

Abstract
We tested the hypothesis that adenosine and nitric oxide can be sensed by capillaries and are implicated in the remote arteriolar dilatation initiated by muscle contraction. We also explored a role for KATP channel activity in this response. Small bundles of muscle fibres underlying a group of capillaries in cremaster muscles of anaesthetized hamsters were electrically stimulated to contract for 2 min at each of 2, 4 and 8 Hz. Diameter changes were measured in the inflow arteriole to the group of capillaries after muscle contraction in the presence or absence of 10−6m xanthine amine congener (XAC) to block A1 and A2 adenosine receptors, 10−4 or 10−3mNω-nitro-l-arginine (LNNA) to block nitric oxide production, or 10−5m glibenclamide to block KATP channel activity. Dilatations were unchanged with XAC (3.0 ± 0.5, 3.9 ± 0.7 and 6.1 ± 1.0 μm), but were significantly reduced with LNNA (to 1.8 ± 0.6, 3.5 ± 0.7 and 4.9 ± 0.7 μm) or glibenclamide (to 0.4 ± 0.3, 0.8 ± 0.7 and 1.9 ± 0.6 μm). Neither KATP channel activity nor nitric oxide was required for transmission or manifestation of the dilator response. Thus, muscle contraction can be sensed by capillaries and the signalling mechanism for the ensuing remote dilatation depends on KATP channel activity and on NO, but not adenosine. Local application of 10−4m adenosine, 10−4m sodium nitroprusside or 10−5m pinacidil directly to capillaries initiated remote arteriolar dilatations. Thus, capillaries can respond directly to known mediators of metabolic vasodilatation, but these signalling pathways are not invariably implicated in the response to muscle contraction.
Capillary recruitment is a significant early response to increased metabolism in skeletal muscle (Honig & Frierson, 1980; Honig et al. 1982; Klitzman et al. 1982; Hargreaves et al. 1990). The control of capillary recruitment and capillary blood flow redistribution resides in the terminal arteriolar region of the microcirculation (Klitzman et al. 1982; Lindbom & Arfors, 1984; Sarelius, 1986); in arterioles in this region of the microvasculature, dilatations have been observed in direct association with contracting muscle fibres (Gorczynski et al. 1978; Berg et al. 1997; Cohen et al. 2000) as well as in response to changes in vasoactive agents or conditions associated with muscle contraction (for example, increased adenosine (Proctor & Duling, 1982), release of NO (Hester et al. 1993; Lau et al. 1998, 2000) or decrease in PO2 (Duling & Berne, 1970)). However, in order for flow to be increased in a network of capillaries via a metabolically linked dilatation of the upstream arteriole, there must be an appropriate matching between the metabolic needs of the region of tissue perfused by those capillaries, and the flow in the capillaries themselves. When muscle activity is increased, individual motor units are recruited to contract, with the numbers and types of motor units that are recruited being dependent on the output demanded of the whole muscle (for a review, see Burke, 1981). The individual muscle fibres comprising a single motor unit have common metabolic profiles, but are dispersed throughout the muscle (Burke, 1981), hence as motor units are recruited, different local metabolic signals will be generated at any given tissue site. Thus it is unlikely that the metabolic activity of fibres underlying a particular capillary network will be the same as that of the muscle fibres traversed by the controlling arteriole, suggesting that if metabolic needs are sensed and responded to solely at the level of the controlling arteriole, precise matching of capillary blood flow to metabolic needs will not necessarily be achieved. Effectively, this organization of control would require that the perfusion needs of a particular capillary bed would need to be predetermined upstream, where in fact the local metabolic signals may well be different.
Recently, we have demonstrated that muscle contraction under capillaries can generate a vasodilatatory signal that results in remote dilatation of the upstream arterioles (Berg et al. 1997; Cohen et al. 2000). This suggests a mechanism whereby the metabolic needs of a local region of tissue can be matched precisely to the capillary blood flow of that region. Implicit in this conclusion is that the capillaries themselves must be able to sense and respond to metabolic signals generated by contracting muscle fibres. Indeed, it has been established that remote vasomotor responses can be produced in arterioles when neurotransmitters are applied to capillaries (Dietrich, 1989; Dietrich & Tyml, 1992; McGahren et al. 1998; Sarelius et al. 2000), indicating that capillaries have the ability to sense these agents and initiate signals that result in the appropriate response remotely in arterioles. For example, application of noradrenaline to capillaries can produce remote arteriolar vasoconstriction (Dietrich, 1989; Dietrich & Tyml, 1992), whereas acetylcholine application to capillaries results in upstream arteriolar dilatation (Sarelius et al. 2000). We therefore hypothesized that the remote arteriolar vasodilatation initiated by muscle contraction under capillaries would be mediated by vasodilator products of muscle contraction that are known to dilate arterioles directly. Both adenosine (Proctor & Duling, 1982; Proctor, 1984) and NO (Hester et al. 1993; Lau et al. 1998, 2000) are known to contribute to arteriolar dilatation during contraction of cremaster muscle (which is our model system), hence we asked whether these agents could, in addition, be sensed at the capillary level to initiate the remote vasodilatatory signal generated by skeletal muscle contraction. Because both adenosine receptor activation (Jackson, 1993; Kuo & Chancellor, 1995; Danialou et al. 1997; Bryan & Marshall, 1999) and NO dilator capacity (Murphy & Brayden, 1995; Tare et al. 2000) have been linked to ATP-sensitive K+ (KATP) channel function, and because KATP channels have been identified as important in arteriolar dilatations during muscle contraction (Saito et al. 1996; Murrant & Sarelius, 2000b; Hammer et al. 2001), we also sought to determine whether activity of these channels could be linked to the vasodilatatory signal initiated in capillaries by the contraction of muscle fibres.
METHODS
Preparation and muscle fibre stimulation
All procedures were approved by the Animal Care and Use Committee of the University of Rochester and were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, USA).
Male adult Golden Hamsters (HSD Han : Aura, 100–150 g) were anaesthetized with pentobarbital sodium (70 mg kg−1, i.p.). Supplemental anaesthesia was maintained throughout the experiments via a left femoral venous catheter; depth of anaesthesia was assessed by monitoring reflex withdrawal to a toe pinch. Body temperature was maintained at 37 ± 0.5 °C by convective heating. In most animals, blood pressure was monitored (left femoral arterial catheter). The right cremaster muscle was prepared and viewed as described elsewhere (Baez, 1973; Sweeney & Sarelius, 1989; Cohen et al. 2000). The prepared muscle was continuously superfused with warmed (34 °C) bicarbonate-buffered physiological salt solution containing (in mm): NaCl, 131.9; KCl, 4.7; CaCl2, 2.0; MgSO4, 1.2; NaHCO3, 30.0; equilibrated with 5 % CO2-95 % N2 to maintain pH at 7.4 ± 0.05 and PO2 < 10 mmHg. After surgery, the cremaster preparation was allowed to stabilize for 30–60 min. The microcirculation was visualized with a Leitz Laborlux microscope equipped with a 25 × long working distance objective (Leitz, NA 0.35), and displayed via a video camera (RCA SIT E1103 or Dage MTI 72S) on a Hitachi monitor. Final magnification from tissue to monitor was × 1400–2800. Before the start of data collection, viability of the preparation was assessed by confirming in three randomly selected arterioles that there was a brisk vasodilatation to 10−4m local topical adenosine and constriction to transient superfusate equilibration with 10 % oxygen. The small number of preparations that did not display these responses were discarded. At the completion of all experimental protocols, animals were administered a lethal i.v. dose of sodium pentobarbital.
Key aspects of the terminal arteriolar and capillary architecture in this tissue have been described elsewhere (Sweeney & Sarelius, 1989; Berg & Sarelius, 1995) and are schematized in Fig. 1. Briefly, capillaries in this muscle occur in groups (modules) arising from an arteriole (the module inflow arteriole), which is the terminal vessel of the arteriolar tree. Usually, 3–8 capillary modules arise from a common arteriolar branch (Berg & Sarelius, 1995), which in turn branches from a larger vessel that runs approximately transversely across the muscle fibres. Capillary modules in a clear central region of the prepared tissue were identified by their vascular geometry as well as their inability to dilate to locally applied adenosine. Selection criteria included clarity and suitability of the site for electrode or micropipette placement but never included functional criteria such as the number of capillaries in the module or the blood flow rate: many suitable sites are identifiable in each cremaster preparation. Microelectrodes were used to contract small bundles of muscle fibres (usually 4–5 fibres) underlying the selected capillary module, as described previously (Berg et al. 1997; Cohen et al. 2000). Microelectrodes consisted of a 25 μm diameter platinum/iridium wire enclosed in a glass micropipette and connected to the silver wire in a microelectrode holder (WPI, Inc., Sarasota, FL, USA). The pipette tip was insulated with silicone (Dow Corning) so that only ≈2–5 μm of the wire was exposed. Electrodes were placed on the selected muscle fibre bundle ≥ 1000 μm from the selected capillary module; the ground electrode was a silver wire placed around the rim of the pedestal supporting the tissue. Muscle contraction for 2 min was induced by square wave pulses of 0.4 ms duration and 5–30 V at each of 2, 4 and 8 Hz. A recovery period of 3 min was allowed before proceeding to 2 min contraction at the next highest frequency.
Figure 1. Schematic (not to scale) to show the principal features of the experimental site in cremaster muscle of anaesthetized hamsters.
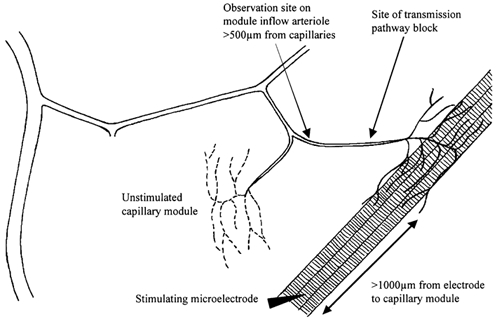
General experimental protocols
In all contraction experiments, muscle fibre bundles that ran underneath the selected capillary module were stimulated to contract for 2 min at each frequency, and the vasodilator response was observed at the proximal end of the inflow arteriole to that module. Contracting muscle fibres were always ≥ 500 μm away from the observation site, thus ruling out diffusion of vasoactive substances from muscle fibres as a primary determinant of the arteriolar response. In previous work, we have also ruled out the contribution of neural or flow-dependent mechanisms to the arteriolar response (Berg et al. 1997; Cohen et al. 2000).
To characterize the initiating mechanism(s) for the remote dilatation of the module inflow arteriole, we first completed the muscle contraction protocol under control conditions before repeating the sequence of 2 min muscle contractions in the presence of selected test substances. The test substances (see below) were added either to the superfusate solution bathing the cremaster muscle or, in some experiments, were applied directly to a local region of the microvasculature using a micropipette as previously described (Frame & Sarelius, 1995; Cohen et al. 2000). Briefly, micropipettes with tip diameter ≈10 μm were filled with the test solution and placed close to (∼15–25 μm), and directed towards, the selected arteriolar region or the selected capillaries. Flow out of the micropipette was achieved by raising pressure in the water manometer to which it was connected. A tracer of fluorescein isothiocyanate dextran (40 kDa, 10−4m) was added to each pipette solution so that brief epifluorescence could be used to confirm that there was flow out of the micropipette, and to verify the flow direction of the micropipette contents. It has been confirmed that this tracer concentration is not vasoactive (Frame & Sarelius, 1995). Care was taken to ensure that convective flow of pipette contents was directed only over the selected region of interest, and away from other components of the microvasculature that were being studied. Furthermore, local application sites were chosen so that they were always ≥500 μm away from the site at which responses were being monitored. Thus, responses of the module inflow arteriole could not be attributed to either convection or diffusion of the micropipette contents between the application and observation sites.
Materials
Xanthine amine congener (XAC, 10−6m) was added to the superfusate to block A1 and A2 adenosine receptors (Fredholm et al. 1994). We used the nitric oxide synthase inhibitor Nω-nitro-l-arginine (LNNA, 10−4 or 10−3m) and the NO donor sodium nitroprusside (SNP, 10−4m) to test for involvement of NO, and we explored KATP channel activity by using the KATP channel antagonist glibenclamide (10−5m) and channel opener pinacidil (10−5m). For both routes of application (in the superfusate or locally using a micropipette), XAC and LNNA were applied for 10 min and glibenclamide for 40 min before re-testing the arteriolar response. These exposure times were determined in preliminary experiments in which we identified the concentration of antagonist required to block at least 80 % of the vasodilatation to each agonist. In experiments using micropipettes to produce direct local application of adenosine, SNP or pinacidil to capillaries, we used 2 min exposures to mimic the 2 min muscle contraction periods.
Adenosine, SNP and LNNA were made as 10−2m stock solutions in distilled water and stored at 4 °C. On experimental days, they were diluted to 10−3 or 10−4m with control superfusate. XAC was frozen (-70 °C) as 10−4m aliquots in 0.1 n NaOH and diluted with control superfusate to 10−6m. Glibenclamide and pinacidil were made as stock solutions with DMSO and diluted to 10−5m with control superfusate. We (Cohen et al. 2000) and others (Jackson, 1993) have shown that this concentration of DMSO in the working superfusate (0.04 %) has no effect on vessel reactivity. All chemicals were purchased from Sigma Chemical Co.
Data collection and statistical analyses
Arteriolar responses were videotaped (SonyVO9600 or Panasonic AG6300) and analysed off-line, using a videotaped stage micrometer for reference. We measured the average (resting) diameter in the 15–30 s before the start of each contraction frequency. For each contraction frequency, diameters were measured in the 10 s immediately following cessation of the 2 min muscle contraction, because tissue movement during muscle contraction often precluded measurements during the contraction period. Maximal arteriolar diameter was measured at the end of the experiment after 5 min exposure to 10−4m adenosine added to the superfusate.
Results were expressed either as diameter change from baseline (Dtest - Dbaseline), or, where resting arteriolar diameter was significantly changed by the test condition (e.g. in some experiments with glibenclamide in the superfusate), we also expressed the diameter change as a percentage of the dilator capacity, where this latter was calculated as (Dmax - Dbaseline). There were no differences in the conclusions drawn from the statistical analyses when data were expressed in either form. All data are reported as mean ± standard error with n representing the number of arterioles in each data set. In most experiments, one arteriole was observed per animal: occasionally in experiments using local drug application, two arterioles were observed per animal. Group means were compared using Student's paired t test (Snedecor & Cochran, 1967), with differences judged significant at P ≤ 0.05.
RESULTS
The mean maximal diameter of arterioles used in this study was 18.3 ±3.1 μm, with a range of 9.2–26.6 μm.
Role of A1 and A2 adenosine receptors in remote dilatations
To test whether adenosine contributed to the signal sensed by the capillaries during muscle contraction, 10−6m xanthine amine congener (XAC) was added to the superfusate to block A1 and A2 adenosine receptors. This addition of 10−6m XAC to the superfusate did not affect the remote dilatation of the module inflow arteriole in response to muscle contraction (Fig. 2). Because addition of XAC did not inhibit the dilator response, we verified the efficacy of the XAC by measuring the arteriolar diameter change in response to 10−4m adenosine locally applied to a capillary module for 2 min in the absence or presence of XAC. XAC significantly decreased adenosine-induced remote dilatations from 4.5 ± 0.9 μm to 1.1 ± 0.4 μm (n = 9), indicating not only that XAC in the superfusate was able to produce significant antagonism of adenosine receptors, but also that capillaries have the capacity to respond to adenosine if it is present. Thus A1 and A2 receptor activation appears not to be involved in initiation of the remote dilatations induced in the module inflow arteriole by muscle contraction, and a role for adenosine in the contraction-dependent stimulation of capillaries to initiate remote vasodilator signals was not explored further.
Figure 2. Diameter changes of module inflow arterioles (n = 7) in the absence (control, open bars; recovery, hatched bars) or presence (filled bars) of the adenosine A1 and A2 receptor antagonist xanthine amine congener (XAC, 10−6m), in response to 2 min of remote muscle contraction under capillaries at 2, 4 and 8 Hz.
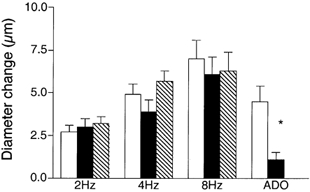
Also shown are responses of module inflow arterioles when adenosine (ADO, 10−4m) was applied topically to capillary modules (n = 9), in the absence (open bar) or presence (filled bar) of XAC. Bars are means ± s.e.m. All changes during muscle contraction are significantly different from rest, but XAC is not different from controls at any stimulation frequency. * Significantly different from the control for that condition.
Contibution of NO to the remote dilatation
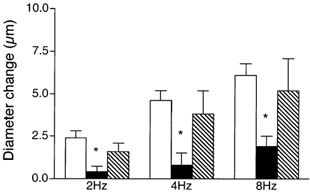
To explore a role for NO in contributing to the remote dilatation, the nitric oxide synthase inhibitor Nω-nitro-l-arginine (LNNA, 10−4 or 10−3m) was added to the superfusate and the arteriolar response to muscle contraction was observed. Baseline diameters were not significantly changed in the presence of LNNA (12.2 ± 1.0 μm with LNNA versus 10.5 ± 1.0 μm in controls, n = 9). Figure 3A shows that dilatation of the module inflow arteriole during remote muscle contraction was significantly attenuated during addition of LNNA to the superfusate (10−4m, n = 5, 10−3m, n = 4; there was no difference between data sets so the data were pooled). Control dilatations of 2.6 ± 0.6, 5.5 ± 0.8 and 7.1 ± 0.7 μm at 2, 4 and 8 Hz were significantly reduced to 1.8 ± 0.6, 3.5 ± 0.7 and 4.9 ± 0.7 μm with LNNA, a significant overall reduction to 30.9 ± 5.7 % of the dilator capacity.
Figure 3. Diameter changes of module inflow arterioles in the absence (control, open bars; recovery, hatched bars) or presence (filled bars) of the nitric oxide synthase inhibitor Nω-nitro-l-arginine (LNNA, 10−4 or 10−3m), in response to 2 min of remote muscle contraction under capillaries at 2, 4 and 8 Hz.
A, addition of LNNA to the superfusate, (n = 9); B, local application of LNNA to the observation site via micropipette (n = 5). Bars are means ± s.e.m. * Significantly different from the control for that condition.
However, a substance added to the superfusate has the potential to affect the arteriolar dilatation via a number of different scenarios, by affecting any or all of (i) the initiation of the dilator signal at the capillaries in the vicinity of the contracting muscle fibres; (ii) the transmission of the dilator signal along the vessel wall; (iii) the ability of the arteriole to dilate at the observation site. Thus if addition of a test substance to the superfusate modifies the remote arteriolar dilatation, it is necessary to undertake additional experiments to determine whether the response could be attributed either to inhibition of transmission of the dilator signal along the vessel wall, or to direct inhibition of the arteriolar dilator machinery at the observation site. In a previous study (Cohen et al. 2000) we showed that local micropipette-application of LNNA to the transmission pathway between the capillaries and the upstream observation site did not affect the remote contraction-induced arteriolar dilatation, indicating that NO is not involved in transmission of this dilator signal along the blood vessel wall. To determine whether LNNA directly affected the ability of the module inflow arteriole to dilate, we completed separate experiments in which LNNA was directly micropipette-applied to the observation site during the muscle contraction protocol. LNNA had no significant effect on this arteriolar response (Fig. 3B), thus we conclude that NO is also not required for manifestation of the arteriolar dilatation. In previous work (Frame & Sarelius, 1995; Cohen, 2000) we have confirmed the efficacy of LNNA delivery from micropipettes. Taken together, these findings indicate that as NO is neither required for manifestation of the arteriolar dilatation itself, nor is it required for transmission of the dilator signal along the vessel wall, its site of action must be at the capillaries, where the dilator signal is initiated.
To confirm that capillaries have the ability to respond directly to NO, we used a micropipette to apply the NO donor sodium nitroprusside (SNP, 10−4m) directly to module capillaries for 2 min (to match the 2 min period of muscle stimulation) and monitored the diameter of the upstream arteriole. There was an increase in diameter of 5.0 ± 1.4 μm (n = 6) in upstream arterioles in response to SNP applied locally across the capillary module.
KATP channel involvement in remote arteriolar dilatation
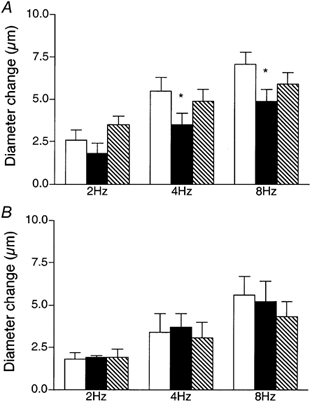
To determine if remote contraction-induced dilatations in the module inflow arteriole were dependent on KATP channel activity, the KATP channel antagonist glibenclamide (10−5m) was added to the superfusate. As shown in Fig. 4, this resulted in significant attenuation of the contraction-induced remote dilatation of the module inflow arteriole. Control dilatations of 2.4 ± 0.4, 4.6 ± 0.6 and 6.1 ± 0.7 μm at 2, 4 and 8 Hz respectively were significantly reduced to 0.4 ± 0.3, 0.8 ± 0.7 and 1.9 ± 0.6 μm in the presence of glibenclamide (n = 7). As outlined above for NO, significant attenuation of the remote dilatation under these conditions could reflect inhibition of KATP channel activity at the capillaries, or in the transmission pathway along the blood vessel wall, or at the site of arteriolar response. Hence, in separate sets of experiments, we used a micropipette to locally apply 10−5m glibenclamide to either the transmission pathway or the observation site, as described earlier for NO. When glibenclamide was applied via a micropipette directly to the transmission pathway (Fig. 5A), the remote dilatation of the module inflow arteriole was unaffected, indicating that the inhibition of dilatation observed when glibenclamide was added to the superfusate was not due to block of the transmission of the dilator signal from its origination at the capillaries to the site of arteriolar response. Similarly, micropipette-application of glibenclamide directly to the observation site in the module inflow arteriole did not block the dilator response (Fig. 5B). We confirmed that glibenclamide was indeed able to reverse a local pinacidil-induced dilatation (Fig. 6), verifying the efficacy of the glibenclamide. To confirm that capillaries can respond to a KATP channel opener by producing a remote arteriolar dilatation, we micropipette-applied the KATP channel activator pinacidil (10−5m) directly to module capillaries for 2 min while module inflow arteriolar diameter was monitored. This application of pinacidil produced a remote dilator response of 3.4 ± 0.8 μm (n = 5).
Figure 4. Diameter changes of module inflow arterioles (n = 7) in the absence (control, open bars; recovery, hatched bars) or presence (filled bars) of the KATP channel antagonist glibenclamide (10−5m), in response to 2 min of remote muscle contraction under capillaries at 2, 4 and 8 Hz.
Bars are means ± s.e.m. * Significantly different from the control for that condition.
Figure 5. Diameter changes of module inflow arterioles in response to 2 min of remote muscle contraction under capillaries at 2, 4 and 8 Hz, in the absence (control, open bars; recovery, hatched bars) or presence (filled bars) of the KATP channel antagonist glibenclamide (10−5m), locally applied via micropipette to the transmission pathway (A, n = 5) or the upstream observation site (B, n = 5).
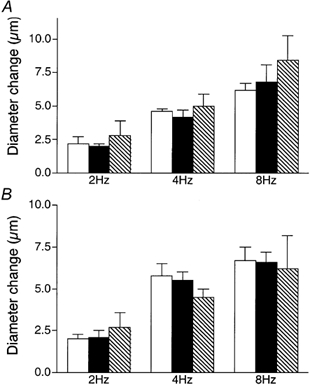
Bars are means ± s.e.m. Responses in the presence of glibenclamide are not different from controls.
Figure 6. Diameter changes of module inflow arterioles induced by local micropipette application of the KATP channel opener pinacidil (10−5m, open bars) are reversed by addition of glibenclamide (10−5m, filled bars) added either to the superfusate (n = 6) or locally via micropipette (n = 4).
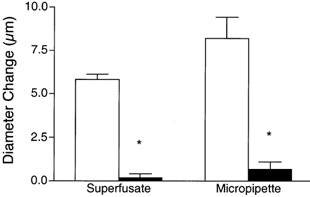
Bars are means ± s.e.m. * Significantly different from the control for that condition.
Is the response to NO linked to KATP channel activity?
There is evidence that NO can stimulate KATP channel activity (Murphy & Brayden, 1995; Tare et al. 2000), hence an obvious question for us to ask is whether the observed contribution of NO to the remote dilatation was via a KATP channel-dependent mechanism. To test this we measured remote arteriolar dilatations with both glibenclamide and LNNA added to the superfusate (Fig. 7). This combination significantly attenuated the response to muscle stimulation from 16.6 ± 2.6, 42.7 ± 3.6 and 60.7 ± 10.8 % of the dilator capacity at 2, 4 and 8 Hz to 5.8 ± 2.7, 16.3 ± 4.2 and 24.3 ± 4.1 % respectively in the presence of both glibenclamide and LNNA (n = 9). This attenuation was not different from that with glibenclamide alone (to 4.0 ± 3.4, 5.4 ± 7.7 and 17.2 ± 4.1 % respectively at 2, 4 and 8 Hz, n = 7), but was a greater attenuation than with LNNA alone (to 19.8 ± 9.6, 31.1 ± 12.1 and 41.7 ± 6.2 %, n = 9), implying that the response to LNNA could be accounted for by an effect on KATP channel activity.
Figure 7. Diameter changes of module inflow arterioles (n = 9) in the absence (control, open bars; recovery, hatched bars) or presence (filled bars) of the KATP channel antagonist glibenclamide (10−5m) together with the nitric oxide synthase inhibitor Nω-nitro-l-arginine (LNNA, 10−4m), in response to 2 min of remote muscle contraction under capillaries at 2, 4 and 8 Hz.
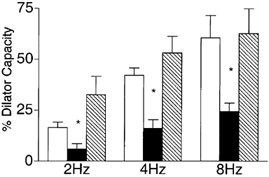
Bars are means ± s.e.m. * Significantly different from the control for that condition.
To further explore the possibility of a mechanistic link between NO and KATP channel activity, we micropipette-applied 10−4m SNP directly to capillaries for 2 min as described earlier, and measured the ensuing remote arteriolar dilatation in the absence or presence of 10−5m glibenclamide in the superfusate. There was no significant difference in the response between the two conditions (69 ± 9 % of the dilator capacity with SNP alone versus 63 ± 9 % for SNP plus glibenclamide), representing diameter changes of 5.0 ± 0.9 and 5.9 ± 0.9 μm respectively (n = 10).
Taken together, these results suggest that in this system KATP channel activity may be affected by NO, but in a complex way that is beyond the scope of the present study to resolve.
DISCUSSION
The principal finding from this study is that skeletal muscle contraction activates vasodilator pathways that can be sensed by capillaries, which then initiate signals that are transmitted upstream to remote arteriolar sites, causing them to dilate. Further, capillaries are differently responsive to different putative metabolic dilators, as illustrated by the finding that while both adenosine and NO can be sensed by capillaries and cause initiation of a remote vasodilator response, only NO, and not adenosine, is implicated in the remote dilatation induced by muscle contraction.
We conclude that KATP channel activity is a major component of the response initiated by muscle contraction because glibenclamide applied to the tissue via the superfusion solution substantially inhibited the remote dilatation, while glibenclamide applied either directly to the transmission pathway for the remote response or directly to the upstream observation site had no effect on the remote dilatation. This indicates that the transmission pathway and dilator machinery for the response were intact when glibenclamide was present in the superfusate, and therefore indicates that inhibition must have occurred at the capillaries, i.e. at the site of initiation of the response. The force developed during contraction of isolated cremaster muscles is the same in the presence or absence of 10−5m glibenclamide (Murrant & Sarelius, 2002), thus we conclude that KATP channel activity is necessary for the initiation of the remote dilatation. Because glibenclamide was added to the superfusate and therefore applied to the entire tissue, we are not able to identify whether the relevant location for this KATP channel activity is on the skeletal muscle fibres or on the capillaries themselves. We confirmed that a remote arteriolar dilatation can be initiated in the region of the capillaries by locally applying the KATP channel opener pinacidi using a micropipette. We were not able to test whether glibenclamide applied locally to the capillaries could inhibit the upstream dilatation produced by muscle contraction because this required close apposition of the micropipette tip and the contracting muscle fibres, usually resulting in broken pipette tips. Thus KATP channels on the capillary endothelial cells (Brayden, 1990), the skeletal muscle fibres (Renaud et al. 1996) or possibly intracellularly on mitochondrial membranes (Garlid et al. 1996) remain as candidates for involvement in this response.
Our study also indicates that NO is involved in initiation of the remote dilatation. We found an overall reduction in the remote arteriolar dilatation in the presence of LNNA, and were able to localize this inhibition to the site of initiation of the dilator response, i.e. the capillaries themselves. Force development during contraction of isolated cremaster muscle is not affected by LNNA (Cohen, 2000; Murrant & Sarelius, 2002), hence this reduced arteriolar dilatation cannot be attributed to reduced metabolic workload. NO has been implicated in vasodilator responses to muscle contraction (Murrant & Sarelius, 2002), and it is known that NO of skeletal muscle origin can play a role in functional hyperaemia (Lau et al. 1998, 2000). In the present study we demonstrated that direct application of the NO donor SNP to the capillary region could initiate a remote dilatation, suggesting that these vessels do indeed have the capacity to respond to NO such as could be released by contracting muscle fibres. We could not demonstrate a significant effect of glibenclamide on the remote dilatation produced by SNP. However, we found that the substantial glibenclamide-dependent attenuation of the muscle contraction-dependent remote response was not different when LNNA was present with the glibenclamide, suggesting that the NO-dependent signalling pathway is not independent of a pathway involving KATP channels, and implying that NO does, in some way, influence KATP channel activity in this system. Clearly, however, it seems unlikely that all of the response attributable to KATP channel activity can be accounted for by a direct action of NO on these channels.
Our data do not exclude the possibility that NO could be a signalling intermediate generated at the capillaries themselves (in response to some other as yet unidentified signal from contracting muscle fibres) and subsequently involved in initiation of the remote dilator signal. We have shown elsewhere that NO is not involved in the transmission of the remote response to the module inflow arteriole (Cohen et al. 2000), hence we conclude that NO released by skeletal muscle and sensed by capillaries, or NO generated by the capillaries, must contribute to initiation of the dilator signal that is transmitted upstream.
Adenosine has been implicated in local arteriolar dilatations produced by muscle contraction (Proctor & Duling, 1982; Proctor, 1984; Murrant & Sarelius, 2002), and is implicated in the production of remote dilatations when those dilatations are initiated at the arteriolar level (Murrant & Sarelius, 2002). The present study shows, in contrast, that although capillaries do indeed have the ability to respond to adenosine by producing a remote dilatation, adenosine is not involved in stimulation of capillaries by muscle contraction to produce a remote dilatation, indicating that different signalling pathways are involved in the contraction-dependent remote response.
It has been established that while transmission of the remote dilator signal initiated at the capillary level can involve gap junctions (Berg et al. 1997; Cohen et al. 2000), transmission of the remote dilator signal initiated in arterioles does not (Murrant & Sarelius, 2000a), again indicating that the remote response pathways initiated by muscle contraction are complex and must involve more than one mechanism. We speculate that a component of the remote response initiated by muscle contraction might involve another transmission route along the blood vessel wall, such as the local paracrine route mediated by cellular calcium waves that has been described for endothelium and other cell types (Charles, 1998; Sauer et al. 2000; Ohata et al. 2001). Although we have shown that local muscle fibre contraction in the region of capillaries or arterioles does not induce measurable whole cell calcium changes in endothelial cells (Murrant et al. 2000; Sarelius et al. 2000), this does not rule out involvement of phenomena such as local calcium oscillations or waves as contributors to the signalling pathway for remote dilator mechanisms. Clearly, studies addressing such possibilities are beyond the scope of the present work.
Capillary recruitment and capillary blood flow redistribution in an exercising skeletal muscle involve integration of many vasodilatatory mechanisms, both centrally and locally mediated, that together contribute to the whole organ response. In the present study, we have taken advantage of the spatial relationships between the microvasculature and skeletal muscle fibres in cremaster muscle to identify a direct role for capillaries in local sensing of, and response to, acute changes in skeletal muscle contractile activity. Clearly, the relative importance of this response in an intact exercising muscle will depend on the balance of many inputs, including this one.
In summary, we have shown that muscle contraction underneath capillaries can be sensed by the capillaries themselves, which initate remote dilatations in upstream arterioles. These dilatations are dependent on KATP channel activity and on NO, although it is not yet clear how the response to NO is related to KATP channel activity. Adenosine, while implicated in other local dilator responses, and which can indeed be sensed by capillaries, is not involved in the capillary-initiated remote dilatation to muscle contraction. Thus, not all established mediators of functional hyperaemia contribute to this flow increase by directly signalling capillaries to initiate remote dilatations. Although aspects of these signalling pathways remain to be described, it is clear that initiation of upstream arteriolar dilatations can control capillary recruitment and contributes to functional hyperaemia by locally increasing blood flow to those capillaries that are directly associated with contracting skeletal muscle fibres.
Acknowledgments
We thank Ms Patricia A. Titus for her technical support, and Dr Coral L. Murrant for helpful discussions. This work was supported by NIH grant HL 56574.
REFERENCES
- Baez S. An open cremaster preparationfor the study of blood vessels by in vivo microscopy. Microvascular Research. 1973;5:384–394. doi: 10.1016/0026-2862(73)90054-x. [DOI] [PubMed] [Google Scholar]
- Berg BR, Cohen KD, Sarelius IH. Direct coupling between blood flow and metabolism at the capillary level in striated muscle. American Journal of Physiology. 1997;272:H2693–2700. doi: 10.1152/ajpheart.1997.272.6.H2693. [DOI] [PubMed] [Google Scholar]
- Berg BR, Sarelius IH. Functional capillary organization in striated muscle. American Journal of Physiology. 1995;268:H1215–1222. doi: 10.1152/ajpheart.1995.268.3.H1215. [DOI] [PubMed] [Google Scholar]
- Brayden JE. Membrane hyperpolarization is a mechanism of endothelium-dependent cerebral vasodilation. American Journal of Physiology. 1990;259:H668–673. doi: 10.1152/ajpheart.1990.259.3.H668. [DOI] [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. Journal of Physiology. 1999;514:163–175. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE. Motor units: anatomy, physiology, and functional organization. In: Brooks VB, editor. Handbook of Physiology, section 1, The Nervous System, Motor Control. II. Bethesda, MD, USA: American Physiological Society; 1981. pp. 345–422. part 1, chap. 10. [Google Scholar]
- Charles A. Intercellular calcium waves in glia. Glia. 1998;24:39–49. doi: 10.1002/(sici)1098-1136(199809)24:1<39::aid-glia5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Cohen KD. Transmission and initiation of remote arteriolar dilations in response to muscle contraction underneath capillaries. Rochester, NY, USA: University of Rochester; 2000. PhD Dissertation. [DOI] [PubMed] [Google Scholar]
- Cohen KD, Berg BR, Sarelius IH. Remote arteriolar dilations in response to muscle contraction under capillaries. American Journal of Physiology - Heart and Circulatory Physiology. 2000;278:H1916–1923. doi: 10.1152/ajpheart.2000.278.6.H1916. [DOI] [PubMed] [Google Scholar]
- Danialou G, Vicaut E, Sambe A, Aubier M, Boczkowski J. Predominant role of A1 adenosine receptors in mediating adenosine induced vasodilatation of rat diaphragmatic arterioles: involvement of nitric oxide and the ATP-dependent K+ channels. British Journal of Pharmacology. 1997;121:1355–1363. doi: 10.1038/sj.bjp.0701247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich HH. Effect of locally applied epinephrine and norepinephrine on blood flow and diameter in capillaries of rat mesentery. Microvascular Research. 1989;38:125–135. doi: 10.1016/0026-2862(89)90021-6. [DOI] [PubMed] [Google Scholar]
- Dietrich HH, Tyml K. Microvascular flow response to localized application of norepinephrine on capillaries in rat and frog skeletal muscle. Microvascular Research. 1992;43:73–86. doi: 10.1016/0026-2862(92)90007-c. [DOI] [PubMed] [Google Scholar]
- Duling BR, Berne RM. Longitudinal gradients in periarteriolar oxygen tension. A possible mechanism for the participation of oxygen in local regulation of blood flow. Circulation Research. 1970;27:669–678. doi: 10.1161/01.res.27.5.669. [DOI] [PubMed] [Google Scholar]
- Frame MD, Sarelius IH. l-Arginine-induced conducted signals alter upstream arteriolar responsivity to l-arginine. Circulation Research. 1995;77:695–701. doi: 10.1161/01.res.77.4.695. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M. Nomenclature and classification of purinoceptors. Pharmacological Reviews. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial KATP channel as a receptor for potassium channel openers. Journal of Biological Chemistry. 1996;271:8796–8799. doi: 10.1074/jbc.271.15.8796. [DOI] [PubMed] [Google Scholar]
- Gorczynski RJ, Klitzman B, Duling BR. Interrelations between contracting striated muscle and precapillary microvessels. American Journal of Physiology. 1978;235:H494–504. doi: 10.1152/ajpheart.1978.235.5.H494. [DOI] [PubMed] [Google Scholar]
- Hammer LW, Ligon AL, Hester RL. Differential inhibition of functional dilation of small arterioles by indomethacin and glibenclamide. Hypertension. 2001;37:599–603. doi: 10.1161/01.hyp.37.2.599. [DOI] [PubMed] [Google Scholar]
- Hargreaves D, Egginton S, Hudlicka O. Changes in capillary perfusion induced by different patterns of activity in rat skeletal muscle. Microvascular Research. 1990;40:14–28. doi: 10.1016/0026-2862(90)90003-a. [DOI] [PubMed] [Google Scholar]
- Hester RL, Eraslan A, Saito Y. Differences in EDNO contribution to arteriolar diameters at rest and during functional dilation in striated muscle. American Journal of Physiology. 1993;265:H146–151. doi: 10.1152/ajpheart.1993.265.1.H146. [DOI] [PubMed] [Google Scholar]
- Honig CR, Frierson JL. Role of adenosine in exercise vasodilation in dog gracilis muscle. American Journal of Physiology. 1980;238:H703–715. doi: 10.1152/ajpheart.1980.238.5.H703. [DOI] [PubMed] [Google Scholar]
- Honig CR, Odoroff CL, Frierson JL. Active and passive capillary control in red muscle at rest and in exercise. American Journal of Physiology. 1982;243:H196–206. doi: 10.1152/ajpheart.1982.243.2.H196. [DOI] [PubMed] [Google Scholar]
- Jackson WF. Arteriolar tone is determined by activity of ATP-sensitive potassium channels. American Journal of Physiology. 1993;265:H1797–1803. doi: 10.1152/ajpheart.1993.265.5.H1797. [DOI] [PubMed] [Google Scholar]
- Klitzman B, Damon DN, Gorczynski RJ, Duling BR. Augmented tissue oxygen supply during striated muscle contraction in the hamster. Relative contributions of capillary recruitment, functional dilation, and reduced tissue PO2. Circulation Research. 1982;51:711–721. doi: 10.1161/01.res.51.6.711. [DOI] [PubMed] [Google Scholar]
- Kuo L, Chancellor JD. Adenosine potentiates flow-induced dilation of coronary arterioles by activating KATP channels in endothelium. American Journal of Physiology. 1995;269:H541–549. doi: 10.1152/ajpheart.1995.269.2.H541. [DOI] [PubMed] [Google Scholar]
- Lau KS, Grange RW, Chang WJ, Kamm KE, Sarelius I, Stull JT. Skeletal muscle contractions stimulate cGMP formation and attenuate vascular smooth muscle myosin phosphorylation via nitric oxide. FEBS Letters. 1998;431:71–74. doi: 10.1016/s0014-5793(98)00728-5. [DOI] [PubMed] [Google Scholar]
- Lau KS, Grange RW, Isotani E, Sarelius IH, Kamm KE, Huang PL, Stull JT. nNOS and eNOS modulate cGMP formation and vascular response in contracting fast-twitch skeletal muscle. Physiological Genomics. 2000;2:21–27. doi: 10.1152/physiolgenomics.2000.2.1.21. [DOI] [PubMed] [Google Scholar]
- Lindbom L, Arfors KE. Interrelationship between arteriolar blood flow distribution and capillary perfusion in skeletal muscle. Progress in Applied Microcirculation. 1984;5:84–92. [Google Scholar]
- McGahren ED, Beach JM, Duling BR. Capillaries demonstrate changes in membrane potential in response to pharmacological stimuli. American Journal of Physiology. 1998;274:H60–65. doi: 10.1152/ajpheart.1998.274.1.H60. [DOI] [PubMed] [Google Scholar]
- Murphy ME, Brayden JE. Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. Journal of Physiology. 1995;486:47–58. doi: 10.1113/jphysiol.1995.sp020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrant CL, Kim MB, Cohen KD, Sarelius IH. Skeletal muscle contraction-induced arteriolar dilations are independent of changes in endothelial cell calcium. Journal of Vascular Research. 2000;37:33. (abstract) [Google Scholar]
- Murrant CL, Sarelius IH. Local and remote arteriolar dilations initiated by skeletal muscle contraction. American Journal of Physiology - Heart and Circulatory Physiology. 2000a;279:H2285–2294. doi: 10.1152/ajpheart.2000.279.5.H2285. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Sarelius IH. Role of adenosine receptors and ATP-sensitive potassium channels in local and remote arteriolar dilation initiated by muscle contraction. FASEB Journal. 2000b;14:A28. doi: 10.1152/ajpheart.2000.279.5.H2285. (abstract) [DOI] [PubMed] [Google Scholar]
- Murrant CL, Sarelius IH. Multiple dilator pathways in skeletal muscle contraction-induced arteriolar dilations. American Journal of Physiology - Regulatory and Integrative Physiology. 2002 doi: 10.1152/ajpregu.00405.2001. (in the Press) [DOI] [PubMed] [Google Scholar]
- Ohata H, Ikeuchi T, Kamada A, Yamamoto M, Momose K. Lysophosphatidic acid positively regulates the fluid flowinducedlocal Ca2+ influx in bovine aortic endothelial cells. Circulation Research. 2001;88:925–932. doi: 10.1161/hh0901.090300. [DOI] [PubMed] [Google Scholar]
- Proctor KG. Reduction of contraction-induced arteriolar vasodilation by adenosine deaminase or theophylline. American Journal of Physiology. 1984;247:H195–205. doi: 10.1152/ajpheart.1984.247.2.H195. [DOI] [PubMed] [Google Scholar]
- Proctor KG, Duling BR. Adenosine and free-flow functional hyperemia in striated muscle. American Journal of Physiology. 1982;242:H688–697. doi: 10.1152/ajpheart.1982.242.4.H688. [DOI] [PubMed] [Google Scholar]
- Renaud JM, Gramolini A, Light P, Comtois A. Modulation of muscle contractility during fatigue and recovery by ATP sensitive potassium channel. Acta Physiologica Scandinavica. 1996;156:203–212. doi: 10.1046/j.1365-201X.1996.210000.x. [DOI] [PubMed] [Google Scholar]
- Saito Y, McKay M, Eraslan A, Hester RL. Functional hyperemia in striated muscle is reduced following blockade of ATP-sensitive potassium channels. American Journal of Physiology. 1996;270:H1649–1654. doi: 10.1152/ajpheart.1996.270.5.H1649. [DOI] [PubMed] [Google Scholar]
- Sarelius IH. Cell flow path influences transit time through striated muscle capillaries. American Journal of Physiology. 1986;250:H899–907. doi: 10.1152/ajpheart.1986.250.6.H899. [DOI] [PubMed] [Google Scholar]
- Sarelius IH, Cohen KD, Murrant CL. Role for capillaries in coupling blood flow with metabolism. Clinical and Experimental Pharmacology and Physiology. 2000;27:826–829. doi: 10.1046/j.1440-1681.2000.03340.x. [DOI] [PubMed] [Google Scholar]
- Sauer H, Hescheler J, Wartenberg M. Mechanical strain-induced Ca2+ waves are propagated via ATP release and purinergic receptor activation. American Journal of Physiology — Cell Physiology. 2000;279:C295–307. doi: 10.1152/ajpcell.2000.279.2.C295. [DOI] [PubMed] [Google Scholar]
- Snedecor GW, Cochran WG. Statistical Methods. 6. Ame, IA, USA: The Iowa State Unversity Press; 1967. [Google Scholar]
- Sweeney TE, Sarelius IH. Arteriolar control of capillary cell flow in striated muscle. Circulation Research. 1989;64:112–120. doi: 10.1161/01.res.64.1.112. [DOI] [PubMed] [Google Scholar]
- Tare M, Parkington HC, Coleman HA. EDHF, NO and a prostanoid: hyperpolarization-dependent and -independent relaxation in guinea-pig arteries. British Journal of Pharmacology. 2000;130:605–618. doi: 10.1038/sj.bjp.0703332. [DOI] [PMC free article] [PubMed] [Google Scholar]