

Abstract
Reliable biomarkers of toxicity are necessary both for the safe conduct of pre-clinical and clinical trials, and are increasingly needed for accurate clinical evaluation of treatment regimens with the potential to cause tissue injury. Recent advances in technology have added several new tools to the biomarker screening toolkit and improved the throughput of existing quantitative assays. Genomics, proteomics, and metabolomics have provided a wealth of data in the search for predictive, specific biomarkers. Multiplexed ELISA-based assay systems, silicon nanowire arrays, and patterned paper present unique abilities for fast, efficient sample analysis over a broad dynamic range. Powerful integrative systems biology software and growing open-source data repositories offer new ways to share, reduce, and analyze data from multiple sources. Novel technologies reviewed here have the potential to significantly reduce assay time and cost and improve the sensitivity of screening methods for candidate biomarkers of toxicity.
1. Introduction
Reliable biomarkers of toxicity are necessary both for the safe conduct of pre-clinical and clinical trials, as well as for clinical evaluation of treatment regimens with the potential to cause tissue injury. Useful biomarkers of toxicity should be present in biofluids such as blood or urine to allow minimally invasive collection and rapid quantitation and should sensitively and reproducibly indicate potential adverse health effects at a time or dose prior to overt or irreversible tissue damage, toxicity, or disease onset (Tugwood et al. 2003). The FDA’s Critical Path Initiative has reinforced the need for additional biomarkers to predict drug toxicity in preclinical studies, specifically biomarkers that can act as surrogate endpoints and/or aid in making efficacious and cost-saving decisions or terminating drug development more quickly. In response to the Critical Path Initiative, in October 2006, a biomarker consortium including the FDA, the Foundation for the National Institutes of Health, and the Pharmaceutical Research and Manufacturers of America was launched, which focuses on developing biomarkers for use in regulatory decision making, as well as biomarker discovery (Wagner et al. 2007).
Recent advances in technology have not only improved the throughput and sensitivity of existing biomarker assays, but have also added tools to the biomarker screening toolkit. Genomic, proteomic, and metabolomic approaches have been used successfully both alone and in combination to screen for useful biomarkers of toxicity. Gene microarrays and protein chip arrays can generate thousands of useful data points from a single sample. These expression profiles, which can themselves be used grossly as biomarkers, can be compared across multiple time points, doses, or experimental populations to look for molecules predictive of toxic injury. Integrative systems biology software applications have been successful in compiling and analyzing information from these and other more nascent “-omics” fields. When combined with data on the array of traditional pathophysiological responses to toxicants, these applications can be powerful search engines for novel biomarkers of toxicity.
In order to qualify a biomarker, extensive quantitation of the protein is necessary to demonstrate the stability, sensitivity, specificity and reproducibility of the biomarker in multicentric collaborative studies using a variety of preclinical and clinical disease models (Figure 1). To facilitate this qualification process, sensitive, specific and high-throughput assays are critical for effective measurement of the biomarker either alone or in combination to allow comparison with traditional markers that are considered “gold standards.” Recent advances in ELISA-based technologies and the creation of novel protein assays have made the process of evaluating a biomarker from benchtop to bedside faster and more sensitive. Improved data processing capabilities of integrative systems biology applications have shrunk the gap between data generation and data analysis technologies considerably, so that multiple data sources can be combined to inform the presence of potential biomarkers.
Figure 1.
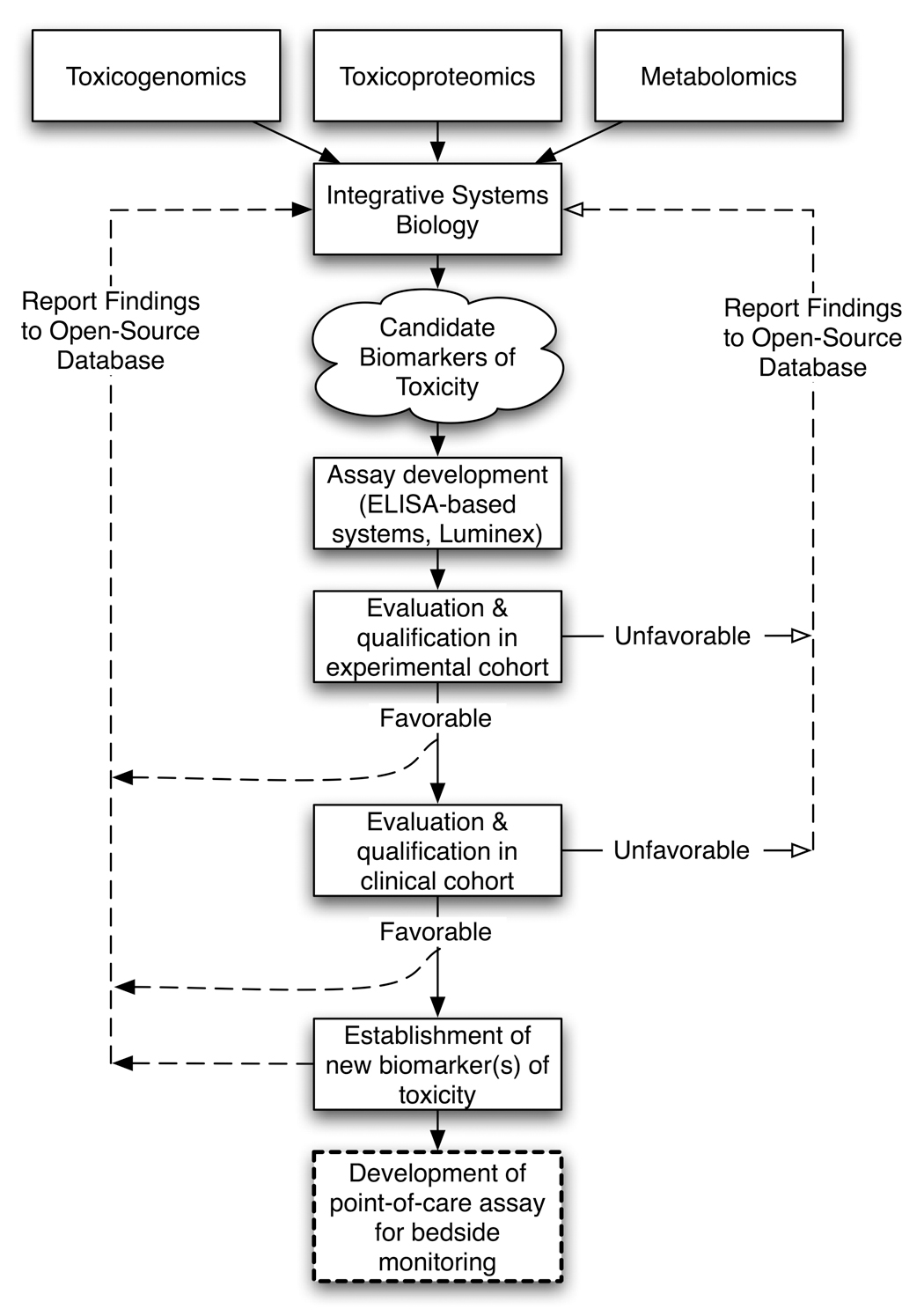
Biomarker discovery and evaluation process
The limiting factor in biomarker discovery and quantitation – as in every developing field – is cost, both in terms of dollar cost of the assay reagents and equipment and the man-hours required to perform the assay (Table 1). Novel technologies reviewed here have the potential to significantly reduce assay time and/or assay cost, which may help the discovery and evaluation of biomarkers proceed apace.
Table 1.
Characteristics of assays for biomarker quantitation
| Assay | Sensitivity | Dynamic Range (Logs) | # of Epitopically Distinct Antibodies Required | Current Multiplexing Capability | Adaptable to Bedside Monitoring | Assay Duration | Assay Cost |
|---|---|---|---|---|---|---|---|
| ELISA | 15 pg/ml | 3 | 2 | None | No | 5–7 hr | + + |
| FACCT ELISA | 0.08 fM | 3 | 2 | None | No | 8–10 hr | + + |
| Mesoscale® | 1.0 pg/ml | 4 | 2 | Up to 10 analytes | No | 4 hr | + + + |
| Searchlight® | 0.78 pg/ml | 4 | 2 | Up to 24 analytes | No | 3 hr | + + + |
| FAST Quant® | 1.2 pg/ml | 4 | 2 | Up to 9 analytes | No | Overnight | + + + |
| Luminex® | 0.68 pg/ml | 5 | 2 | Up to 27 analytes | No | 3.5 hr | + + + |
| Nanowire | 90 fg/ml | 5 | 1 | Up to 3 analytes | Yes | 30 min | + + |
| Patterned Paper | 0.38 µM | 2 | Antibody-based assay not yet available | 2 analytes | Yes | 10 min | + |
2. Approaches for Discovery
2.1 Toxicogenomics
If we assume there is some change in gene expression in response to toxicity, then expression profiling is an extremely powerful tool for identifying potential biomarkers of toxicity (Tugwood et al. 2003). DNA microarray is the most prevalent and powerful genomic assay for identifying potential new biomarkers. The entire human genome – about 25,000 genes – can now be contained on a single DNA microarray to generate a complete profile of the genomic response to a toxicant (Arcellana-Panlilio and Robbins 2002; Geschwind 2003; Ishkanian et al. 2004). Representational difference analysis (RDA) of tissue-specific or cell-specific microarrays is used to screen for potential biomarkers based on differential gene expression; protein-coding genes whose transcription is differentially upregulated in the setting of tissue injury are considered candidate biomarkers (Hubank and Schatz 1994). The shortcoming of using only genomic data in the search for protein biomarkers of toxicity lies in the difficulty of associating a change in transcript profile with an actual change in protein abundance (Tugwood et al. 2003). DNA microarray detects only stably-expressed mRNA transcripts, which does not necessarily inform the presence of a candidate protein biomarker, and so subsequent semi-quantitative assays (ie. immunoblot, immunostaining) must be performed before microarray data can be considered truly indicative.
Using RDA, Ichimura et al. identified the most differentially upregulated gene in the postischemic rat kidney, KIM-1, and confirmed its utility as a biomarker using immunoblot, immunostaining, and RNA in situ hybridization (Ichimura et al. 1998). KIM-1 protein is shed from the proximal tubular epithelium into the urine following ischemic or toxic kidney injury, and its concentration in the urine is able to discriminate injured vs. healthy kidneys (Ichimura et al. 1998).
2.2 Toxicoproteomics
Estimates for the number of proteins in the proteome range as high as 100,000, excluding post-translational modifications, which push the range higher still. To accommodate this massive data source, proteomics-driven biomarker discovery employs several robust and well-characterized technologies, including 2D-MS (2-dimensional gel electrophoresis-mass spectrometry), RC-MS (retentate chromatography-mass spectrometry), and commercial derivatives thereof, such as SELDI (surface-enhanced laser desorption ionization), as well as more targeted investigative tools like antibody arrays (Elrick et al. 2006).
SELDI-TOF-MS combines data on time of flight (TOF) in a constant electric field with mass spectrometry to sequentially determine the charge and mass of proteins in a single sample (Issaq et al. 2003). An algorithm can then be used to identify trends in mass/charge ratio peaks across a spectrum of proteins, and use the resultant expression profiles as biomarkers for specific disease types, as has been done for ovarian (Conrads et al. 2004), prostate (Li et al. 2004), breast (Vlahou et al. 2003), lung (Xiao et al. 2003; Zhukov et al. 2003), pancreas (Koopman et al. 1999; Rosty et al. 2002), and liver cancers (Melle et al. 2004; Poon et al. 2003). SELDI-TOF-MS is especially attractive for disease biomarker identification because of its quick assay time and low required sample volumes. Differential expression of proteins analyzed by SELDI-TOF-MS can be used to determine the approximate size of a putative biomarker; the identity of the protein can then be resolved by SDS-PAGE and confirmed by immunoblot (Mazzatti et al. 2007). ProteinChip arrays (Ciphergen, Fremont, CA) can be used to investigate the response of a proteome to a toxicant in both biological fluids and in specific tissues.
Matrix-assisted laser desorption/ionization-time of flight-mass spectrometry (MALDI-TOF-MS) is similar to SELDI, except that protein solutions are mixed with a UV-absorbing matrix and then cocrystallized on a matrix before being ionized. This makes MALDI highly sensitive to small peptides, but reduces the range of peptide detection as compared to SELDI and results in some degree of interference from matrix ions (Chensong Pan 2006; Mazzatti et al. 2007). Addition of ammonium salts or cetriamonium bromide (CTAB) to matrices has been shown to improve the resolution of MALDI spectra by reducing their sensitivity to salts and contaminants, as has addition of diamond nanoparticles to samples prior to matrix deposition (Kong et al. 2005). Carbon nanotubes have been used as an inorganic matrix, eliminating interference issues associated with the matrix and allowing analytes to be well desorbed/ionized during analysis (Xu et al. 2003), thus improving the quantitative reproducibility of the assay (Chensong Pan 2006).
A novel microfluidic approach to proteomic profiling on chips – the Experion system (Bio-Rad Laboratories, Hercules, CA) – has recently been employed for urinary proteome analysis. The chip uses an electrical current applied across a microfluidic channel to first direct samples from their respective wells and then draw the sample across a fluorescent-dye-containing polymer gel matrix to separate and label charged species within the samples (Thongboonkerd et al. 2007). Waste is drawn off by an intersecting channel prior to separation to improve the resolution of the assay. Using a sample volume of 4 µl, data gathered from the Experion microfluidic system differentiated patients with diabetic nephropathy (DN) or IgA nephropathy from healthy controls and DN and IgA nephropathy patients from each other based on their proteomic spectra (Thongboonkerd et al. 2007).
Advances in microfluidic device design have also improved analysis of the glycoproteome. Zhuang et al. describe a novel microfluidic device that improves the efficiency of separation over previous microchip gel electrophoresis devices as much as 50-fold by subjecting a sample to a 750V/cm electric field as it travels along a spiral microfluidic channel (Zhuang et al. 2007). The sample is labeled with the charged fluorophore, APTS (8-amino-1,3,6-trisulfonic acid), to impart a charge to the glycans and permit fluorescence detection of the complete separation. Assay time was reduced to under 3 minutes with very good run-to-run reproducibility, and the improved separation efficiency allowed for discrimination of positional and linkage isomers (Zhuang et al. 2007).
2.3 Metabolomics
Metabolomics is uniquely suited to the discovery of new biomarkers of toxicity in that the metabolome is much smaller than the proteome – representing at most about 10,000 compounds – and many metabolites have known functions and participate in well-characterized pathways. Furthermore, metabolites are the last step in the molecular response to a toxicant; unlike gene and protein expression that can be altered and regulated by homeostatic mechanisms, metabolite levels respond to the gross effect of differences in gene and protein expression and represent a more holistic response (van Ravenzwaay et al. 2007).
Data on changes in metabolite expression profiles are traditionally gathered from biofluids using 1H NMR. Magic angle spinning (MAS) NMR, in which a tissue sample is spun about an axis at 54.7° to the direction of the magnetic field, improves the resolution of NMR spectra by eliminating the effect of dipolar couplings and chemical shift anisotropies, and reducing anisotropic magnetic susceptibility (Vaidya et al. 2005). NMR data is compressed and simplified to generate a segmentation of each NMR spectrum containing approximately 250 intensity values each. These metabolic descriptors are then analyzed by pattern recognition/multivariate statistical software, which can classify the data by toxin type and dose using principal components analysis (PCA) or partial least squares discriminant analysis (PLSDA) (Vaidya et al. 2005). These data can also be displayed over a time course to better visualize the physiologic response to a toxicant.
In the past three years, gas chromatography (GC)- and liquid chromatography (LC)-, and hydrophilic interaction chromatography (HILIC)-mass spectrometry (MS) have increasingly been used for screening over NMR, due to their improved speed, sensitivity, and specificity. A recent study employed multiple techniques, including GC-MS, reversed phase (RP)-LC-MS, and HILIC-MS, to determine the urinary metabolite profile of a small clinical cohort comprising healthy patients (n=6) and patients with renal cell carcinoma (RCC, n=6). Using 50 important features from profiles derived from any single technique, the authors were able to differentiate RCC vs. healthy patients. As in proteomics, metabolite MS spectra peaks at these 50 features can be considered grossly as a putative biomarker for RCC (Kind et al. 2007).
2.4 Integrative Systems Biology Software Applications
With so many sources of data available for screening and analysis of potential biomarkers, a need has emerged for comprehensive, accessible databases that integrate multiple data sources and powerful software programs to access them. Even taken alone, “omics” discovery methods are extremely data-intensive and generate reams of information that are not easily distributed or shared via publication in a journal (Howes and Foster 2007). The depth of complexity involved in correlating multiple “omics” datasets (ie. genomics, proteomics and metabolomics) is greater still, but the recent development of open databases like CEBS (Chemical Effects in Biological Systems, http://cebs.niehs.nih.gov) and PhenoGen (http://phenogen.uchsc.edu), along with commercial databases like ArrayTrack and ArrayExpress, has made it easier to synthesize these large data sets with other growing data sources, such as metabolomics (Bhave et al. 2007; Elrick et al. 2006; Xirasagar et al. 2004). A proteome-specific open-source data repository, PrestOMIC, is also being developed that enables researchers to upload and share data with the scientific community via a user-friendly interface (Howes and Foster 2007). The open-source systems biology application, SysBio-OM, integrates information from the CEBS database with other open source projects, including MAGE-OM (Micro-Array Gene Expression Object Model) and PEDRo (Proteomics Experiment Data Repository), to model changes in gene, protein, and metabolite expression and protein-protein interactions following insult (Xirasagar et al. 2004).
SysTox-OM is a more specific application that models changes to the genome, proteome, and metabolome following introduction of a toxicant. Furthermore, it incorporates conventional toxicology information – clinical chemistry, hematology, observations and histopathology – making it possible to evaluate the changes to the phenome across multiple time points and to target specific tissues for analysis (Xirasagar et al. 2006). For example, a user could identify a single phenotype of toxicity, identify all the drugs that are known to result in it, and compare gene and protein expression profiles in the kidney following administration of each drug to look for a common pathway, mechanism, or biomarker. The growth of these open databases and the expansion of the analytical toolkit are critical to the discovery of new biomarkers of toxicity.
3. Technologies for Quantitation
3.1 ELISA
Enzyme-linked immunosorbent assay (ELISA) is the most prevalent method of measuring biomarker concentrations and yields reproducible results in sample volumes as small as 200 µl, assuming that a sample is run in duplicate. Most protein biomarker quantitation assays are based on a “sandwich” principle, whereby the biomarker of interest is sandwiched between a primary antibody immobilized on the surface of a 96- or 384-well plate and a biotinylated secondary antibody. The biotin moiety is then bound by streptavidin linked to horseradish peroxidase or alkaline phosphatase, which reliably converts a colorless substrate to a colored product that can be quantitated by measuring its absorbance at a specific wavelength. The drawbacks of the ELISA assay in the setting of toxicity analysis are chiefly that 1) it can only measure a single biomarker per assay, 2) five to seven hours are required for results, 3) its dynamic range is only reliable across three orders of magnitude, and 4) it requires two highly avid and epitopically distinct antibodies, which limits the speed and affordability of development of assays for emerging biomarkers.
FACTT (Fluorescent Amplification Catalyzed by T7 polymerase Technique) ELISA can achieve subfemtomolar sensitivity (~0.08fM) by coupling a dsRNA amplification module to streptavidin, which then binds through biotin to the sandwiched antigen. T7 RNA polymerase amplifies the RNA, which is visualized by the fluorescent intercalating dye, RiboGreen (Zhang et al. 2006). Fluorescence intensity of the amplified product is proportional to the amount of antigen originally present in the sample. While this technique increases the range of the conventional ELISA, it increases the overall assay time to 8–11 hours (Zhang et al. 2006). Neither FACCT nor traditional ELISA address the emerging need for multiplexing capability – the ability to detect several different biomarkers simultaneously in the same sample. The final drawback of this and several other assay systems discussed here are its unsuitability for the point-of-care monitoring of toxicity that is needed for the safe treatment of patients undergoing chemotherapy, antibiotic regimens, and other potentially toxic courses of therapy.
3.2 Solid-Phase ELISA Variants
Several variations on the traditional ELISA have emerged in recent years to address the need for detection of multiple analytes in the same biological sample. FAST Quant® utilizes multiple capture antibodies microspotted on a nitrocellulose slide to achieve multiplexing capability (up to 9 analytes), and uses streptavidin-Cy5 to enable fluorescence detection of the antigen-antibody complex (Whatman, Kent, UK). An overnight incubation is required, however, which far exceeds the assay time of comparable ELISA-based multiplex systems.
MesoScale® uses electrochemiluminescence to visualize antigen concentration. A trapping antibody is adsorbed to a carbon electrode in a microwell, which binds antigen in a sample via traditional immunochemistry (MesoScale, Gaithersburg, MD). A rubidium-labeled detection antibody (Ru(bpy)32+) is used in combination with an enhancer, tripropylamine, which emits light at 620 nm when excited by an electrical impulse from the carbon electrode. Up to 10 analytes can be detected in a single sample by this method.
Searchlight® employs microspotted capture antibodies on a 96-well plate. Antigen is trapped by the spotted antibodies, then bound to a biotinylated secondary antibody specific to a distinct antigenic epitope (Thermo Fisher Scientific, Waltham, MA). The antigen-antibody complex is finally bound to streptavidin-horseradish peroxidase and is visualized using a chemiluminescent substrate, luminol. Up to 16 analytes can be detected in a single sample by this method. An infrared detection method is also available in which streptavidin-DyLight™ 800 binds to the antigen-antibody complex, enabling the detection of up to 24 analytes in a single sample.
Microspot ELISA-based assays like FAST Quant®, Searchlight®, and MesoScale® reduce the sample volume necessary for duplicate quantitation to 50–100 µl and provide multiplexing capability for measurement of up to 24 analytes per well. Assay time is reduced to three to four hours for MesoScale® and Searchlight® and the dynamic range of detection is increased to 4 orders of magnitude, making these assay systems ideal for pre-clinical biomarker validation and large cohort clinical studies, but not for bedside monitoring.
3.3 Luminex
Luminex is a microsphere-based assay system that combines FACS sorting, counting, and microfluidics with a solution-phase variant of the ELISA ‘sandwich’ principle, yielding reproducible results in sample volumes as small as 60 µl. A primary antibody is bound via its N-terminus to a carboxyl group on a polystyrene microsphere. The microsphere contains a combination of red and orange fluorochromes, identifiable by a 635 nm laser, that gives each set of microspheres a unique spectral ID. The biomarker bound by this microsphere-antibody complex is then incubated with a biotinylated secondary antibody. Biotin reliably binds to the final reactant – streptavidin-phycoerythrin – which can be quantitated by exciting phycoerythrin with a 572 nm laser and measuring mean fluorescence intensity. By combining in a single well several unique sets of beads, each bound to a distinct primary antibody, as many as 27 biomarkers have been measured in the same sample.
This technology reduces overall assay time to under four hours, can measure multiple biomarkers simultaneously in the same sample, and has a dynamic range comprising 5 orders of magnitude. However, discrete antibody pairs are still required and, while the assay is valuable for pre-clinical and clinical high throughput toxicity evaluation, the large size and high cost of the Luminex equipment make it unsuitable for bedside monitoring.
3.4 Nanowire
Although the technology is still in the early stages of development, silicon nanowires with a primary antibody covalently bound to their surface have been shown capable of detecting biomarkers by registering a change in conductance proportional to the amount of antigen bound (Patolsky et al. 2006). The sample can be passed over an array of nanowires, each bearing an antibody to a distinct protein, to multiplex the assay (Zheng et al. 2005). Once loaded onto the nanowire, a quantitative read-out is available in minutes. Zheng et al. have used a multiplex nanowire array to simultaneously detect three cancer biomarkers, prostate specific antigen (PSA), carcinoembryonic antigen (PSA-ACT), and mucin-1, in the femtomolar range (Zheng et al.).
The technology has been validated for proteins in solution and pretreated (desalted) serum, but its utility may be affected in biofluids like urine or cerebrospinal fluid for which differences in pH and electrolyte concentrations induce changes in ionic conductance. This could have a confounding effect when measuring biomarker concentrations, as nonspecific conductance changes create a high signal-to-noise ratio and compromise the sensitivity of the assay. As with serum, it may be possible to pretreat such samples, for example, by passing them through a size exclusion column.
Although the conductance change must be externally amplified by bulky equipment and as such is sensitive to the placement of external leads, nanowire technology is a potential candidate for bedside adaptation due to the small size of the nanowire array and relatively inexpensive fabrication cost.
3.5 Patterned paper
A novel type of paper patterned with a hydrophobic polymer has been developed by the Whitesides group as a low-cost, high-throughput technology for the detection of multiple biomarkers in sample volumes as low as 5 µl (Martinez et al. 2007). Chromatography paper is soaked in SU-8 2010 photoresist, a hydrophobic, UV-sensitive polymer. An opaque mask is laid over the paper, which is then exposed to UV light. After developing and washing with isopropanol, a hydrophobic pattern remains in the shape of the overlay, which is used to direct the flow of the sample along the hydrophilic paper to regions within the overlay that have been spotted with enzymatic detecting reagents. Although this technology is in the early stages of development, it has already been demonstrated to detect multiple biomarkers (glucose and protein) in a biological sample at levels consistent with currently available assays. The sample is channeled to the testing area by capillary action, preventing the contamination of the area by sediment, dirt, or foreign substances, which are too large to pass through the pores in the paper. Patterned paper preloaded with reagents has been shown to be stable when stored at 4°C for up to 15 days (Martinez et al. 2007).
Although this technology has not yet been validated as a quantitative assay, a quantitative method should be forthcoming. Furthermore, patterned paper eliminates many of the current drawbacks of ELISA-based systems. It allows for the rapid (10–11 minutes), inexpensive detection of multiple biomarkers in a single 5 µl drop of sample and requires no external equipment or electricity to operate, making it a potential bedside technology for determining whether toxicity thresholds have been exceeded.
4. Summary
Although ELISA is a powerful method of quantitating a protein present in serum, urine, and other biofluids, its utility is limited by its ability to identify only one protein at a time. Recently developed commercial ELISA-based platforms expand the range of this technology to detect up to 24 distinct antibodies within each well, allowing for detection of multiple analytes in a single sample. Luminex® technology expands the range of analytes measured in a single sample to as many as 27 by using microspheres in solution phase as the solid support for trapping antibodies. These assays are high-throughput and offer detection levels as low as the femtomolar range, but they are currently unsuitable for development of a bedside assay of toxicity. Nanowire-based assays are an attractive direction for future development in this area, although several technical hurdles must still be overcome before the assay can be sufficiently miniaturized. Patterned paper, while not yet validated as a quantitative tool, is the only assay system reviewed here capable of biomarker detection at the bedside. Further studies using non-enzymatic reaction chemistry (ie. antibody-based detection) must still be done to prove its efficacy.
The data generated from these assay systems and “-omics” discovery technologies have limited usefulness if they stand alone. The advent of microarrays and protein chips has made gleaning information from biological systems extraordinarily easy and quick, but this mountain of data has not yet been mined. Integration of information from these sources using powerful analytical computing platforms, open-source databases, and advances in technology thereof represent the next phase in the identification and qualification of novel biomarkers of toxicity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Fitz B. Collings, Brigham and Women’s Hospital, Harvard Medical School, Harvard Institutes of Medicine, Renal Division, Rm 550, 4 Blackfan Circle, Boston, MA 02115, Tel: 617-525-5974, Fax: 617-525-5965, Email: fcollings@partners.org.
Vishal S. Vaidya, Brigham and Women’s Hospital, Harvard Medical School, Harvard Institutes of Medicine, Renal Division, Rm 550, 4 Blackfan Circle, Boston, MA 02115, Tel: 617-525-5974, Fax: 617-525-5965, Email: vvaidya@partners.org.
References
- Tugwood JD, Hollins LE, Cockerill MJ. Genomics and the search for novel biomarkers in toxicology. Biomarkers. 2003;8:79–92. doi: 10.1080/1354750031000070103. [DOI] [PubMed] [Google Scholar]
- Challenge and opportunity on the critical path to new medical products. 2004 US FDA http//www.fda.gov/oc/initiatives/criticalpath/whitepaper.html.
- Wagner JA, Williams SA, Webster CJ. Biomarkers and surrogate end points for fit-for-purpose development and regulatory evaluation of new drugs. Clin Pharmacol Ther. 2007;81:104–107. doi: 10.1038/sj.clpt.6100017. [DOI] [PubMed] [Google Scholar]
- Arcellana-Panlilio M, Robbins SM. Cutting-edge technology. I. Global gene expression profiling using DNA microarrays. Am J Physiol Gastrointest Liver Physiol. 2002;282:G397–G402. doi: 10.1152/ajpgi.00519.2001. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. DNA microarrays: translation of the genome from laboratory to clinic. Lancet Neurol. 2003;2:275–282. doi: 10.1016/s1474-4422(03)00379-x. [DOI] [PubMed] [Google Scholar]
- Ishkanian AS, Malloff CA, Watson SK, DeLeeuw RJ, Chi B, Coe BP, Snijders A, Albertson DG, Pinkel D, Marra MA, Ling V, MacAulay C, Lam WL. A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet. 2004;36:299–303. doi: 10.1038/ng1307. [DOI] [PubMed] [Google Scholar]
- Hubank M, Schatz DG. Identifying differences in mRNA expression by representational difference analysis of cDNA. Nucleic Acids Res. 1994;22:5640–5648. doi: 10.1093/nar/22.25.5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem. 1998;273:4135–4142. doi: 10.1074/jbc.273.7.4135. [DOI] [PubMed] [Google Scholar]
- Elrick MM, Walgren JL, Mitchell MD, Thompson DC. Proteomics: recent applications and new technologies. Basic Clin Pharmacol Toxicol. 2006;98:432–441. doi: 10.1111/j.1742-7843.2006.pto_391.x. [DOI] [PubMed] [Google Scholar]
- Issaq HJ, Conrads TP, Prieto DA, Tirumalai R, Veenstra TD. SELDI-TOF MS for diagnostic proteomics. Anal Chem. 2003;75:148A–155A. [PubMed] [Google Scholar]
- Conrads TP, Fusaro VA, Ross S, Johann D, Rajapakse V, Hitt BA, Steinberg SM, Kohn EC, Fishman DA, Whitely G, Barrett JC, Liotta LA, Petricoin EF, 3rd, Veenstra TD. High-resolution serum proteomic features for ovarian cancer detection. Endocr Relat Cancer. 2004;11:163–178. doi: 10.1677/erc.0.0110163. [DOI] [PubMed] [Google Scholar]
- Li J, White N, Zhang Z, Rosenzweig J, Mangold LA, Partin AW, Chan DW. Detection of prostate cancer using serum proteomics pattern in a histologically confirmed population. J Urol. 2004;171:1782–1787. doi: 10.1097/01.ju.0000119823.86393.49. [DOI] [PubMed] [Google Scholar]
- Vlahou A, Laronga C, Wilson L, Gregory B, Fournier K, McGaughey D, Perry RR, Wright GL, Jr, Semmes OJ. A novel approach toward development of a rapid blood test for breast cancer. Clin Breast Cancer. 2003;4:203–209. doi: 10.3816/cbc.2003.n.026. [DOI] [PubMed] [Google Scholar]
- Xiao X, Liu D, Tang Y, Guo F, Xia L, Liu J, He D. Development of proteomic patterns for detecting lung cancer. Dis Markers. 2003;19:33–39. doi: 10.1155/2003/278152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukov TA, Johanson RA, Cantor AB, Clark RA, Tockman MS. Discovery of distinct protein profiles specific for lung tumors and pre-malignant lung lesions by SELDI mass spectrometry. Lung Cancer. 2003;40:267–279. doi: 10.1016/s0169-5002(03)00082-5. [DOI] [PubMed] [Google Scholar]
- Koopman LA, Szuhai K, van Eendenburg JD, Bezrookove V, Kenter GG, Schuuring E, Tanke H, Fleuren GJ. Recurrent integration of human papillomaviruses 16, 45, and 67 near translocation breakpoints in new cervical cancer cell lines. Cancer Res. 1999;59:5615–5624. [PubMed] [Google Scholar]
- Rosty C, Christa L, Kuzdzal S, Baldwin WM, Zahurak ML, Carnot F, Chan DW, Canto M, Lillemoe KD, Cameron JL, Yeo CJ, Hruban RH, Goggins M. Identification of hepatocarcinoma-intestine-pancreas/pancreatitis-associated protein I as a biomarker for pancreatic ductal adenocarcinoma by protein biochip technology. Cancer Res. 2002;62:1868–1875. [PubMed] [Google Scholar]
- Melle C, Kaufmann R, Hommann M, Bleul A, Driesch D, Ernst G, von Eggeling F. Proteomic profiling in microdissected hepatocellular carcinoma tissue using ProteinChip technology. Int J Oncol. 2004;24:885–891. [PubMed] [Google Scholar]
- Poon TC, Yip TT, Chan AT, Yip C, Yip V, Mok TS, Lee CC, Leung TW, Ho SK, Johnson PJ. Comprehensive proteomic profiling identifies serum proteomic signatures for detection of hepatocellular carcinoma and its subtypes. Clin Chem. 2003;49:752–760. doi: 10.1373/49.5.752. [DOI] [PubMed] [Google Scholar]
- Mazzatti DJ, Pawelec G, Longdin R, Powell JR, Forsey RJ. SELDI-TOF-MS ProteinChip array profiling of T-cell clones propagated in long-term culture identifies human profilin-1 as a potential bio-marker of immunosenescence. Proteome Sci. 2007;5:7. doi: 10.1186/1477-5956-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chensong Pan SX, Houjiang Zhou, Yu Fu, Mingliang Ye, Hanfa Zou. Recent developments in methods and technology for analysis of biological samples by MALDI-TOF-MS. Analytical and Bioanalytical Chemsitry. 2006;387:193–204. doi: 10.1007/s00216-006-0905-4. [DOI] [PubMed] [Google Scholar]
- Kong XL, Huang LC, Hsu CM, Chen WH, Han CC, Chang HC. High-affinity capture of proteins by diamond nanoparticles for mass spectrometric analysis. Anal Chem. 2005;77:259–265. doi: 10.1021/ac048971a. [DOI] [PubMed] [Google Scholar]
- Xu S, Li Y, Zou H, Qiu J, Guo Z, Guo B. Carbon nanotubes as assisted matrix for laser desorption/ionization time-of-flight mass spectrometry. Anal Chem. 2003;75:6191–6195. doi: 10.1021/ac0345695. [DOI] [PubMed] [Google Scholar]
- Thongboonkerd V, Songtawee N, Sritippayawan S. Urinary proteome profiling using microfluidic technology on a chip. J Proteome Res. 2007;6:2011–2018. doi: 10.1021/pr060586+. [DOI] [PubMed] [Google Scholar]
- Zhuang Z, Starkey JA, Mechref Y, Novotny MV, Jacobson SC. Electrophoretic analysis of N-glycans on microfluidic devices. Anal Chem. 2007;79:7170–7175. doi: 10.1021/ac071261v. [DOI] [PubMed] [Google Scholar]
- van Ravenzwaay B, Cunha GC, Leibold E, Looser R, Mellert W, Prokoudine A, Walk T, Wiemer J. The use of metabolomics for the discovery of new biomarkers of effect. Toxicol Lett. 2007;172:21–28. doi: 10.1016/j.toxlet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Vaidya VS, Nicholson JK, Mehendale HM. Metabonomics. Encyclopedia of Toxicology. 2005:41–46. [Google Scholar]
- Kind T, Tolstikov V, Fiehn O, Weiss RH. A comprehensive urinary metabolomic approach for identifying kidney cancerr. Anal Biochem. 2007;363:185–195. doi: 10.1016/j.ab.2007.01.028. [DOI] [PubMed] [Google Scholar]
- Howes CG, Foster LJ. PrestOMIC, an open source application for dissemination of proteomic datasets by individual laboratories. Proteome Sci. 2007;5:8. doi: 10.1186/1477-5956-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave SV, Hornbaker C, Phang TL, Saba L, Lapadat R, Kechris K, Gaydos J, McGoldrick D, Dolbey A, Leach S, Soriano B, Ellington A, Ellington E, Jones K, Mangion J, Belknap JK, Williams RW, Hunter LE, Hoffman PL, Tabakoff B. The PhenoGen Informatics website: tools for analyses of complex traits. BMC Genet. 2007;8:59. doi: 10.1186/1471-2156-8-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xirasagar S, Gustafson S, Merrick BA, Tomer KB, Stasiewicz S, Chan DD, Yost KJ, 3rd, Yates JR, 3rd, Sumner S, Xiao N, Waters MD. CEBS object model for systems biology data, SysBio-OM. Bioinformatics. 2004;20:2004–2015. doi: 10.1093/bioinformatics/bth189. [DOI] [PubMed] [Google Scholar]
- Xirasagar S, Gustafson SF, Huang CC, Pan Q, Fostel J, Boyer P, Merrick BA, Tomer KB, Chan DD, Yost KJ, 3rd, Choi D, Xiao N, Stasiewicz S, Bushel P, Waters MD. Chemical effects in biological systems (CEBS) object model for toxicology data, SysTox-OM: design and application. Bioinformatics. 2006;22:874–882. doi: 10.1093/bioinformatics/btk045. [DOI] [PubMed] [Google Scholar]
- Zhang H, Cheng X, Richter M, Greene MI. A sensitive and high-throughput assay to detect low-abundance proteins in serum. Nat Med. 2006;12:473–477. doi: 10.1038/nm1378. [DOI] [PubMed] [Google Scholar]
- Patolsky F, Zheng G, Lieber CM. Fabrication of silicon nanowire devices for ultrasensitive, label-free, real-time detection of biological and chemical species. Nat Protoc. 2006;1:1711–1724. doi: 10.1038/nprot.2006.227. [DOI] [PubMed] [Google Scholar]
- Zheng G, Patolsky F, Cui Y, Wang WU, Lieber CM. Multiplexed electrical detection of cancer markers with nanowire sensor arrays. Nat Biotechnol. 2005;23:1294–1301. doi: 10.1038/nbt1138. [DOI] [PubMed] [Google Scholar]
- Martinez AW, Phillips ST, Butte MJ, Whitesides GM. Patterned paper as a platform for inexpensive, low-volume, portable bioassays. Angew Chem Int Ed Engl. 2007;46:1318–1320. doi: 10.1002/anie.200603817. [DOI] [PMC free article] [PubMed] [Google Scholar]