

Abstract
The functioning of midbrain dopaminergic neurones is closely involved in mental processes and movement. In particular the modulation of the inhibitory inputs on these cells might be crucial in controlling firing activity and dopamine (DA) release in the brain. Here, we report a concentration-dependent depressant action of dopamine on the GABAB IPSPs intracellularly recorded from dopaminergic neurones. Such effect was observed in spite of the presence of D1/D2 dopamine receptor antagonists. A reduction of the GABAB IPSPs was also caused by noradrenaline (norepinephrine) and by l-β-3,4-dihydroxyphenylalanine (l-DOPA), which is metabolically transformed into DA. The DA-induced depression of the IPSPs was partially antagonised by the α2 antagonists yohimbine and phentolamine. DA did not change the postsynaptic effects of the GABAB agonist baclofen, suggesting a presynaptic site of action. Furthermore, DA did not modulate the GABAA-mediated IPSP. The DA-induced depression of the GABAB IPSP occluded the depression produced by serotonin and was not antagonized by serotonin antagonists. The DA- and 5-HT-induced depression of the GABAB IPSP persisted when calcium and potassium currents were reduced in to the presynaptic terminals. These results describe an unconventional presynaptic, D1 and D2 independent action of DA on the GABAB IPSP. This might have a principal role in determining therapeutic/side effects of l-DOPA and antipsychotics and could be also involved in drug abuse.
Different physiological actions of DA on midbrain dopaminergic neurones have been reported so far. These actions are mainly inhibitory and attributed to membrane hyperpolarization, activated by postsynaptic D2 autoreceptors (Lacey et al. 1987; Mercuri et al. 1992), and enhancement of the GABAB IPSPs, mediated by D1 presynaptic receptors (Cameron & Williams, 1993). In addition, DA might cause presynaptic D2 or 5-HT receptor-mediated inhibition of excitatory inputs (Koga & Momiyama, 2000; Jones et al. 2000) and postsynaptic noradrenergic (α1) receptor-mediated reduction of glutamate metabotropic IPSPs (Paladini et al. 2001).
Although it is well established that acute stimulation of D1 presynaptic receptors enhances the GABAB IPSP (Cameron & Williams, 1993), after chronic treatment with cocaine and morphine, D1 receptor activation decreases rather than increases the amplitude of this potential (Bonci & Williams, 1996). This D1-mediated negative modulation of the GABAB synaptic inputs, having striatal/accumbal origin (Johnson et al. 1992; Sugita et al. 1992; Cameron & Williams, 1993), is caused by the co-activation of presynaptic adenosine A1 receptors. It has been suggested that it is determinant in regulating drug-related phenomena, such as sensitization and withdrawal (Bonci & Williams, 1996; Shoji et al. 1999; Fiorillo & Williams, 2000). Interestingly, other abused drugs might exert a modulation of the GABAB synaptic inputs on the dopaminergic neurones by mechanisms principally involving 5-HT release (Johnson et al. 1992; Cameron & Williams, 1994). On the contrary, no clear actions of DA, 5-HT and psychostimulants on the GABAA IPSPs have yet been reported. Considering, the rather complex regulation of DA of the inhibitory potentials on the dopaminergic neurones, we re-examined the action of this cathecolamine on GABA release. Here, we describe a selective, non D1/D2-mediated presynaptic inhibition of GABA release on GABAB synapses.
METHODS
Preparation and recordings
Intracellular recordings with sharp microelectrodes were made from midbrain dopaminergic neurones in horizontal slices (250–300 μm thick) prepared from male Wistar rats (150–300 g) (Mercuri et al. 1995). The animals were anaesthetized with halothane and decapitated. The Comitato Etico of Tor Vergata University approved the experimental procedures. The brain was rapidly removed from the skull and horizontal slices of the ventral midbrain were cut using a vibratome. A single slice containing the substantia nigra (SN) and the ventral tegmental area (VTA) was transferred to a recording chamber, immobilized with titanium mesh and perfused at a rate of 2.5 ml min−1, with a solution maintained at 35 °C and equilibrated with a mixture of 95 % O2-5 % CO2. The standard solution contained (mm): NaCl 126, KCl 2.5, NaH2PO4 1.2, MgCl2 1.2, CaCl2 2.4, glucose 10 and NaHCO3 19, (pH 7.4). The dopaminergic neurones of the VTA and substantia nigra pars compacta were identified by their electrical properties (Lacey et al. 1987; 1989; Grace & Onn, 1989; Johnson et al. 1992; Johnson & North, 1992; Mercuri et al. 1995; Liss et al. 1999), which included the presence of a regular spontaneous firing activity, relaxation of hyperpolarizing electrotonic potentials mediated by the activation of Ih and the GABAB IPSP. No differences were observed between neurones of the VTA and SN, the data were pooled. To prevent spontaneous action potentials the membrane potential was adjusted to between −65 and −70 mV by hyperpolarizing current injection. The recording electrodes were filled with 2 m KCl and had a tip resistance of 30–80 MΩ.
Synaptic potentials
GABAB inhibitory synaptic potentials were evoked in dopaminergic cells using bipolar tungsten stimulating electrodes with a tip separation of 300–700 μm (Johnson et al. 1992; Johnson & North, 1992; Sugita et al. 1992; Cameron & Williams, 1993; Wu et al. 1995; Bonci & Williams, 1996; Shoji et al. 1999; Fiorillo & Williams, 2000). A train of four to eight stimuli of 70 μs at 8–20 V was delivered at 70 Hz every 30 s. Stimulating electrodes were placed within 500–700 μm rostral or caudal of the recording electrode. The amplitude of the evoked synaptic potential was measured from recordings that represent the average of four responses. To isolate these potentials pharmacologically, experiments were done in the presence of the dopamine D2 receptor antagonist sulpiride (3–30 μm), the D1 receptor antagonist SCH 23390 (1–10 μm), bicuculline methiodide (30–50 μm, GABAA and mGluR IPSP), strychnine (1 μm, glycine) and prazosin (300 nm, α1 noradrenaline). In some experiments we also perfused either apamin (100 nm, small-conductance calcium-activated potassium (SK) channel antagonist; Ishii et al. 1997; n = 4) or MCPG (500 μm, mGluR antagonist; Conn & Pin, 1997; n = 3).
In addition, the ionotropic glutamate AMPA and NMDA receptors were blocked by using 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μm) and 2-amino-5-phosphonopentanoic acid (AP-5, 50 μm), respectively. The GABAB receptor antagonists CGP 55845 (300 nm) and 2-hydroxy-saclofen (100–300 μm) were perfused to block the GABAB IPSP.
Drugs
All drugs were bath applied at a known concentration. Only baclofen (300 μm) was delivered by pressure ejection (20–30 p.s.i.) from a micropipette positioned a few micrometres above the slice. The following drugs were used: cyanopindolol, pindolol, methysergide, CNQX, clozapine, MAP4, prazosin, yohimbine, sulpiride, SKF 38393, SCH 23390, 1,3-dipropyl-8-cyclopentylxanthine (DPCPX), UK 14304, quinpirole hydrochloride, alpha-methyl-4-carboxyphenylglycine (MCPG), from Tocris Cookson, 4-aminopiridine (4-AP), dopamine hydrochloride, noradrenaline (norepinephrine) (NA), AP-5, l-β-3,4-dihydroxyphenylalanine (l-DOPA), carbidopa, 5-hydroxytryptamine hydrochloride (serotonin, 5-HT), bicuculline methiodide, nifedipine, strychnine, tolbutamide, spiperone hydrochloride, chlorpromazine, atropine, phentolamine from Sigma, apamin and ω-conotoxin MVIIC (ω-CgTx), from Alomone Labs, baclofen from Roche. CGP 55845 was a gift from Novartis.
Data analysis
Numerical data were expressed as means ± standard error of the mean (s.e.m.). Student's t test for paired observations was used to compare data. A P < 0.05 was considered significant. The percentage change produced by a drug was calculated from mean amplitude of four responses before and after equilibrium had been reached. To estimate the IC50 and maximal response, concentration-response curves were fitted with a least squares regression using a logistic equation: y = ax/(x + b). Where y is the magnitude of effect, a is maximum effect, x is drug concentration and b is the concentration that inhibits the effects by 50 %.
RESULTS
Dopamine reduces the GABAB IPSP in a concentration-dependent manner
Intracellular recordings were made from dopaminergic neurones in slices of the rat substantia nigra and ventral tegmental area maintained in vitro (Lacey et al. 1989; Johnson & North, 1992; Mercuri et al. 1995). GABAB IPSPs (10–20 mV) (Johnson et al. 1992; Sugita et al. 1992; Cameron & Williams, 1993; Wu et al. 1995; Bonci & Williams, 1996; Shoji et al. 1999; Fiorillo & Williams, 2000) were evoked by a local short train of stimuli (holding potential, −65 to −70 mV), in the presence of a cocktail of ionotropic and metabotropic glutamate-, GABAA-, glycine-, noradrenaline α1-receptor antagonists and SK channel antagonists. These IPSPs were reduced or abolished by either saclofen (300 μm) or CGP 55845 (300 nm), GABAB receptor antagonists (Fig. 1).
Figure 1. Dopamine inhibits the GABAB IPSP.
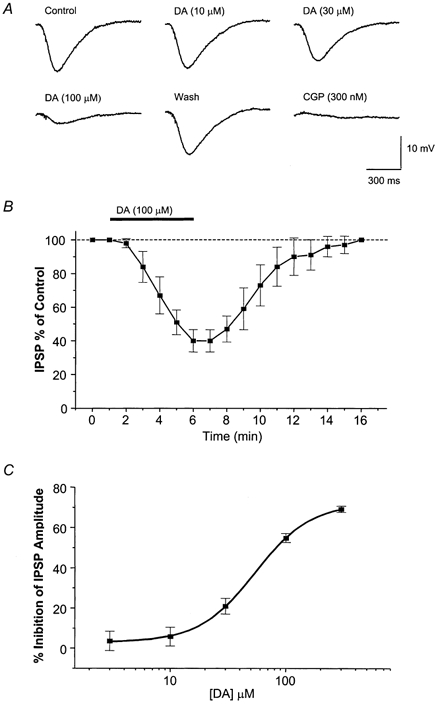
A, example of the concentration-dependent and reversible inhibition caused by DA in the slow IPSP which was subsequently reduced by the GABAB receptor antagonist CGP (300 nm). B, time course of DA effects. Here and in subsequent graphs, the amplitudes of the IPSPs were normalized to the control amplitude determined for at least 5 min before application of DA. Each data point in this graph represents the mean ± s.e.m. of 7 cells. C, concentration-dependent curve of the reducing effects of DA on the GABAB-mediated IPSP. Each point represents the mean ± s.e.m. of 25 cells.
Even in the presence of D1- and D2-selective antagonists (SCH 23390 (1–10 μm) and sulpiride (3–30 μm), respectively), dopamine (3–300 μm) reduced the amplitude of the GABAB IPSP (115 of 121 neurones) (Fig. 1). This action had a slow onset of 2–3 min, peaked in 4–5 min, and recovered in 7–10 min of washing (Fig. 1B) and was reproducible. The depressant effect of dopamine was concentration dependent (IC50, 54.1 ± 1.3 μm, n = 25) (Fig. 1C). The decrease in IPSP amplitude was 54.8 ± 15.3 % (n = 25) when a concentration of 100 μm DA was superfused while no changes in membrane input resistance and IPSP time course were observed. During the effects of DA a small depolarization of the membrane potential (2.1 ± 0.8 mV, n = 22) was observed in 22 of 35 cells. This depolarization required the injection of 10–30 pA hyperpolarizing current to hold the membrane potential constant (at approximately −65 to −70 mV) throughout the experimental session. No current was required in the remaining cells. In some experiments we perfused either apamin (100 nm, SK channel antagonist; n = 4) or MCPG (500 μm, mGluR antagonist; n = 3) to reduce further the possible metabotropic glutamate component in the slow IPSP. Under these conditions the depressant effects of DA on the GABAB IPSPs were not affected (not shown).
In the absence of the D1 antagonist SCH 23390 and in the presence of the DA-uptake inhibitor nomifensine (10 μm), DA (3 μm) slightly increased the amplitude of the IPSPs by 2.2 ± 1 % (P > 0.05, n = 4), while in three neurones decreased it by 4 ± 0.8 % (P < 0.05). In the attempt to counteract the depressant effect of DA we used other DA antagonists besides sulpiride and SCH 23390. High concentrations of haloperidol (10 μm), clozapine (10 μm), chlorpromazine (10–30 μm) and spiperone (10 μm) (DA and also 5-HT1A/5-HT2 antagonist) did not significantly affect the action of DA (100 μm) (Fig. 2A) (P > 0.05 for each compound). Furthermore, we tested the possibility that, in spite of the presence of their respective antagonists, the D1 agonist SKF 38393 (3–10 μm) (n = 4) and/or the D2 agonist quinpirole (10 μm) (n = 3) might have a depressant action on the IPSP. However, these compounds were ineffective (P > 0.05). The D1/D2 receptor agonist apomorphine (3 μm) was also without effect on the IPSP (n = 3) (P > 0.05). Next, we searched for possible effects of noradrenaline (NA) and the invertebrate biogenic amine octopamine on the GABAB IPSP. NA (10–100 μm) (n = 4) but not octopamine (30–100 μm) (n = 3) reversibly decreased the amplitude of the GABAB IPSP (Fig. 2B and C). Interestingly, the maximal inhibition caused by NA (100 μm) was only 45 % of that caused by an equimolar concentration of DA. The depressant effect induced by DA (100 μm) was not altered by specific adrenergic antagonists propanolol (20 μm, β1) (n = 3), pindolol (300 nm, β1/2 and also 5-HT1A) (n = 2) and prazosin (300 nm, α1)(n = 3), P > 0.05 for each compound). Interestingly, it was partially reduced by yohimbine (10 μm, α2) (42.5 ± 13.6 % of control) (n = 5, P < 0.05) and by relative high concentrations of the α1-α2 antagonist phentolamine (30 μm) (53.1 ± 26.2 % of control (n = 4); P < 0.05) (Fig. 2D). Conversely, the specific α2 agonist UK 14304 (10 μm) had no detectable effects on the amplitude of GABAB potentials (n = 3) (P > 0.05) (not shown).
Figure 2. Dopaminergic antagonists do not change the DA-mediated reduction of the GABAB IPSP, NA mimics DA action while several noradrenergic antagonists are ineffective.
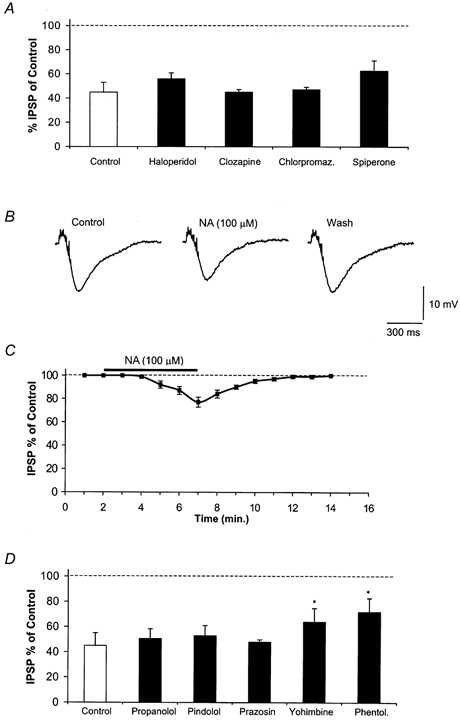
A, the bars show that haloperidol (10 μm), clozapine (10 μm), chlorpromazine (Chlorpromaz, 10 μm) and spiperone (10 μm) had no significant antagonizing effect (P > 0.05 for each compound) on the DA-induced (100 μm) depression of the GABAB potentials. Each column shows the average of three to four experiments. B, reversible inhibition of an IPSP by noradrenaline (100 μm). C, time course of NA-induced depression of the IPSP. Each data point represents the mean ± s.e.m. of four cells. D, the graph shows the lack of antagonizing effect (P > 0.05) of several NA antagonists, propanolol (20 μm), pindolol (300 nm), prazosin (300 nm), on the DA-induced depression (100 μm) of the IPSP. Note that yohimbine (10 μm) and phentolamine (Phentol, 30 μm) caused an incomplete but significant (P < 0.05) reduction in the DA effect. Each column shows an average of four to five cells. Asterisks indicate significant reduction of the DA-induced effect.
l-DOPA acts throughout its metabolic transformation
Next we tested whether the DA precursor l-DOPA also reduced the amplitude of the GABAB IPSP. l-DOPA (100 μm) depressed this inhibitory potential to 70.2 ± 7.7 % (n = 4) of control (Fig. 3). It took longer than dopamine to act, 7–12 versus 2–4 min. The reduction of IPSP amplitude was due to transformation of l-DOPA into DA because it was selectively diminished to 97 ± 5 % (n = 4, P < 0.05) of control by a pretreatment of the slices with the DOPA-decarboxylase inhibitor carbidopa (300 μm, 15–25 min) (Mercuri et al. 1990).
Figure 3. l-DOPA suppresses the GABAB IPSP through its metabolic transformation.
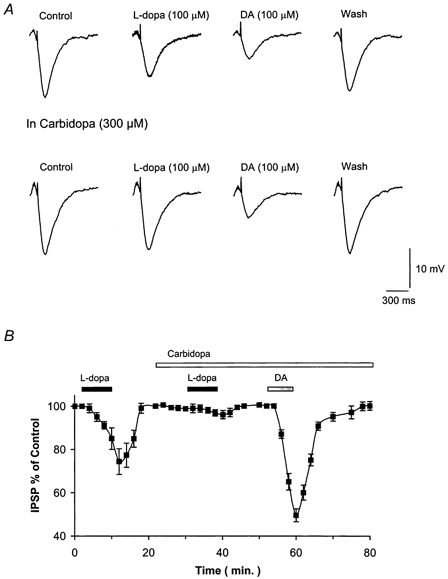
A, reversible inhibition of the IPSP caused by l-DOPA and DA. Only the attenuation caused by l-DOPA but not that caused by DA is blocked by pretreatment of the slices with carbidopa (300 μm). B, time course of l-DOPA (100 μm) and DA (100 μm) effects and modification of l-DOPA action by carbidopa (300 μm). Each data point in this figure represents the mean ± s.e.m. of four cells.
The effects of dopamine on the GABAB inputs are presynaptic and specific
Dopamine did not affect the postsynaptic hyperpolarizations caused by the local application of baclofen (300 μm) (10–20 p.s.i, 8–10 ms) while it reduced the GABAB IPSP evoked on the same cells (n = 5) (Fig. 4A). This supports a presynaptic site of action. Next, we examined the possible effects of DA on the GABAA-mediated IPSP. DA (30–100 μm) had no significant effect on the amplitude of the GABAA IPSP (Fig. 4B) (n = 3 P > 0.05).
Figure 4. Dopamine selectively decreases the GABAB-mediated IPSP by a presynaptic mechanism leaving the GABAA IPSP unaffected.
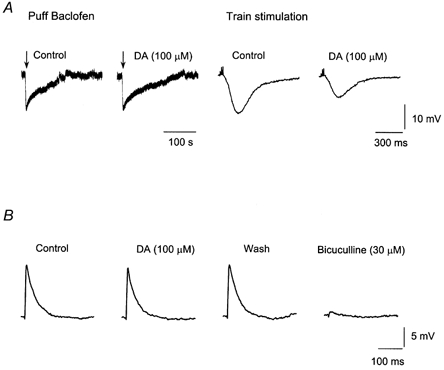
A, dopamine (100 μm) reduces the GABAB IPSP without changing the membrane hyperpolarization caused by puffer applications of the GABAB agonist baclofen (300 μm, 10–20 p.s.i., 8–10 ms). B, lack of DA effects on the GABAA IPSP.
Lack of effect of antagonists for presynaptic neuromodulators
We also tested whether a DA-induced release of adenosine underlies the depressant action of DA on the GABAB IPSP (Bonci & Williams, 1996). However the selective A1 receptor antagonist DPCPX (3 μm) (Wu et al. 1995) did not affect DA action (n = 3, P > 0.05) on the GABAB IPSP (not shown). In addition a combination of atropine (3 μm) and MAP4 (500 μm) (n = 2) (to block muscarinic and group III presynaptic glutamate receptors, respectively) did not prevent the DA-induced depression of the GABAB IPSP (not shown).
Dopamine and serotonin have occluding presynaptic effects
In agreement with Johnson et al. (1992) the application of serotonin (5-HT; 100 μm) also inhibited the GABAB IPSP by 41.5 % ± 9.2 % (n = 7) throughout the activation of 5-HT1B receptors (Fig. 5A). Thus, the 5-HT1B receptor antagonist cyanopindolol (300 nm) reduced the depression of the GABAB IPSP caused by serotonin to 10 ± 0.9 % (n = 5, P < 0.05) (Johnson et al. 1992) while it did not affect DA action (Fig. 5). In addition, the depression of the IPSP caused by DA was not affected by pindolol (300 nm, 5-HT1A and β1/2; 3 cells) and methysergide (10 μm; a serotoninergic antagonist that was reported to reduce the depressant action of DA on the excitatory synaptic events; Jones et al. 2000; 3 cells) (P > 0.05, for each compound). If the signalling pathway operated by 5-HT is also that engaged by DA, the IPSP inhibition caused by these neurotransmitters when bath applied in combination should be approximately the same as when each is applied alone. Thus, we observed that the inhibition caused by DA (100 μm) was greatly reduced by the previous application of a maximal concentration of 5-HT (100 μm) (Fig. 5C). The lack of an additive effect supports the hypothesis that DA and 5-HT operate throughout the same signalling pathway activated by 5-HT1B receptors and unconventional DA-recognising sites located on the GABA terminals impinging on the dopaminergic neurones.
Figure 5. Activation of 5-HT1B presynaptic receptors occludes the effects of DA on the GABAB IPSP.
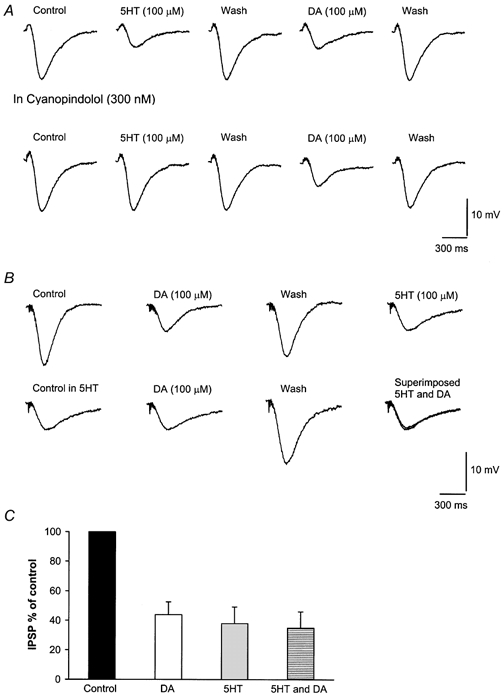
Examples of IPSP inhibition caused by 5-HT and DA. Only the 5-HT but not the DA effects are antagonized by the 5-HT1B receptor antagonist cyanopindolol (A). The inhibition of the IPSP caused by 5-HT is very similar to that induced by DA (B). The graph shows that the inhibition of the GABAB IPSP caused by DA and 5HT is not additive. Each column shows the average of four to eight cells (C).
The presynaptic actions of DA and 5-HT are independent of the modulation of high voltage-activated calcium, 4-AP and tolbutamide-sensitive potassium channels
The DA- and 5-HT-induced depression of GABA release on GABAB synapses could result either from the depression of calcium currents or the opening of potassium channels in presynaptic terminals. In the presence of the non-selective antagonist of the N/Q- and P-type calcium channels ω-CgTx MVIIC (2 μm) (Wheeler et al. 1994), a steady-state reduction of the amplitude of the GABAB IPSP to 30 ± 12 % (n = 6 of control) was obtained in 10–15 min.
Subsequently, increasing the intensity of the stimulating current restored the initial amplitude of the GABAB IPSP. In the presence of ω-CgTx MVIIC, either DA (100 μm) or 5-HT (100 μm) still depressed the GABAB IPSP by 68 ± 5 % (n = 3) and 61 ± 8 % (n = 3), respectively (Fig. 6). Remarkably, a similar degree of depression was observed in control conditions. In fact, DA (100 μm) and 5-HT (100 μm) reduced the GABAB IPSP by 65 ± 8 % (n = 3) (P > 0.05) and 60 ± 5 % (n = 3), (P > 0.05) of control, respectively. We also examined the possibility that DA and 5-HT could modulate L-type calcium channels to decrease GABA transmission by perfusing the L-type antagonist nifedipine (10 μm) (Bonci et al. 1998). In the presence of nifedipine, the amplitude of the GABAB IPSP did not significantly change and DA (100 μm) (n = 3) and 5-HT (100 μm) (n = 3) still suppressed the GABAB potential in a manner not different with respect to control conditions (P > 0.05 for both neurotransmitters) (Fig. 6). Alternatively, DA and 5-HT could control GABA release by modulating potassium channels in the presynaptic terminals. The depression of voltage-dependent potassium currents by the application of 4-AP (300 μm) rapidly (3–6 min) increased the amplitude of the GABAB potential by 300 ± 30 % (n = 3) of control. However, by decreasing the intensity of the stimulating current we restored the initial amplitude of the inhibitory potential. In the presence of 4-AP, DA (100 μm) (n = 3) and 5-HT (100 μm) (n = 3) both reduced the GABAB potential to 48 ± 5 % (n = 3) and 60 ± 10 % (n = 3) as in control conditions (P > 0.05 for each compound) (Fig. 6). In the presence of the K-ATP channel blocker, tolbutamide (1 mm), DA (100 μm) (n = 3) and 5-HT (100 μm) (n = 3) also caused an inhibition of the GABAB IPSP as in control conditions (P > 0.05 for each compound) (Fig. 6). Tolbutamide alone had no effect on the amplitude of the GABAB potential.
Figure 6. The superfusion of calcium and potassium blockers did not prevent DA and 5HT presynaptic actions.
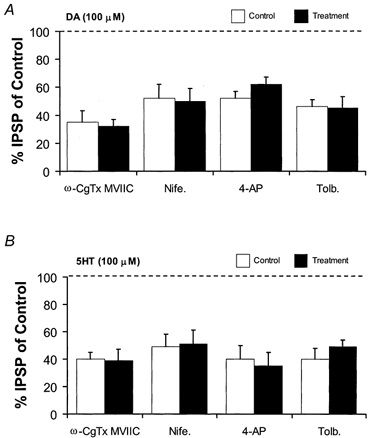
Dopamine (A) and serotonin (B) presynaptic actions in control and drug-treated slices (Nife, nifedipine; Tolb, tolbutamide). Columns show means of three experiments Note that either the DA- or the 5-HT-mediated inhibition of neurotransmitter release are not significantly different from control during the different experimental conditions (P > 0.05).
DISCUSSION
This paper describes an unconventional and novel depressant effect of dopamine on the GABAB IPSP, which is not mediated via the activation of D1- and D2-like receptors. In addition, the observation that we could antagonize the depressant effects of l-DOPA by blocking the DOPA-decarboxylase enzymes supports the specificity of the DA-induced depression of the GABAB IPSPs. In an area, reach of dopaminergic neurones l-DOPA mainly produces DA. However, the scant noradrenergic innervation of the substantia nigra pars compacta also suggests that DA and not NA mediates the depression of the GABAB IPSP.
While it is generally accepted that DA facilitates GABA release on GABAB synapses in the ventral midbrain by stimulating presynaptic D1 receptors (Cameron & Williams, 1993), our results demonstrate that, at least under particular conditions, DA strongly depresses (in a concentration-dependent manner), the GABAB potentials by activating sites with a new pharmacological profile. Therefore, these unconventional sites are activated by DA and NA and not by the invertebrate biogenic amine octopamine (Degen et al. 2000). In addition, they are not stimulated by D1- and D2-like agonists and not affected by a series of DA antagonists.
Although most NA antagonists were ineffective, the α2 adrenergic-blocking drugs yohimbine and phentolamine partially reduced DA action on the GABAB potentials. Conversely, the α2 adrenergic agonist UK 14304 was ineffective. This suggests the existence of functional DA-recognising sites, which are affected by α2-adrenergic antagonists but not by agonists.
The clear-cut depression exerted by DA on the slow GABAB IPSP without changing the fast inhibitory component (GABAA) favours a specialised action of this catecholamine at the GABAB synapses in the ventral midbrain. The functional distinction is eventually sustained by anatomical differences between GABAergic terminals making separate connections on GABAA and GABAB receptors on the dopaminergic cells, being the GABAB responses probably caused by synapses arising from the forebrain and the GABAA from local interneurones (Lacey et al. 1989; Johnson et al. 1992; Sugita et al. 1992; Shoji et al. 1999). Interestingly, the novel sites of DA action are very likely located on the same GABAergic terminals bearing 5-HT1B receptors. This is supported by the fact that the DA-induced depression of the GABAB IPSPs can be occluded by the application of 5-HT, suggesting that DA, which is released from dopaminergic cells dendrites (Cheramy et al. 1981), uses the same machinery operated by presynaptic 5-HT1B receptors on the GABAergic afferents (Johnson et al. 1992).
Based on the experimental evidence that the DA- and the 5-HT-induced depression of the GABAB IPSPs are neither sensitive to the inhibition of N-, P/Q- and L type calcium channels nor to the depression of 4-AP- and K-ATP-sensitive potassium channels, we suggest that different subtypes of presynaptic channels could be involved in the inhibition of GABA release. Alternatively, DA and 5-HT could mediate inhibition of neurotransmitter release one step beyond an increased permeability to ions by directly regulating exocytotic fusion through the activation of G-proteins (Blackmer et al. 2001).
The presynaptic modulation of GABA release caused by DA might be an important phenomenon for the development of addiction, since it has also been demonstrated that the psychostimulant cocaine also causes a specific depression of the GABAB synaptic transmission mainly stimulating presynaptic 5-HT receptors (Cameron & Williams, 1994). The absence of any effect of 5-HT-, adenosine-, muscarine- and metabotropic glutamate receptor antagonists increases the possibility that DA activates unique sites to reduce GABAB IPSPs. There are conflicting results in the literature descibing the net effect of dopamine and dopaminergic agonists on GABA efflux in the ventral mesencephalon, namely that D1 receptors are mainly stimulatory and D2 receptors are mainly inhibitory (Arbilla et al. 1981; Kelly et al. 1985; Mattuszewich & Yamamoto, 1999). However, relatively high doses of DA preferentially cause a depression of GABA release in in vivo experiments (Korf et al. 1981; Chesselet, 1984). It is not difficult to imagine circumstances where DA could reduce, instead of increase, the release of GABA in the ventral midbrain. Thus, chronic treatment with high doses of neuroleptics, which block the classical D2-/D1-like receptors (Sedvall et al. 1995), could facilitate the depression of the GABAB IPSPs mediated by endogenously released DA. Additionally, the larger and sustained efflux of DA either caused by l-DOPA treatment of parkinsonian patients or by the compulsive intake of amphetamine/cocaine in addicts might inhibit the release of GABA at the GABAB synapses in the ventral midbrain. This might activate, under particular circumstances, the mesostriatal and mesocortical dopaminergic systems (Gonon, 1988) and could be involved in the production of extrapyramidal side effects and/or therapeutic actions of antipsychotics (Grace, 1991; Abi-Dargham et al. 2000; Kegeles et al. 2000). Alternatively, the presynaptic actions of DA described here could be involved in determining the beneficial/side effects of l-DOPA in parkinsonian patients (Chase et al. 1998) and might be involved in the addictive effects of psychostimulants (Wise & Bozarth, 1987; White, 1996).
Acknowledgments
We thank Drs Paolo Calabresi and Antonello Bonci for comments on the manuscript. This work was supported by a CNR grant (biomolecole per la salute umana) to NBM.
REFERENCES
- Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M. From the cover: increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proceedings of the National Academy of Sciences of the USA. 2000;97:8104–8109. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbilla S, Kamal LA, Langer SZ. Inhibition by apomorphine of the potassium-evoked release of [3H]-gamma-aminobutyric acid from the rat substantia nigra in vitro. British Journal Pharmacology. 1981;74:389–397. doi: 10.1111/j.1476-5381.1981.tb09983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Takahashi M, Martin TF, Alford S, Hamm HE. G protein betagamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science. 2001;292:293–297. doi: 10.1126/science.1058803. [DOI] [PubMed] [Google Scholar]
- Bonci A, Grillner P, Mercuri NB, Bernardi G. L-Type calcium channels mediate a slow excitatory synaptic transmission in rat midbrain dopaminergic neurons. Journal of Neuroscience. 1998;18:6693–6703. doi: 10.1523/JNEUROSCI.18-17-06693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron. 1996;3:631–639. doi: 10.1016/s0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- Cameron DL, Williams JT. Cocaine inhibits GABA release in the VTA through endogenous 5-HT. Journal of Neuroscience. 1994;14:6763–6767. doi: 10.1523/JNEUROSCI.14-11-06763.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase TN, Oh JD, Blanchet PJ. Neostriatal mechanisms in Parkinson's disease. Neurology. 1998;51(2):S30–35. doi: 10.1212/wnl.51.2_suppl_2.s30. [DOI] [PubMed] [Google Scholar]
- Cheramy A, Leviel V, Glowinski J. Dendritic release of dopamine in the substantia nigra. Nature. 1981;289:537–542. doi: 10.1038/289537a0. [DOI] [PubMed] [Google Scholar]
- Chesselet MF. Presynaptic regulation of neurotransmitter release in the brain: facts and hypothesis. Neuroscience. 1984;12:347–375. doi: 10.1016/0306-4522(84)90058-7. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annual Review Pharmacology Toxicology. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Degen J, Gewecke M, Roeder T. Octopamine receptors in the honey bee and locust nervous system: pharmacological similarities between homologous receptors of distantly related species. British Journal of Pharmacology. 2000;130:587–594. doi: 10.1038/sj.bjp.0703338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo CD, Williams JT. Selective inhibition by adenosine of mGluR IPSPs in dopamine neurones following cocaine treatment. Journal of Neurophysiology. 2000;83:1307–1314. doi: 10.1152/jn.2000.83.3.1307. [DOI] [PubMed] [Google Scholar]
- Gonon FG. Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurones as studied by in vivo electrochemistry. Neuroscience. 1988;24:19–28. doi: 10.1016/0306-4522(88)90307-7. [DOI] [PubMed] [Google Scholar]
- Grace AA. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience. 1991;41:1–24. doi: 10.1016/0306-4522(91)90196-u. [DOI] [PubMed] [Google Scholar]
- Grace AA, Onn SP. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurones recorded in vitro. Journal of Neuroscience. 1989;9:3463–3481. doi: 10.1523/JNEUROSCI.09-10-03463.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii TM, Maylie J, Adelman JP. Determinants of apamin and d-tubocurarine block in SK potassium channels. Journal Biological Chemistry. 1997;272:23195–23200. doi: 10.1074/jbc.272.37.23195. [DOI] [PubMed] [Google Scholar]
- Johnson SW, Mercuri NB, North RA. 5-Hydroxytryptamine 1B receptors block the GABAB synaptic potential in rat dopamine neurones. Journal of Neuroscience. 1992;12:2000–2006. doi: 10.1523/JNEUROSCI.12-05-02000.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurones in the rat ventral tegmental area and their synaptic inputs. Journal of Physiology. 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Kornblum JL, Kauer JA. Amphetamine blocks long-term synaptic depression in the ventral tegmental area. Journal of Neuroscience. 2000;20:5575–5580. doi: 10.1523/JNEUROSCI.20-15-05575.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegeles LS, Abi-Dargham A, Zea-Ponce Y, Rodenhiser-Hill J, Mann JJ, Van Heertum RL, Cooper TB, Carlsson A, Laruelle M. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biological Psychiatry. 2000;48:627–640. doi: 10.1016/s0006-3223(00)00976-8. [DOI] [PubMed] [Google Scholar]
- Kelly E, Jenner P, Marsden CD. The effects of dopamine and dopamine agonists on the release of 3H-ABA and 3H-5HT from rat nigral slices. Biochemical Pharmacology. 1985;34:2655–2662. doi: 10.1016/0006-2952(85)90563-5. [DOI] [PubMed] [Google Scholar]
- Koga E, Momiyama T. Presynaptic dopamine D2-like receptors inhibit excitatory transmission onto rat ventral tegmental dopaminergic neurones. Journal of Physiology. 2000;523:163–173. doi: 10.1111/j.1469-7793.2000.t01-2-00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf J, Van Der Heyden JA, Venema K, Postema F. Distribution and release of GABA in the basal ganglia. Advances in Biochemical Psychopharmacology. 1981;30:105–117. [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. Journal of Physiology. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and the actions of dopamine and opioids. Journal of Neuroscience. 1989;4:1233–1241. doi: 10.1523/JNEUROSCI.09-04-01233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B, Bruns R, Roeper J. Alternative sulfonylurea receptor expression defines metabolic sensitivity of K-ATP channels in dopaminergic midbrain neurones. EMBO Journal. 1999;18:833–846. doi: 10.1093/emboj/18.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matuszewich L, Yamamoto BK. Modulation of GABA release by dopamine in the substantia nigra. Synapse. 1999;32:29–36. doi: 10.1002/(SICI)1098-2396(199904)32:1<29::AID-SYN4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Mercuri NB, Bonci A, Calabresi P, Stefani A, Bernardi G. Properties of the hyperpolarization-activated cation current Ih in rat midbrain dopaminergic neurones. European Journal of Neuroscience. 1995;7:462–469. doi: 10.1111/j.1460-9568.1995.tb00342.x. [DOI] [PubMed] [Google Scholar]
- Mercuri NB, Calabresi P, Bernardi G. Responses of rat substantia nigra compacta neurones to L-DOPA. British Journal of Pharmacology. 1990;100:257–260. doi: 10.1111/j.1476-5381.1990.tb15792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri NB, Calabresi P, Bernardi G. The electrophysiological actions of dopamine and dopaminergic drugs on neurones of the substantia nigra pars compacta and ventral tegmental area. Life Science. 1992;51:711–718. doi: 10.1016/0024-3205(92)90479-9. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Fiorillo CD, Morikawa H, Williams JT. Amphetamine selectively blocks inhibitory glutamate transmission in dopamine neurones. Nature Neuroscience. 2001;3:275–281. doi: 10.1038/85124. [DOI] [PubMed] [Google Scholar]
- Sedvall G, Pauli S, Farde L, Karlsson P, Nyberg S, Nordstrom AL. Recent developments in PET scan imaging of neuroreceptors in schizophrenia. Israel Journal of Psychiatry and Related Sciences. 1995;32:22–29. [PubMed] [Google Scholar]
- Shoji Y, Delfs J, Williams JT. Presynaptic inhibition of GABAB-mediated synaptic potentials in the ventral tegmental area during morphine withdrawal. Journal of Neuroscience. 1999;19:2347–2355. doi: 10.1523/JNEUROSCI.19-06-02347.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Johnson SW, North RA. Synaptic inputs to GABAA and GABAB receptors originate from discrete afferent neurones. Neuroscience Letters. 1992;134:207–211. doi: 10.1016/0304-3940(92)90518-c. [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- White FJ. Synaptic regulation of mesocorticolimbic dopamine neurones. Annual Review of Neuroscience. 1996;19:405–436. doi: 10.1146/annurev.ne.19.030196.002201. [DOI] [PubMed] [Google Scholar]
- Wise RA, Bozarth MA. A psychomotor stimulant theory of addiction. Psychological Review. 1987;94:469–492. [PubMed] [Google Scholar]
- Wu YN, Mercuri NB, Johnson SW. Presynaptic inhibition of gamma-aminobutyric acidB-mediated synaptic current by adenosine recorded in vitro in midbrain dopamine neurones. Journal of Pharmacology and Experimental Therapeutics. 1995;273:576–581. [PubMed] [Google Scholar]