

Abstract
Like most extracellular bacterial proteases, Streptomyces griseus protease B (SGPB) and α-lytic protease (αLP) are synthesized with covalently attached pro regions necessary for their folding. In this article, we characterize the folding free energy landscape of SGPB and compare it to the folding landscapes of αLP and trypsin, a mammalian homolog that folds independently of its zymogen peptide. In contrast to the thermodynamically stable native state of trypsin, SGPB and αLP fold to native states that are thermodynamically marginally stable or unstable, respectively. Instead, their apparent stability arises kinetically, from unfolding free energy barriers that are both large and highly cooperative. The unique unfolding transitions of SGPB and αLP extend their functional lifetimes under highly degradatory conditions beyond that seen for trypsin; however, the penalty for evolving kinetic stability is remarkably large in that each factor of 2.4–8 in protease resistance is accompanied by a cost of ~105 in the spontaneous folding rate and ~5–9 kcal/mole in thermodynamic stability. These penalties have been overcome by the coevolution of increasingly effective pro regions to facilitate folding. Despite these costs, kinetic stability appears to be a potent mechanism for developing native-state properties that maximize protease longevity.
Keywords: Streptomyces griseus protease B, protein folding, pro region, kinetic stability, protein evolution
Extracellular bacterial proteases, as well as vacuolar and lysosomal proteases, are generally synthesized with covalently attached pro regions required for their folding. Studies on α-lytic protease (αLP) revealed that its pro region acts as a folding catalyst, accelerating the conversion of an on-pathway molten-globule folding intermediate to the native state from a t1/2 of 1800 years to seconds (Sohl et al. 1998; Fig. 1 ▶). This catalysis of αLP folding requires two discrete functions of its pro region: stabilization of the folding transition state, which accelerates folding by a factor of 3 × 109 (Sohl et al. 1998), and tight binding to the native state (Ki = 0.32 nM) (Peters et al. 1998), which drives the folding reaction toward the folded protease. Once folding is complete, the pro region is proteolytically degraded, releasing mature, native protease (Cunningham and Agard 2003). Interestingly, the αLP native state (NatαLP) is thermodynamically less stable than either the folding intermediate or the fully unfolded molecule. Only a large and highly cooperative unfolding barrier (t1/2 = 1.2 years) (Sohl et al. 1998) prevents the active protease from spontaneously unfolding. Recent experiments suggest that such kinetic stability provides a potent mechanism by which proteins attain remarkable degrees of resistance to proteolysis, ensuring maximal lifetimes under harsh degradatory conditions (Jaswal et al. 2002).
Figure 1.

The folding free energy landscape for αLP at 0°C (modified from Peters et al. 1998; Sohl et al. 1998; Derman and Agard 2000; S. Jaswal, unpubl.). Solid lines and dashed lines depict the free energy barriers encountered by αLP in the absence and presence of its pro region (P), respectively. The αLP native state (N) is thermodynamically less stable than either the intermediate state (I) or the fully unfolded molecule (U). Free energies of activation were calculated from folding and unfolding rates using the transition state theory (Glasstone et al. 1941). The stability of the αLP intermediate state was measured at 4°C, and pro region binding to the native state was measured at 25°C.
Although αLP folding is well characterized (Baker et al. 1992; Sohl et al. 1998), little is known about the details of the folding mechanisms of related pro-proteases. Furthermore, while there is a great deal of conservation in the protease domains of αLP family members, the pro regions are much less conserved in both size (76–166 residues) and sequence (Cunningham et al. 1999; Serkina et al. 2001). For αLP, both the N- and C-terminal domains of its pro region (ProαLP; Sauter et al. 1998) are critically important in stabilizing the folding transition state (Cunningham et al. 2002). However, for the close homolog Streptomyces griseus protease B (SGPB), sequence alignments based on the ProαLP structure indicate that the pro region N-terminal domain is completely absent (Fig. 2 ▶), yet the remaining single-domain pro region (ProSGPB) is sufficient for proper folding of its protease (Baardsnes et al. 1998). Of central interest to understanding the evolution of kinetic stability is to discover how SGPB can fold with only a single-domain pro region. Specifically, do large free energy barriers, which result in slow folding and unfolding rates, similarly define the SGPB folding landscape? If so, how does the smaller ProSGPB facilitate rapid folding?
Figure 2.

Sequence alignment of the αLP and SGPB pro regions. Homology models of ProSGPB show that it is compatible with the α-helical (h) and β-strand (s) ProαLP structure (Sauter et al. 1998) indicated above its sequence. Pro region sequence identity (*) and similarity (•) are indicated below the sequences.
This paper explores the folding mechanism of SGPB and compares it to the previously characterized αLP folding landscape, revealing the commonality of the pro-mediated folding pathway and elements that may be critical in the evolution of kinetic stability. Additionally, comparison of the SGPB and αLP folding landscapes with that of trypsin, a thermodynamically stable mammalian homolog that does not require its zymogen peptide for folding (Wang et al. 1997), has further elucidated the advantages and penalties associated with the evolution of kinetic stability.
Results
Expression and purification of recombinant SGPB
Unlike previous studies with recombinant SGPB that used Bacillus subtilis strains for expression, we developed a high-level expression system using Escherichia coli to make genetic manipulations and functional studies more efficient. The construct was based on that used in the high-level αLP expression system, and it uses a T7 PA1 early gene promotor combined with two lac operators to ensure tight, regulated expression (Deuschle et al. 1986), and a phoA signal sequence for efficient secretion into the periplasmic space (Chang et al. 1986). As with αLP expression (Mace et al. 1995), the strain of E. coli used for expression of SGPB was critical, with strain D1210 (Sadler et al. 1980) giving the best expression. Unlike αLP (Mace et al. 1995), SGPB was not expressed when grown in LB growth medium. The best conditions were found in a modified YT growth medium, where tryptone was replaced with NZ amine, but with twice the normal concentration (YZZ; see Materials and Methods). Buffering the media at pH 6.3 with 60 mM ACES also increased SGPB expression levels. Optimal expression was found when the E. coli grew to an OD600 of 1–1.2 at 30°C and then were induced at 18°C. As judged by enzyme activity (Sidhu et al. 1994), the result was ≥5 mg/L SGPB expressed in the culture media, and a final yield of ~2 mg/L of pure protein following the described purification (see Materials and Methods).
ProSGPB-catalyzed refolding of SGPB
A hallmark of αLP folding is the ability of the 166-residue ProαLP, supplied as a separate polypeptide chain, to catalyze the refolding of denatured αLP. While ProαLP is fully structured in solution, it is only marginally stable (ΔGu = 2 kcal/mole) (Anderson et al. 1999; Cunningham et al. 2002). One concern is the stability of the 76-residue ProSGPB, because a ProαLP mutant with a deleted N-terminal domain was unstable (Cunningham et al. 2002). Furthermore, many of the pro regions of comparable size to ProSGPB in the subtilisin family of serine proteases are also intrinsically unstable (Bryan et al. 1995; Bhattacharjya et al. 2000). Initial characterization of the ProSGPB by circular dichroism (CD) revealed that it is unstructured under conditions where ProαLP shows folded spectra. Titration of the subtilisin pro region with the stabilizing agent ethylene glycol proved to be a successful technique for the study of unstable pro regions (Bryan et al. 1995). Similarly, ProSGPB becomes structured in the presence of 50% ethylene glycol (see Supplemental Material), displaying a combination of α-helical and β-sheet signatures, as expected from homology with ProαLP (Fig. 2 ▶). Therefore, the stability of ProSGPB was determined by titrating it with ethylene glycol, revealing that the unfolded state of ProSGPB is favored by 2.4 ± 0.2 kcal/mole (Fig. 3A ▶). Despite this instability, ProSGPB folds upon binding to the SGPB native state (NatSGPB; Fig. 3B ▶), where it adopts essentially the identical secondary structure as observed in the presence of 50% ethylene glycol (see Supplemental Material). The affinity of the interaction between ProSGPB and NatSGPB was quantified via the ability of ProSGPB to inhibit NatSGPB substrate cleavage activity. This analysis showed that ProSGPB acts as a classic competitive inhibitor of NatSGPB, binding with an affinity of 19 ± 2 nM (Fig. 3C ▶).
Figure 3.

Characterization of ProSGPB by circular dichroism (CD) and inhibition of SGPB. (A) The molar ellipticity at 220 nm of ProSGPB as a function of ethylene glycol concentration was fit as described (see Materials and Methods) to determine the thermodynamic stability of ProSGPB (ΔGf = 2.4 ± 0.3 kcal/mole). (B) The CD spectrum, plotted as molar ellipticity versus wavelength, of SGPB (open circles) displays primarily β-sheet secondary structure, whereas the spectrum of ProSGPB (open triangles) appears to be almost completely random coil. The spectrum of the noninteracting proteins (filled squares) is significantly different from the spectrum of the mixed proteins (filled circles), which displays increased α-helical signal, consistent with the formation of the predicted helical structure of ProSGPB, upon binding to SGPB (B, inset). Every fifth data point is shown to simplify the spectra. (C) SGPB proteolytic activity was measured in the presence of increasing amounts of ProSGPB, and the data were fit to the competitive inhibitor equation to determine the affinity of the interaction between ProSGPB and the SGPB native state (Ki = 19 ± 2 nM).
Because ProSGPB was capable of interacting with folded SGPB, we explored its ability to facilitate SGPB folding in vitro. The production of NatSGPB, monitored using activity assays, was measured upon mixing purified recombinant ProSGPB with chemically denatured SGPB. Dilution of this unfolded SGPB from denaturant results in a molten-globule folding intermediate (IntSGPB; data not shown), similar to that observed for αLP (Baker et al. 1992; Jaswal 2000). Furthermore, as with αLP (Peters et al. 1998), the ProSGPB-catalyzed folding reaction showed Michaelis-Menten-like kinetics (Fig. 4A ▶), providing a measure of ProSGPB affinity for IntSGPB (KM = 530 ± 66 μM) and its ability to stabilize the rate-limiting folding transition state (kcat = 0.0010 ± 0.0001 sec−1).
Figure 4.

Kinetic analysis of SGPB folding and unfolding. (A) The concentration dependence of the ProSGPB-catalyzed refolding of SGPB shows Michaelis-Menten type behavior (kcat = 0.0010 ± 0.0001 sec−1, KM = 530 ± 66 μM). (B) The rate of spontaneous SGPB folding (kf = 2.7 × 10−6 ± 0.1 × 10−6 sec−1) was calculated from a linear fit of the fraction of folded SGPB as a function of refolding time. (C) The rate of SGPB unfolding in the absence of denaturant (GndHCl) was calculated from a linear fit of the denaturant dependence of the unfolding rates (ku = 7.4 × 10−7 ± 0.4 × 10−7 sec−1).
Uncatalyzed refolding of SGPB
To fully evaluate the catalytic efficiency of the ProSGPB-mediated folding reaction it is necessary to determine the rate of spontaneous refolding of IntSGPB to NatSGPB. The uncatalyzed refolding rate is so slow that it was critical to use a highly sensitive thiobenzyl ester substrate to quantify the small amounts of refolded active protease (see Materials and Methods). This analysis shows that the half-life for spontaneous SGPB folding is 3 days (Fig. 4B ▶; Table 1). Although this is a very slow rate for a protein folding reaction, it is nonetheless 2 × 105 times faster than that found for αLP (Table 1).
Table 1.
Kinetic and equilibrium constants
| Trypsin | SGPB | αLP | |
| kf (sec−1) | 1 ± 4a | 2.7 × 10−6 ± 0.1 × 10−6 | 1.3 × 10−11 ± 0.06 × 10−11b |
| ku (sec−1) | 8.4 × 10−9 ± 0.4 × 10−9 | 7.4 × 10−7 ± 0.4 × 10−7 | 2.1 × 10−8 ± 0.2 × 10−8c |
| kinact (sec−1) | 4.2 × 10−7 ± 1.5 × 10−7 | 1.0 × 10−6 ± 0.2 × 10−6 | 7.8 × 10−6 ± 0.2 × 10−6 |
| Ku | 1 × 10−8 ± 4 × 10−8 | 0.27 ± 0.02 | 1600 ± 200 |
| ΔGu (kcal/mole) | 10 ± 2 | 0.71 ± 0.04 | −4.00 ± 0.07 |
| pro region size (amino acids) | 0 | 76 | 166 |
a Calculated from measured Ku and ku values.
bkf for αLP taken from Derman and Agard 2000.
cku for αLP taken from S. Jaswal, unpublished data.
SGPB unfolding and stability
To determine whether the differences in the spontaneous folding rates of SGPB and αLP extend to the unfolding reaction, the rate of SGPB unfolding was measured using intrinsic tryptophan fluorescence to probe the loss of tertiary structure. This rate is sufficiently slow that it was measured in the presence of various concentrations of denaturant and then determined from a linear extrapolation of the logarithm of the observed unfolding rate constants as a function of denaturant concentration. The half-life for SGPB unfolding in the absence of denaturant is 11 days (Fig. 4C ▶), which is ~35 times shorter than that for αLP unfolding (Table 1).
Due to the slow SGPB folding and unfolding rates, it is impractical to directly measure the thermodynamic equilibrium between the folded and unfolded states. Instead, we can derive the overall equilibrium constant from the spontaneous folding and unfolding rates (Sohl et al. 1998), with the assumption that the small change in pH between the experiments (pH 5.0–5.6, see Materials and Methods) has a negligible effect on the stability of SGPB. This analysis indicates that SGPB is marginally stable with a ΔG favoring the native state, over the intermediate state, by only ~1 kcal/mole. Urea titration measurements (data not shown) indicate that the stability of IntSGPB is ~0.5 kcal/mole, which is comparable to the ~1 kcal/mole value obtained for the αLP intermediate state (Sohl et al. 1998). Thus, NatSGPB is ~5 kcal/mole more stable than NatαLP.
Survival assay
The functional consequences of the faster SGPB unfolding behavior were tested, under harsh proteolytic conditions, in a survival assay. In this assay, equimolar amounts of trypsin, SGPB, and αLP were mixed and incubated at pH 7, 37°C, and allowed to digest one another. The substrate specificities of the different proteases are sufficiently orthogonal to one another that the concentration of each active protease within the mixture can be readily determined. A previous study comparing αLP, chymotrypsin, and trypsin indicated that activity measurements precisely paralleled the levels of uncleaved protease (Jaswal et al. 2002). The normalized protease activity for each protease throughout the time course (Fig. 5 ▶) was fit using either a single exponential decay model (trypsin) or, if the rate was sufficiently slow, to a linear decay model (αLP, SGPB). This analysis determined the inactivation rates for αLP, SGPB, and trypsin (Table 1). As with αLP (Jaswal et al. 2002; S. Jaswal, unpubl.), SGPB is inactivated at the same rate as it globally unfolds at this temperature (data not shown).
Figure 5.

Protease resistance of αLP, SGPB, and trypsin. Coincubation of αLP, SGPB, and trypsin under strongly proteolytic conditions at 37°C reveals that SGPB (filled circles) is inactivated 2.4 times faster than αLP (open squares) (kinact, SGPB = 1.0 × 10−6 ± 0.2 × 10−6 sec−1, kinact,αLP = 4.2 × 10−7 ± 1.5 × 10−7 sec−1). SGPB (data not shown) and αLP (S. Jaswal, unpubl.) are inactivated at approximately the same rate as their global unfolding at this temperature. Both SGPB and αLP significantly outlast trypsin (open triangles) (kinact, trypsin = 7.8 × 10−6 ± 0.2 × 10−6 sec−1).
Trypsin stability and unfolding
To better understand the implications of the accelerated destruction of trypsin, we sought to characterize its thermodynamic stability and its unfolding rate. To avoid complications from autolysis at higher temperatures, this analysis was carried out only at 0°C. Monitoring tryptophan fluorescence over a range of denaturant concentrations, we used equilibrium unfolding measurements to determine the stability of the trypsin native state (10 ± 2 kcal/mole, Fig. 6A ▶). Because trypsin is inactivated faster than both SGPB and αLP, we would expect that it also unfolds more rapidly than the other two proteases. The calculated rate for trypsin unfolding, extrapolated from denaturant dependent kinetic unfolding data, is 8.4 × 10−9 ± 0.4 × 10−9 sec−1 (t1/2 = 2.6 years, Fig. 6B ▶). Surprisingly, this rate is approximately the same as that for αLP unfolding (ku = 2.1 × 10−8 sec−1, Table 1); however, this need not be the case at higher temperatures.
Figure 6.
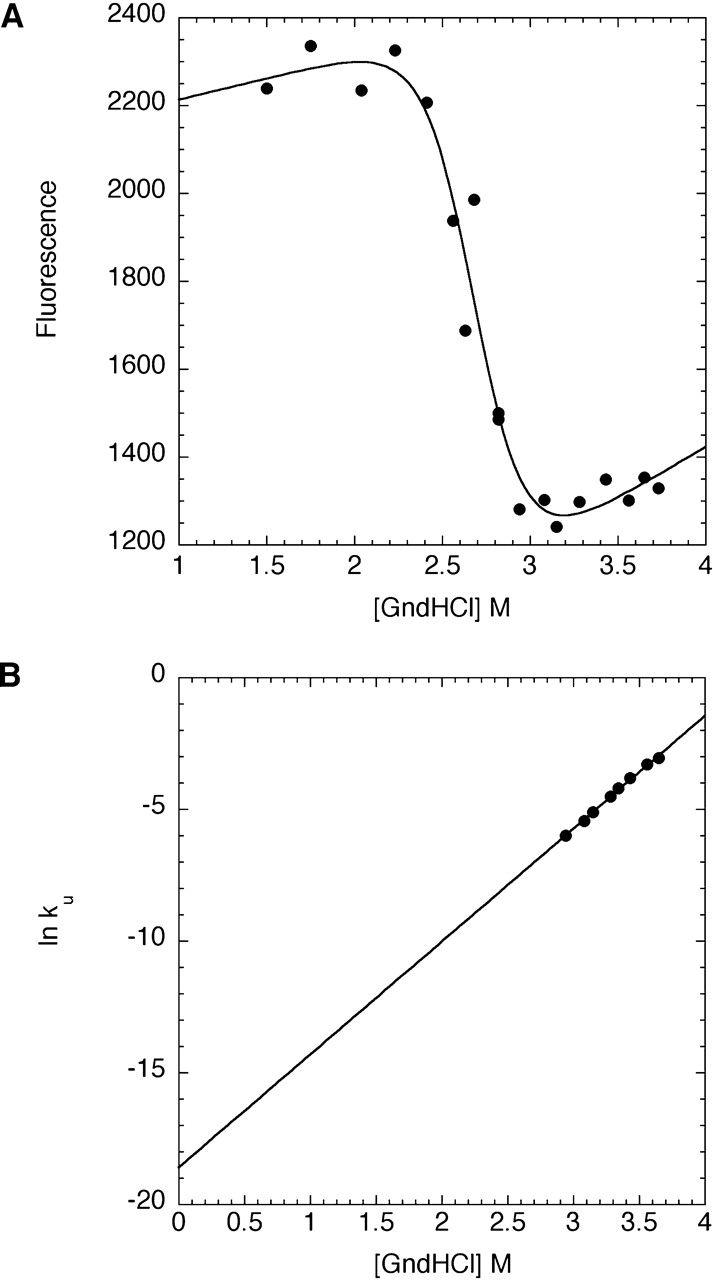
Equilibrium and kinetic analysis of trypsin unfolding. (A) The equilibrium fluorescence of trypsin as a function of denaturant was fit to the linear free energy model to determine the thermodynamic stability of trypsin (ΔGu = 10 ± 2 kcal/mole). (B) Each symbol denotes the logarithm of the rate constant for trypsin unfolding at a given denaturant (GndHCl) concentration. Linear extrapolation of the data yields the unfolding rate in the absence of GndHCl (ku = 8.4 × 10−9 ± 0.4 × 10−9 sec−1).
Discussion
Elucidation of a generalized pro-mediated folding mechanism is critical for reaching a mechanistic understanding of the evolution of kinetic stability. Our detailed analysis of SGPB folding demonstrates the commonality of pro region-catalyzed folding pathways. Furthermore, it also reveals significant energetic differences between the folding landscapes of related proteases, providing important insights into the development of functional longevity via kinetic stability.
Similar to αLP folding, SGPB folding is catalyzed by its pro region; however, characterization of the ProSGPB-catalyzed folding of SGPB indicates that ProSGPB (kcat/KM = 1.9 M−1sec−1) is a less efficient catalyst compared to ProαLP (kcat/KM = 1300 M−1sec−1) (Sohl et al. 1998). Furthermore, because the rate of spontaneous folding of SGPB is 2 × 105 times faster than that for αLP (Table 1), ProSGPB is a far weaker catalyst than is apparent from the ratio of kcat/KM. Where ProαLP accelerates folding by a factor of 2 × 109, the much smaller ProSGPB only accelerates folding of its protease by a factor of ~400. Thus, there seems to be a strong connection between pro region size and efficacy. In part, this may be due to the lower stability of the smaller pro region, as increasingly stable subtilisin pro region mutants were found to be more potent folding catalysts (Bryan 2002). Additionally, a comparison of the SGPB and αLP folding landscapes (Figs. 1 ▶, 7 ▶) reveals a striking difference in the mechanisms of folding catalysis. Although ProαLP preferentially stabilizes the αLP folding transition state, ProSGPB binds more tightly to NatSGPB, thus primarily serving as a native-state template. Given the sequence homology between SGPB and αLP, the energetic difference in their folding free energy barriers (7 kcal/mole) is remarkable (Figs. 1 ▶, 7 ▶). In contrast, the unfolding free energy barriers only differ by 2 kcal/mole (Figs. 1 ▶, 7 ▶). Thus, the minor differences in sequence and structure between these two closely related homologs disproportionately affect folding energetics, resulting in a marginally stable SGPB native state (~1 kcal/mole), whereas NatαLP is thermodynamically unstable.
Figure 7.
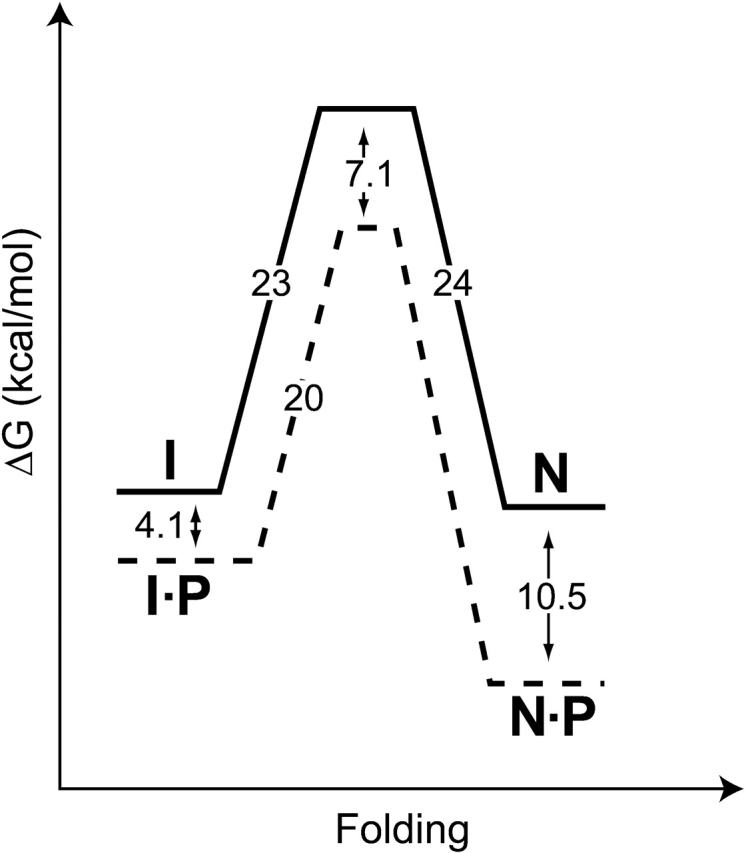
SGPB folding free energy landscape at 0°C. Solid lines depict the free energies of activation for SGPB folding and unfolding (from Fig. 4B,C ▶). Dashed lines illustrate the effect of ProSGPB (P) on the free energy profile (from Figs. 3C ▶, 4A ▶). Unlike αLP, the SGPB native state (N) is marginally stable compared to its intermediate state (I). Free energies of activation were calculated from folding and unfolding rates using transition state theory (Glasstone et al. 1941). Pro region binding to the native state was measured at 25°C.
Although the SGPB folding landscape allows for folding of the protease with a less effective folding catalyst than ProαLP, it results in a native state that has less kinetic stability than NatαLP. The kinetic stability of NatαLP maximizes its functional lifetime by limiting its unfolding to proteolytically sensitive states (Jaswal et al. 2002); thus, we predicted that the decreased free energy barrier to SGPB unfolding will result in a shorter lived protease. This was tested using a protease survival assay (Fig. 5 ▶), which revealed that SGPB is indeed inactivated 2.4 times faster than αLP. We expected from the prior comparison of αLP, chymotrypsin, and trypsin survival (Jaswal et al. 2002) that both SGPB and αLP would outlive trypsin. This is indeed the case, with SGPB and αLP outlasting trypsin by a factor of 8 and 20, respectively. Furthermore, SGPB and αLP are both inactivated at the same rate as their global unfolding at this temperature, suggesting a direct relationship between protease resistance and the height of the unfolding free energy barrier.
Because trypsin is inactivated faster than both SGPB and αLP, we expected the trypsin unfolding free energy barrier to be significantly smaller than that for either SGPB or αLP. Surprisingly, we found that the rate of trypsin unfolding (Fig. 6B ▶) is approximately the same as that for αLP. Thus, optimal protease resistance requires more than just a high free energy barrier to unfolding. Hydrogen-deuterium exchange experiments (Englander et al. 1996; Chamberlain and Marqusee 1997) have demonstrated that proteins are highly dynamic, spontaneously unfolding and refolding subdomains. To avoid proteolytic destruction, it is also necessary to suppress the formation of these partially unfolded species. In this regard, trypsin appears to be less optimized than either SGPB or αLP. This partial unfolding may be important for biological regulation of trypsin activity because it provides a mechanism for its inactivation. In the case of extracellular bacterial proteases, such as αLP and SGPB, once secreted, precise regulation of their proteolytic activity is unnecessary.
These findings indicate that the extended functional lifetimes of αLP and SGPB result primarily from the highly cooperative nature of their unfolding transitions, something that the unfolding of trypsin lacks. As illustrated in Figure 8 ▶, each step of improvement in protease resistance, corresponding to a factor of 2.4–8 gained in protease resistance, is associated with a factor of ~105 decrease in the rate of spontaneous folding and a ~5–9 kcal/mole decrease in thermodynamic stability of the native state. Circumventing these challenges has necessitated the coevolution of increasingly effective pro regions that are capable not only of accelerating the folding process by factors approaching 1010, but also of shifting the folding equilibrium in favor of the folded protease through tight-binding to the native state.
Figure 8.

Comparative analysis of protease resistance and the folding energetics of trypsin, SGPB, and αLP. By arranging the three proteases in order of their proteolytic sensitivity we can compare the consequences of evolving increased protease resistance (black bars, normalized protease resistance). A dramatically increased barrier to folding (white bars, kcal/mole) and a loss of thermodynamic stability (striped bars, kcal/mole) seem directly correlated with an increase in proteolytic resistance. Circumventing these difficulties requires increasingly complex pro regions. Trypsin folds without a pro region, whereas SGPB requires a single-domain pro region and αLP requires a two-domain pro region to facilitate folding.
The cooperativity of the unfolding transition appears to be a critical determinant of protease longevity. Previous studies of αLP revealed that its native state is extremely rigid, with hydrogen-deuterium exchange protection factors that are among the highest observed, and are distributed throughout the protein (Huyghues-Despointes et al. 1999; Jaswal et al. 2002), rather than isolated to a small core, as is the case for most proteins (Li and Woodward 1999). This suggests a potential mechanism for the evolution of increased protease resistance in which kinetically stable proteases achieve essentially perfect cooperativity in their unfolding transitions by restricting the conformational dynamics of their native states. Our results show that the evolution of such native-state rigidity is accompanied by a thermodynamic cost, such that NatαLP is actually thermodynamically unstable. Despite this trade-off, kinetic stability appears to be the preferred evolutionary strategy for developing native-state properties that maximize protease longevity.
Materials and methods
SGPB and ProSGPB constructs
An EagI site was engineered into the SGPB containing PEB-B8 vector (Baardsnes et al. 1998) between the signal sequence and ProSGPB sequence by Chameleon mutagenesis (Stratagene). The gene encoding ProSGPB and SGPB as a single polypeptide was subcloned into the pPA1phoAlp12 vector (Mace et al. 1995) via EagI and XbaI restriction sites. The EagI site was removed using QuikChange mutagenesis (Stratagene) to correct the ProSGPB N-terminal residues altered by the introduction of the EagI site.
The protease gene and a small C-terminal portion of ProSGPB were removed from the PEB-B8 vector (with the engineered EagI site) by digestion with SmaI and BamHI. The C-terminal end of ProSGPB was rebuilt by ligating a synthetic oligonucleotide cassette between the SmaI and BamHI sites and the complete ProSGPB was subcloned into the pT7XmaPro vector (Sohl et al. 1997). The EagI site was, again, removed by QuikChange mutagenesis and an N-terminal His tag (6× His) was added by standard PCR techniques to aid in ProSGPB purification.
SGPB and ProSGPB production and purification
E. coli strain D1210 (Sadler et al. 1980) containing the described SGPB plasmid was grown in 5 mL of growth medium, YZZ (16 g NZ amine, 5 g yeast extract, 2.5 g sodium chloride per L) supplemented with 20 μg/mL streptomycin and 100 μg/mL carbenicillin, at 37°C shaking at 250 rpm for 6–8 h. This culture was used to inoculate 50 mL of growth medium that was grown overnight, shaking at 30°C, 250 rpm. One liter of growth medium supplemented with 60 mM ACES buffer (pH 6.3) in a 4-L beveled flask was inoculated with the overnight culture and grown at 30°C, shaking at 150 rpm, until the absorbance at 600 nm was between 1–1.2. The flask was equilibrated to a temperature of 18°C over 2 h, and then it was induced with 100 μM IPTG. The culture was harvested after approximately 24 h. Cells were removed from the culture by centrifugation at 8000g for 10 min.
The supernatant from 2 L of culture was diluted twofold with deionized water and the pH was adjusted to 5.0 with glacial acetic acid. Approximately 50 mL of SP-sepharose fast-flow resin was used to batch bind SGPB. This resin was transferred to a column, washed with 10 mM sodium acetate (pH 4.8), and SGPB was eluted with 200 mM sodium chloride. Fractions containing SGPB, determined by activity assay (Sidhu et al. 1994), were pooled, and exchanged into 10 mM sodium acetate (pH 4.8). Partially purified SGPB was further purified on a Mono-S H/R HPLC column (Amersham Biosciences Corp., Piscataway, NJ) in 10 mM sodium acetate (pH 4.8) with a 0–200 mM sodium chloride gradient over 15 column volumes. Protease purity was checked by MALDI and SDS-PAGE, and the HPLC step was repeated if necessary.
The His-tagged ProSGPB was expressed in inclusion bodies in E. coli strain BL21(DE3)/pLysS as described for ProαLP (Derman and Agard 2000) and purified using a denatured Ni-NTA purification (Cunningham et al. 2002). ProSGPB was dialyzed into 10 mM sodium phosphate (pH 7.2).
Circular dichroism (CD) of ProSGPB and SGPB
CD spectra of 1.5 μM ProSGPB and SGPB, alone, nonmixed, and mixed, were recorded, at 5°C, on a Jasco (Easton, MD) J-715 spectropolarimeter with a 1-cm path length mixing cuvette.
Titration of ProSGPB with ethylene glycol was performed at 5°C on a Jasco J-715 spectropolarimeter using 4 μM ProSGPB in 10 mM sodium phosphate (pH 7.2). The CD signal at 220 nm was followed as a function of ethylene glycol concentration, after a 5-min equilibration, with 60 averaged accumulations of 1 sec each. The resulting titration was fit to a two-state folding model that assumes a linear dependence of ΔGf on ethylene glycol concentration (Bryan et al. 1995; Henkels et al. 2001).
ProSGPB inhibition of SGPB
SGPB (0.5 nM) was preincubated with increasing amounts of ProSGPB (0–500 nM) for 5 min at room temperature in 20 mM potassium succinate (pH 5.6) and then assayed for activity as described (Sidhu et al. 1994). The data were fit to the competitive inhibitor variation of the Michaelis-Menten equation.
ProSGPB-catalyzed refolding
Catalyzed refolding experiments were performed as described (Derman and Agard 2000), except the ProSGPB to SGPB concentration ratio was ≥10 : 1 and activity assays were performed as described (Sidhu et al. 1994). Refolding progress curves were generated for a range of ProSGPB concentrations (20–225 μM). The rate constants (kobs) from these monophasic progress curves were determined by a single exponential fit, and the variation of these rate constants as a function of ProSGPB concentration was fit with a modified Michaelis-Menten equation (Peters et al. 1998; Derman and Agard 2000) to determine the kinetic constants kcat and KM. In an alternative analysis, the refolding progress curves were simultaneously fit to the equation:
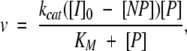 |
resulting in the same kcat and KM values, within error (Berkeley Madonna v. 8.0.1, Berkeley Madonna, Inc., www.berkeleymadonna.com).
Uncatalyzed refolding
Uncatalyzed refolding of SGPB (0.5–1.5 μM) was carried out as described (Derman and Agard 2000), except the assay solution contained 1.6 μM succinyl-Ala-Ala-Pro-Phe-thiobenzyl ester (Bachem Bioscience Inc.) substrate, 250 μM aldrithiol-4, 5% DMSO, and 50 mM Tris (pH 8.0).
SGPB unfolding
Unfolding reactions contained SGPB (1.75 μM) in 10 mM potassium acetate (pH 5.0) at various guanidine hydrochloride (GndHCl) concentrations (determined by refractometry; Pace 1986). The unfolding at 0°C was measured in a Fluoromax-3 (J.Y. Horiba), connected to an external water bath, with excitation at 290 nm and emission at 350 nm. SGPB unfolding data were fit to the equation y = a − bexp−kobst. The unfolding rate in the absence of GndHCl was extrapolated from the logarithm of the observed rate constants plotted as a function of denaturant.
Survival assay
A mixture of 6.5 μM αLP, SGPB, and bovine trypsin (TLCK (tosyl-L-lysine chloromethyl ketone)-treated; Worthington Biochemical Corp.) was incubated at 37°C in 50 mM HEPES, 10 mM calcium chloride (pH 7.0). Measurements of protease activity were carried out as described (Jaswal et al. 2002), except SGPB was monitored using 1 mM succinyl-Ala-Ala-Pro-Phe-pNA (Bachem Bioscience Inc.) substrate. A standard curve was used to calculate the concentration of SGPB at each time point, and trypsin and αLP activities were adjusted for the small background substrate cleavage by SGPB. The protease activity was then normalized relative to the initial protease activity. The inactivation rates for αLP and SGPB were determined from a linear fit, whereas the inactivation rate for trypsin was determined from a single exponential fit.
Trypsin unfolding
Unfolding reactions contained 2 μM bovine trypsin (TLCK-(tosyl-L-lysine chloromethyl ketone) treated, Worthington Biochemical Corp.) in 10 mM potassium acetate, 10 mM calcium chloride, and various concentrations of GndHCl (determined by refractometry; Pace 1986). Autolysis was minimized by performing all unfolding reactions at pH 5.0 and at 0°C. Equilibrium and kinetic unfolding measurements were made in a Fluoromax-3 (J.Y. Horiba), connected to an external water bath, with excitation at 280 nm and emission at 322 nm. The equilibrium fluorescence of trypsin as a function of denaturant were fit to the linear free energy model (Pace 1986). Equilibrium fluorescence values were determined from the approach to equilibrium as measured in kinetic unfolding experiments. Actual values were obtained from linear fits of the unfolded baseline. Kinetic unfolding data were fit to the equation y = a + bexp−kobst−ct, and the rate of trypsin unfolding in the absence of denaturant was extrapolated from the logarithm of the observed rate constants as a function of denaturant.
Data analysis
Unless otherwise specified, all data analysis was performed using Kaleidagraph v. 3.6 (Synergy Software Technologies Inc.).
Electronic supplemental material
Spectra of ProSGPB in 50% ethylene glycol and ProSGPB from the ProSGPB•NatSGPB complex are shown in the file SuppMat. doc.
Acknowledgments
The PEB-B8 plasmid was generously provided by the Borgford laboratory. We thank Y. Shibata and L. Rice for expression and purification advice; N. Sauter, M. Madhusudhan, and A. Sali for assistance with the analysis of pro region homology; and B. Kelch, A. Derman, A. Shiau, and S. Jaswal for many helpful comments. S.M.E.T. was supported by a National Science Foundation predoctoral fellowship. Research funding was provided by the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
SGPB, Streptomyces griseus protease B
αLP, α-lytic protease
ProαLP, the α-lytic protease pro region
ProSGPB, the S. griseus protease B pro region
NatαLP, the native state of α-lytic protease
NatSGPB, the native state of S. griseus protease B
IntSGPB, the intermediate state of S. griseus protease B
CD, circular dichroism
GndHCl, guanidine hydrochloride
TLCK, tosyl-L-lysine chloromethyl ketone
Supplemental material: See www.proteinscience.org
Article published online ahead of print.
Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03336804.
References
- Anderson, D.E., Peters, R.J., Wilk, B., and Agard, D. 1999. α-Lytic protease precursor: Characterization of a structured folding intermediate. Biochemistry 38 4728–4735. [DOI] [PubMed] [Google Scholar]
- Baardsnes, J., Sidhu, S., MacLeod, A., Elliott, J., Morden, D., Watson, J., and Borgford, T. 1998. Streptomyces griseus protease B: Secretion correlates with the length of the propeptide. J. Bacteriol. 180 3241–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, D., Sohl, J.L., and Agard, D.A. 1992. A protein-folding reaction under kinetic control. Nature 356 263–265. [DOI] [PubMed] [Google Scholar]
- Bhattacharjya, S., Xu, P., and Ni, F. 2000. Sequence-specific 1H, 15N and 13C resonance assignments of the inhibitory prodomain of human furin. J. Biomol. NMR 16 275–276. [DOI] [PubMed] [Google Scholar]
- Bryan, P., Wang, L., Hoskins, J., Ruvinov, S., Strausberg, S., Alexander, P., Almog, O., Gilliland, G., and Gallagher, T. 1995. Catalysis of a protein folding reaction: Mechanistic implications of the 2.0 Å structure of the subtilisin–prodomain complex. Biochemistry 34 10310–10318. [DOI] [PubMed] [Google Scholar]
- Bryan, P.N. 2002. Prodomains and protein folding catalysis. Chem. Rev. 102 4805–4816. [DOI] [PubMed] [Google Scholar]
- Chamberlain, A.K. and Marqusee, S. 1997. Touring the landscapes: Partially folded proteins examined by hydrogen exchange. Structure 5 859–863. [DOI] [PubMed] [Google Scholar]
- Chang, C.N., Kuang, W.J., and Chen, E.Y. 1986. Nucleotide sequence of the alkaline phosphatase gene of Escherichia coli. Gene 44 121–125. [DOI] [PubMed] [Google Scholar]
- Cunningham, E.L. and Agard, D.A. 2003. Disabling the folding catalyst is the last critical step in α-lytic protease folding. Protein Sci. (this issue) [DOI] [PMC free article] [PubMed]
- Cunningham, E.L., Jaswal, S.S., Sohl, J.L., and Agard, D.A. 1999. Kinetic stability as a mechanism for protease longevity. Proc. Natl. Acad. Sci. 96 11008–11014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, E.L., Mau, T., Truhlar, S.M.E., and Agard, D.A. 2002. The pro region N-terminal domain provides specific interactions required for catalysis of α-lytic protease. Biochemistry 41 8860–8867. [DOI] [PubMed] [Google Scholar]
- Derman, A.I. and Agard, D.A. 2000. Two energetically disparate folding pathways of α-lytic protease share a single transition state. Nat. Struct. Biol. 7 394–397. [DOI] [PubMed] [Google Scholar]
- Deuschle, U., Kammerer, W., Gentz, R., and Bujard, H. 1986. Promoters of Escherichia coli: A hierarchy of in vivo strength indicates alternate structures. EMBO J. 5 2987–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander, S.W., Sosnick, T.R., Englander, J.J., and Mayne, L. 1996. Mechanisms and uses of hydrogen exchange. Curr. Opin. Struct. Biol. 6 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasstone, S., Laidler, K., and Eyring, H. 1941. The theory of rate processes: The kinetics of chemical reactions, viscosity, diffusion, and electrochemical phenomena, 1st ed. McGraw-Hill Book Company, Inc., New York.
- Henkels, C.H., Kurz, J.C., Fierke, C.A., and Oas, T.G. 2001. Linked folding and anion binding of the Bacillus subtilis ribonuclease P protein. Biochemistry 40 2777–2789. [DOI] [PubMed] [Google Scholar]
- Huyghues-Despointes, B.M., Scholtz, J.M., and Pace, C.N. 1999. Protein conformational stabilities can be determined from hydrogen exchange rates. Nat. Struct. Biol. 6 910–912. [DOI] [PubMed] [Google Scholar]
- Jaswal, S.S. 2000. “Thermodynamics, kinetics, and landscapes in α-lytic protease: A role for pro regions in kinetic stability.” Ph.D. thesis, University of California, San Francisco.
- Jaswal, S.S., Sohl, J.L., Davis, J.H., and Agard, D.A. 2002. Energetic landscape of α-lytic protease optimizes longevity through kinetic stability. Nature 415 343–346. [DOI] [PubMed] [Google Scholar]
- Li, R. and Woodward, C. 1999. The hydrogen exchange core and protein folding. Protein Sci. 8 1571–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace, J.E., Wilk, B.J., and Agard, D.A. 1995. Functional linkage between the active site of α-lytic protease and distant regions of structure: Scanning alanine mutagenesis of a surface loop affects activity and substrate specificity. J. Mol. Biol. 251 116–134. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Peters, R.J., Shiau, A.K., Sohl, J.L., Anderson, D.E., Tang, G., Silen, J.L., and Agard, D.A. 1998. Pro region C-terminus: Protease active site interactions are critical in catalyzing the folding of α-lytic protease. Biochemistry 37 12058–12067. [DOI] [PubMed] [Google Scholar]
- Sadler, J.R., Tecklenburg, M., and Betz, J.L. 1980. Plasmids containing many tandem copies of a synthetic lactose operator. Gene 8 279–300. [DOI] [PubMed] [Google Scholar]
- Sauter, N.K., Mau, T., Rader, S.D., and Agard, D.A. 1998. Structure of α-lytic protease complexed with its pro region. Nat. Struct. Biol. 5 945–950. [DOI] [PubMed] [Google Scholar]
- Serkina, A.V., Shevelev, A.B., and Chestukhina, G.G. 2001. Structure and functions of bacterial proteinase precursors. Bioorg. Khim. 27 323–346. [DOI] [PubMed] [Google Scholar]
- Sidhu, S.S., Kalmar, G.B., Willis, L.G., and Borgford, T.J. 1994. Streptomyces griseus protease C: A novel enzyme of the chymotrypsin superfamily. J. Biol. Chem. 269 20167–20171. [PubMed] [Google Scholar]
- Sohl, J.L., Shiau, A.K., Rader, S.D., Wilk, B.J., and Agard, D.A. 1997. Inhibition of α-lytic protease by pro region C-terminal steric occlusion of the active site. Biochemistry 36 3894–3902. [DOI] [PubMed] [Google Scholar]
- Sohl, J.L., Jaswal, S.S., and Agard, D.A. 1998. Unfolded conformations of α-lytic protease are more stable than its native state. Nature 395 817–819. [DOI] [PubMed] [Google Scholar]
- Wang, E.C., Hung, S.H., Cahoon, M., and Hedstrom, L. 1997. The role of the Cys191–Cys220 disulfide bond in trypsin: New targets for engineering substrate specificity. Protein Eng. 10 405–411. [DOI] [PubMed] [Google Scholar]