid.1992.8.1301. [DOI] [PubMed] [Google Scholar]
- 17.Merrifield R B. J Am Chem Soc. 1963;8:2149–2154. [Google Scholar]
- 18.Atherton E, Sheppard R C. Solid Phase Peptide Synthesis, a Practical Approach. Oxford: IRL; 1989. [Google Scholar]
- 19.Henklein P, Beyermann M, Bienert M, Knorr R. In: Peptide 1990, Proceedings of the 21st European Peptide Symposium. Giralt E, Andreu D, editors. Leiden: ESCOM; 1990. pp. 67–68. [Google Scholar]
- 20.Wollenberger A, Wallukat G. Biomed Biochim Acta. 1983;42:917–929. [PubMed] [Google Scholar]
- 21.Halle W, Wollenberger A. Am J Cardiol. 1970;25:292–299. doi: 10.1016/s0002-9149(70)80006-6. [DOI] [PubMed] [Google Scholar]
- 22.Kernighan B W, Ritchie D M. The C Programming Language. Englewood Cliffs, NJ: Prentice–Hall; 1988. [Google Scholar]
- 23.Engelman D A, Steitz T A, Goldman A. Annu Rev Biophys Biophys Chem. 1986;15:321–353. doi: 10.1146/annurev.bb.15.060186.001541. [DOI] [PubMed] [Google Scholar]
- 24.Harpaz Y, Gerstein M, Chothia C. Structure. 1994;2:641–649. doi: 10.1016/s0969-2126(00)00065-4. [DOI] [PubMed] [Google Scholar]
- 25.Eigen M, McCaskill J S, Schuster P. J Phys Chem. 1988;92:6881–6891. [Google Scholar]
- 26.Jollife I T. Principal Component Analysis. New York: Springer; 1986. [Google Scholar]
- 27.Sammon J W., Jr IEEE Trans Comput. 1969;C-18:401–409. [Google Scholar]
- 28.Bienfait B, Gasteiger J. J Mol Graph Model. 1997;15:203–215. doi: 10.1016/s0263-7855(97)00078-7. [DOI] [PubMed] [Google Scholar]
- 29.Schneider G, Wrede P. Prog Biophys Mol Biol. 1998;70:175–222. doi: 10.1016/s0079-6107(98)00026-1. [DOI] [PubMed] [Google Scholar]
- 30.Kramer A, Keitel T, Winkler K, Stöcklein W, Höhne W, Schneider-Mergener J. Cell. 1997;91:799–809. doi: 10.1016/s0092-8674(00)80468-7. [DOI] [PubMed] [Google Scholar]
- 31.Devillers J, editor. Neural Networks in QSAR and Drug Design. San Diego: Academic; 1996. [Google Scholar]
- 32.Schneider G, Wrede P. J Mol Evol. 1993;36:586–595. doi: 10.1007/BF00556363. [DOI] [PubMed] [Google Scholar]
- 33.Gnann J W, Jr, McCormick J B, Mitchell S, Nelson J A, Oldstone M B. Science. 1987;237:1346–1349. doi: 10.1126/science.2888192. [DOI] [PubMed] [Google Scholar]