

Abstract
Background
Statins are shown to have cholesterol-independent properties such as anti-inflammation and immunomodulation. Activated hepatic stellate cells (HSCs) acquire the capacity to synthesize matrix proteins in damaged liver. We tested the hypothesis that atorvastatin may be capable of inducing apoptosis in HSCs.
Methods
Primary cultures of rat HSCs were exposed to atorvastatin, mevalonic acid and U0126. Quantification of living, apoptotic and necrotic HSCs was performed by flow cytometry and laser-scan microscopy. Cell-cycle analysis was performed by flow cytometry. Pro- and anti-apoptotic factors were investigated by Western blot and electrophoresis mobility shift assay. Protease activity of caspases was calculated using a colorimetric kit.
Results
Atorvastatin leads to a G2-arrest and induces apoptosis in activated HSCs. Atorvastatin-mediated apoptosis could be blocked by co-administration of mevalonic acid and U0126. No effects of atorvastatin on gene expression of CD95, CD95L, NF-κB, p53 and p21WAF1 could be observed. Atorvastatin-induced apoptosis in activated HSCs is related to an increased protease activity of caspase-9 and -3. Gene expression of the major proteins of the bcl-system shows that truncated Bid is involved in apoptosis mediated by atorvastatin. By blocking the extracellular signal-regulated protein kinase (ERK1/2) activation by adding U0126, we could prevent the apoptosis induced by atorvastatin. By Western blot we could not detect any change in the activation of c-jun N-terminal kinase (JNK).
Conclusions
Atorvastatin induces apoptosis in activated HSCs acting through an ERK-dependent cleavage of Bid and a highly increased protease activity of caspase-9 and -3. JNK is not involved in atorvastatin-mediated apoptosis in HSCs.
Keywords: atorvastatin, apoptosis, hepatic stellate cells
Hepatic stellate cells (HSCs) are one of the mesenchymal cell populations involved in liver fibrosis and may become the main actor in certain forms of cirrhosis like haemochromatosis (1) or alcoholic cirrhosis (2). Liver fibrosis can be classified as a wound healing response to a variety of chronic stimuli that is characterized by an excessive deposition of extracellular matrix (ECM), of which type I and type III collagen predominate. At present, liver fibrogenesis is considered a dynamic process involving complex cellular and molecular mechanisms, resulting from the chronic activation of the tissue repair mechanisms, which follows recurring liver tissue injury. If the chronic damage persists, inflammation and fibrosis can progress to liver cirrhosis, ultimately leading to organ failure and death. During recovery from liver fibrosis in the rat carbon tetrachloride or in the bile duct ligation model, there is a reduction in the number of α-smooth muscle actin (SMA)-positive cells. Further findings strongly suggested that apoptosis of activated HSCs is responsible for mediating cell loss during recovery from fibrosis (3–6). Evolving antifibrotic therapies should therefore focus on treatments of activated HSCs.
3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors, or statins, are widely used in the clinic for their cholesterol-lowering properties. Although the benefits of lowering cholesterol levels in the prevention and treatment of cardiovascular diseases are proven, several clinical trials have shown that statins also have cholesterol-independent properties (7–14), including improving endothelial function (15–18), enhancing plaque stability (19, 20), decreasing vascular inflammation and oxidative stress, inhibiting the thrombogenic response in the vascular wall, and immunomodulatory properties by inhibiting expression of MHC class II proteins (21). These ‘pleiotropic’ effects can be attenuated by addition of the postreductase product, mevalonate (22), and are related to inhibition of the production of other isoprenoids such as farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP) (23).
Although the impact of statin therapy on each of these processes is not fully understood, ongoing studies are likely to shed further light on the potential pleiotropic benefits of statins, some of which are focusing on ECM-producing cells to evaluate a potential antifibrogenic effect. An in vitro study on rat HSCs treated with two different statins showed that the collagen synthesis by activated cells was decreased (24). To establish whether this effect could be because of a reduction of cellular viability through the induction of apoptosis, we studied the effect of one of the statins of the newest generation, atorvastatin, on the cellular life of rat HSCs.
Material and methods
Hepatic stellate cell isolation, characterization, plating and culture conditions
Wistar rats were provided by Charles River (Sulzfeld, Germany) and maintained under 12:12-h light/dark cycles with food and water ad libitum. In conducting the research described in this report, all animals received humane care in compliance with the institution's guidelines and the National Institute of Health (NIH) Guidelines. HSCs were isolated by sequential in situ perfusion with collagenase and pronase, as previously described (25). A mean of 40 × 106 HSCs were obtained per rat. Cells were plated onto 24-well Falcon plates (Becton Dickinson, Heidelberg, Germany), 35 mm Petri dishes (Greiner, Krefeld, Germany), 96-well Falcon plates (Becton Dickinson) and Lab Tek tissue culture slides (Nunc, Naperville, IL, USA) with a density of 30 000 cells/cm2. Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% foetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin and 1%l-glutamine. Culture medium was replaced at day 2 after plating and then every other day. Cells were kept in culture at 37 °C in a 5% CO2 atmosphere and 100% humidity.
To evaluate the purity of the cultures, HSCs were tested by immunofluorescence at day 0, day 2 (quiescent/early activated HSC) and day 7 (activated HSC) after plating as described previously. Contamination with Kupffer cells (ED1 positive) was <2%, and neither endothelial cells nor hepatocytes were detected (26–31). With the use of SMA immunoreactivity as an activation parameter (32), HSCs were fully activated after 7 days of primary culture (100% SMA positive). Fibulin-2-positive cells (liver myofibroblasts) were always <1% (33, 34). HSCs at days 2, 4 and 7 of primary culture were washed two times with Gey's balanced salt solution and incubated for 20 h in serum-reduced (0.3% FCS) culture medium alone or in the presence of atorvastatin (10−3, 10−5, 10−7, 10−9, 10−11 mol/L) and/or mevalonic acid (125 μM) as well as U0126 (10 μM).
Cell-cycle analysis
5 × 105 cells in 200 μl Ca2+, Mg2+-free phosphate-buffered saline were fixed in 4 ml 70% ethanol/30% phosphate-buffered saline at 0 °C, digested with 1000 U RNAse A (Sigma-Aldrich, St Louis, MO, USA), and stained with 1% propidium iodide at 37 °C for 30 min. The DNA profiles were determined within 4 h of staining by flow cytometry (EPICS ML, Coulter, Kerfeld, Germany) (35). Data were analysed using the program multicycle for Windows Ver. 3.0 (Phoenix Flow Systems, San Diego, CA, USA).
Flow cytometric quantification of living, apoptotic and necrotic hepatic stellate cells
To quantify apoptotic cells, flow cytometry was used after trypsination of the HSCs (EPICS ML, Coulter). To detect early apoptotic changes, staining with annexin V–fluorescein isothiocyanate (FITC) was used, because of its known high affinity to phosphatidylserine (36). Phosphatidylserine is normally situated on the inner leaflet of the plasma membrane. In the course of cell death, phosphatidylserine is translocated to the outer layer of the membrane (37) (i.e. the external surface of the cell). This occurs in the early phases of apoptosis, while the cell membrane itself remains intact. In contrast to apoptosis, necrosis is accompanied by loss of cell membrane integrity and leakage of cellular constituents into the environment. To distinguish apoptosis and necrosis, propidium iodide, a common dye exclusion test, and annexin V–FITC were used in parallel to show membrane integrity after annexin V–FITC binding to cells. In all investigated cases, no loss of membrane integrity was observed within 30 min after annexin V–FITC binding was detected.
The fluorescence from endogenous vitamin A was digitally removed using unstained cell samples as negative controls. In addition, we performed a staining with Tdt-mediated X-dUTP nick-end labelling (TUNEL) according to the protocol of the manufacturer (Boehringer, Mannheim, Germany) evaluated using confocal microscopy.
Confocal laser scan microscopic detection of apoptosis in activated hepatic stellate cells using the mitochondrial membrane sensor kit
To investigate induction of apoptosis, blocking apoptosis and apoptosis occurring spontaneously at days 2 and 7, we performed a test using confocal laser scan microscopy (Zeiss, Oberkochen, Germany) in the time-scan mode. HSCs at the seventh day (d7) after plating were incubated for 8 and 20 h in reduced serum medium with atorvastatin, with and without mevalonate, with mevalonate (125 μM) alone, with or without U0126 and with U0126 alone (10 μM). Control wells were left untreated. Apoptosis was detected using the ApoAlert Mitochondrial Membrane Sensor Kit (BD Biosciences, Clonthec, San Jose, CA, USA). This is a fluorescence-based assay where the dye is able to accumulate in the intact mitochondria. In case of apoptosis, it aggregates in the cytosol and alters the fluorescence colour. By this change in the fluorescence colour it is possible to discriminate between apoptotic (green) and living (red) cells.
Western blot analysis
Cells on different days after plating were lysed in hot Laemmli buffer (95 °C) and processed by sodium dodecyl sulphate-polyacrilamide gel electrophoresis under reducing conditions according to Laemmli (38). The protein content of cellular lysates was calculated using the Coomassie Protein Assay (Pierce, Rockford, IL, USA); β-actin was used as loading control. Proteins were transferred onto Hybond-ECL nitrocellulose hybridization transfer membranes according to Towbin et al. (39). Immunodetection was performed according to the ECL Western blotting protocol. Primary antibodies were used at a 1:200 dilution. Anti-mouse and anti-rabbit immunoglobulins were each used at a 1/1000 dilution. Densitometric evaluation of the blots was performed using the program scion image Ver. Beta 2 (NIH).
Electrophoresis mobility shift assay
After appropriate treatment times, cells were trypsinized and washed once with 1 ml Tris-buffered saline (20 mmol/L) and Tris HCl (pH=7.2, 0.15 mol/L NaCl). Then cells were resuspended in 0.5 ml extraction buffer (10 mmol/L HEPES pH=7.9, 10 mmol/L KCl, 1.5 mmol/L MgCl2, PMSF 1 mmol/L) and incubated for 15 min on ice. This was followed by the addition of 30 μl NP-40 and vortexing for 10 s. The pellet was harvested with a brief centrifugation and suspended in 50 μl of the same extraction buffer containing 0.4 mol/L NaCl. Cell nuclear regulatory proteins were extracted with vigorous agitation on ice for 15 min. DNA was pelletted with centrifugation (5 min, 4 °C, 15 000 g). The supernatant was taken for electrophoresis mobility shift assay (EMSA) reaction. The protein concentration was determined with Coomassie Assay (Pierce). The labelling was done with T4 polynucleotide kinase. This enzyme cuts the last phosphate from adenosine triphosphate (ATP) and links it to any polynucleotide: in our case to p53 (Santa Cruz Biotech, Santa Cruz, CA, USA) or to NF-κB-α (Promega, Madison, WI, USA) oligonucleotides. EMSA (p53 and NF-κB) and supershift (NF-κB) reactions were done in 20 μl reaction mixture based on the extraction buffer with NaCl containing 10 μg nuclear extract 105 cpm labelled oligonucleotides and in the case of supershift we used 4 μg of rabbit polyclonal antibody against the p65-NF-κB (Transcruz, Santa Cruz Biotechnology). The binding reaction was performed at 4 °C overnight. DNA–protein complexes were resolved by electrophoresis through a 4% polyacrilamide gel containing 45 mM TRIS-borate and 1 mM ethylene diamine tetraacetic acid (EDTA) at pH 8.0. Gel was dried and exposed to X-ray film overnight.
Colorimetric assay of the protease activity of caspase-3, -8 and -9
Measurement was performed using a kit for each caspase (Calbiochem-Novabiochem Corporation, San Diego, CA, USA), in accordance with the manufacturer's instructions. The kits work on the properties of a colorimetric substrate which, when cut, increase light absorption at 405 nm. The kits contain a specific inhibitor of the colorimetric substrate as a negative control. For these experiments, cells were plated onto 96-well plates and analysed at day 7 after culture, after treatment with atorvastatin and/or mevalonic acid (125 μM), with and without U0126 (10 μM). Control wells were left untreated.
Statistical analysis
Results are expressed as mean±SD, and the significance of the difference between the means was assessed by anova followed by Bonferroni's post-test.
Results
Effect of atorvastatin on the cell cycle of isolated rat hepatic stellate cells
Primary isolated HSCs at the second day after plating (d2) were used as a model for quiescent HSCs, while cells at the d7 after plating were used as a model for activated HSCs. In activated HSCs (d7) treated with atorvastatin (10−9 mol/L), there is an increase of cells in phase G2 (from 61±3.2 to 75±4.1%, P < 0.05) while the percentage of cells in the S and G0/G1 phase decreased (from 17±2.1 to 11±3.1% and from 21±2.7 to 14±1.8%, respectively, P < 0.05). In quiescent HSCs treated with atorvastatin, the percentage of cells in the G0/G1 phase is strongly reduced compared with the control (from 65±5.3 to 21±3.6%, P < 0.01) while the percentage of cells in the G2 phase increased (from 31±4.2 to 72±4.3%, P < 0.01). The percentage of cells in the S phase remains unchanged (Fig. 1). The decrease of the portion of cells being in the S phase of cell cycle could also be shown by 3H-thymidine incorporation in both quiescent and activated HSCs. The effects are dose dependent and first detectable after 20 h.
Fig. 1.
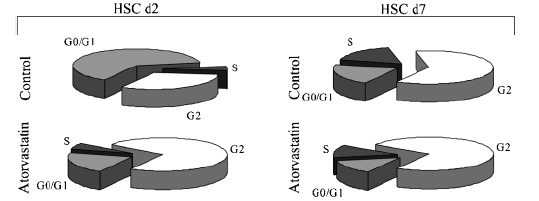
Cell cycle analysis of hepatic stellate cells (HSCs) treated with atorvastatin. In quiescent HSCs the percentage of cells in the S phase remains unchanged from the control. The percentage of cells treated with atorvastatin being in the G2 phase increased, while the percentage of cells being in the G0/G1 phase was reduced. In activated HSCs treated with atorvastatin, the percentage of cells in the S and in the G0/G1 phases decreased while an increase of the cells being in the G2 phase occurred. Values presented are mean percentages of seven independent experiments. Values are means +SD.
Apoptosis detection in hepatic stellate cells
To investigate the fate of HSCs, we performed a study using confocal laser scan microscopy and flow cytometry. In activated HSCs atorvastatin is capable of inducing apoptosis, and this effect is dose dependent (Fig. 2a) and first detectable after 20 h. At dosages with physiological serum concentrations of the order 10−7–10−8 mol/L (40), the fraction of cells undergoing apoptosis increased in a dose-dependent manner from a control value of 19 (spontaneous apoptosis) to 60%. Increasing the dose of atorvastatin, the percentage of apoptotic cells increases proportionally up to 92% apoptosis (P < 0.01) at a dose of 10−3 mol/L atorvastatin. At this time point, <2% of total cells were detected as necrotic. Addition of mevalonate or caspase-9 inhibitor as well as U0126 fully prevented the apoptosis induced by atorvastatin (data not shown). In quiescent HSCs, the fraction of cells undergoing apoptosis at dosages with physiologic serum concentrations was not significantly different from the control. At high concentrations (10−3 and 10−5 mol/L) of atorvastatin, the fraction of apoptotic cells increased from a basal level of 4–10% (Fig. 2b). A toxic effect measured as increased portion of necrotic cells could not be observed.
Fig. 2.

Detection of apoptosis induced by atorvastatin on quiescent hepatic stellate cells (HSCs). HSCs at days 2 and 7 of primary culture were incubated for 20 h in serum-reduced (0.3% foetal calf serum) culture medium alone or in the presence of atorvastatin (10−3, 10−5, 10−7, 10−9, 10−11 mol/L). (A) In activated HSCs (day 7), the percentage of cells undergoing apoptosis increased proportionally from a control value of 19% apoptosis (spontaneous apoptosis) with increasing doses of atorvastatin to 60% at physiological serum concentrations (10−7 and 10−8 mol/L) and to 92% apoptosis at a dosage of 10−3 mol/L atorvastatin. (B) The effects of atorvastatin on quiescent HSCs (day 2) was not significant at physiological serum even though at higher concentrations (10−5 and 10−3 mol/L), the fraction of cells in apoptosis increased from a basal level of 4–10%. Data were obtained by fluorescent-activated cell sorting analysis of cells stained with annexin V–fluorescein isothiocyanate and propidium iodide. Values presented are mean percentages of seven independent experiments. Values are means±SD. Level of significance: *P < 0.05; **P < 0.01.
Effect of atorvastatin on the expression of CD95, CD95L, Bax, Bcl-xl, Bcl2, Bid, t-Bid, JNK, p-JNK, ERK1/2 and p-ERK1/2, in isolated rat hepatic stellate cells
The effects of atorvastatin on important members of pro-apoptotic and anti-apoptotic pathways [CD95, CD95L, Bcl-xl, Bcl-2, Bax, Bid, truncated Bid (t-Bid), c-jun N-terminal kinase (JNK), phospho-JNK (p-JNK), extracellular signal-regulated protein kinase (ERK1/2) and phosphorylated-ERK1/2 (p-ERK1/2)] were evaluated in quiescent and activated HSCs by Western blot analysis (see Fig. 3). To establish if the apoptotic effect of atorvastatin on HSC was mediated by the CD95/CD95L pathway, we evaluated the expression of those proteins in quiescent and activated HSCs, in control and in atorvastatin-treated cultures. No changes in the cells treated with atorvastatin compared with the control could be observed. These results suggest that the CD95/CD95L system is not involved in the apoptotic effect of atorvastatin in activated HSCs. Under the same experimental conditions, we evaluated whether the apoptotic effect of atorvastatin was determined by a pro-apoptotic ratio between the apoptosis-promoting (t-Bid, Bax) and apoptosis-inhibiting (Bcl-2, Bcl-xl) member of the bcl system. While there is, as previously described, an increased expression of Bax and a decreased expression of Bcl-2 and Bcl-xl (pro-apoptotic ratio) in activated HSCs compared with the quiescent HSCs, no significant changes between the control and the cells treated with atorvastatin were detectable (Fig. 3A) other than a decrease of Bid, and an increased occurence of t-Bid, as well as of p-ERK1/2) in activated HSCs treated over 20 h with atorvastatin. This effect could be blocked by adding mevalonate and U0126 (Fig. 3B).
Fig. 3.

Western blot analysis of Bcl-xl, Bcl-2, Bax, truncated Bid (t-Bid), c-jun N-terminal kinase (JNK), phospho-JNK (p-JNK), extracellular signal-regulated protein kinase (ERK1/2), phospho-ERK1/2 (p-ERK1/2), CD95 and CD95L. (A) Sodium dodecyl sulphate-polyacrilamide gel electrophoresis of HSC cell lysates at days 2 and 7 of control cultures and cultures treated over 6 and 20 h with atorvastatin 10−7 mol/L, with and without U0126. Densitometry was performed for the different lanes and normalized on the repective β-actin loading control. Values for the controls at day 2 were set as 100%. (A). An increased expression of Bax and a decreased expression of Bcl-2 and Bcl-xl (pro-apoptotic ratio) in activated hepatic stellate cells (HSCs) compared with the quiescent HSCs were detected. However, no significant changes between the control and the cells treated with atorvastatin were noted. In activated HSCs compared with the quiescent cells, a strongly increased expression of CD95 and a significantly increased expression of CD95L were obtained; however, no changes between the control cells and the cells treated with atorvastatin were noted. (B) On the other hand an increased portion of p-ERK1/2 and t-Bid but not of p-JNK in activated HSCs treated over 20 h with atorvastatin could be observed. This effect could be blocked by adding ERK1/2 inhibitor U0126 as well as by adding mevalonate. We could confirm the data in five different blots of five independent experiments.
Effect of atorvastatin on the expression of NF-κB and p53 in isolated rat hepatic stellate cells
Nuclear factor-κB and p53 are ubiquitous transcription factors involved in the regulation of the apoptotic process. While the upregulation of NF-κB acts on the bcl system enhancing the expression of the apoptosis-promoting members, the upregulation of the transcription factor p53 promotes apoptosis by arresting cells in phase G2 of the cellular cycle. The EMSA and the Western blot analysis for NF-κB (Figs 4 and 5a) and p53 (Figs 4 and 5b) show that, while their expression is increased in activated HSC compared with the quiescent cells, there are no differences in gene expression between the control and cells treated with atorvastatin.
Fig. 4.
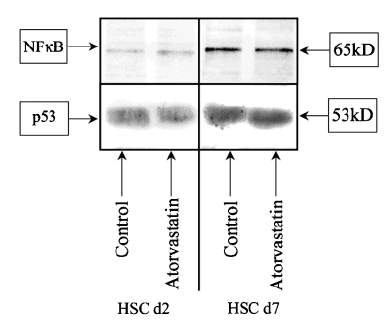
Western blot analysis of nuclear factor (NF)-κB and p53. Sodium dodecyl sulphate–polyacrilamide gel electrophoresis of hepatic stellate cell (HSC) cell lysates at days 2 and 7 of control and atorvastatin 10−7 mol/L. While is possible to detect an increased expression of both nuclear factors in activated HSCs compared with the control, changes between control cells and the cells treated with atorvastatin are not detectable. We could confirm the data in five different blots of five independent experiments.
Fig. 5.
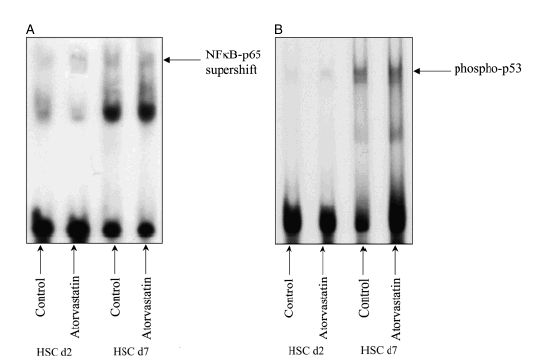
Electrophoretic mobility shift assay to assess nuclear factor (NF)-κB (Fig. 5a, Supershift) and p53 (Fig. 5b) DNA binding activity. Nuclear extracts of hepatic stellate cells (HSCs) at days 2 and 7 in control and in atorvastatin 10−7 mol/L were assessed using a NF-κB and p53 binding site as probe. Please note that in both quiescent and activated HSC, there are no differences between the control and the cells treated with atorvastatin. We could confirm the data in five different blots of five independent experiments.
Activity of caspase-8, -9 and -3
Caspase-3 is a member of the interleukin-1ά converting enzyme (ICE) family of cysteine proteases. It is activated during apoptotic signalling events by upstream proteases including caspase-8 (CD95/CD95L-mediated pathway) and caspase-9 (mitochondrial pathway). Targets of caspase-3 cleavage include poly(ADP-ribose) polymerase (PARP), six nuclear lamins, seven and others that lead to DNA damage and, if no repairing systems are activated, to apoptosis. The protease activity of caspase-8, -9 and -3 was measured in activated HSCs treated with atorvastatin, with and without mevalonate, and in control. As shown in Figure 6, in the cells treated with atorvastatin, the activity of caspase-9 increased to 407%, and the activity of caspase-3 to 540%, while there is no change in the activity of caspase-8. Addition of mevalonate or U0126 fully prevented the atorvastatin-induced increase of caspase-9 and -3 activity. This demonstrates that the pro-apoptotic effect of atorvastatin in HSC is specific and may be mediated by the ERK-dependent mitochondrial pathway, with further activation of the caspase-9 cascade.
Fig. 6.

Percentage changes of caspase-8, -9 and -3 activity. 4 × 106 activated hepatic stellate cells (HSCs) after 20 h incubation with serum-reduced medium control, with atorvastatin 10−7 mol/L, with mevalonic acid 125 mmol, with caspase-9 inhibitors and with atorvastatin and mevalonic acid. Values for HSCs control were set at 100%. In cells treated with atorvastatin, the activity of caspase-9 increased to 407% and the activity of caspase-3 increased to 540%, while in cells treated with mevalonic acid, caspase-9 inhibitors and where mevalonic acid was added to atorvastatin, there were no significant differences to the control. The activity of caspase-8 remained unchanged. We could confirm the data in five independent experiments. Values are means±SD. Level of significance: *P < 0.05; **P < 0.01.
Apoptosis detection using the mitochondrial membrane sensor kit
To prove that the activation of caspase-9 follows the mitochondrial pathway of apoptosis, we performed a fluorescence-based assay that enabled detection of the changes that occur in the mitochondrial membrane potential during apoptosis. The data obtained in quiescent and activated HSCs treated with atorvastatin, with and without mevalonic acid, mevalonic acid alone and in control, confirm the data obtained by fluorescent-activated cell sorting. In addition, apoptosis occurring in activated HSCs at the mitochondrial level could be detected 8 h after administration of atorvastatin, whereas first apoptotic changes at nuclear level (TUNEL positivity) occurred after 20 h. The complete prevention of the atorvastatin-induced apoptosis by mevalonic acid demonstrates that this effect is specific (Fig. 7).
Fig. 7.
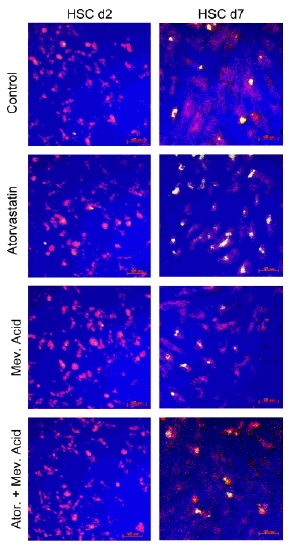
Laser scan microscopy detection of apoptosis in activated hepatic stellate cells (HSCs) by the ApoAlert Mitochondrial Membrane Sensor Kit. At days 2 or 7 of primary culture, cells were incubated for 8 h in serum-reduced (0.3% foetal calf serum) culture medium alone, with atorvastatin 10−7 mol/L, with mevalonic acid 125 μM or with atorvastatin and mevalonic acid. Activated HSCs (d7) treated with atorvastatin, unlike the control and the cells where mevalonate were added, were exhibiting an increase in the green fluorescence owing to the apoptotic changes in the mitochondria. This effect could be inhibited by coadministration of mevalonate. In quiescent HSCs (d2) no significant changes in the fluorescence could be detected among the differently treated cells. We could confirm the data in five independent experiments.
Discussion
In this work we studied the effect of atorvastatin on cell cycle and on the fate of activated rat liver HSCs in primary culture. Atorvastatin dose dependently induces a G2 arrest and an apoptosis rate of 68% at ‘therapeutic’ concentrations in activated HSCs. The apoptotic effect is mediated by the ERK-dependent mitochondrial pathway followed by the increase of caspase-9 activity. This effect can be fully blocked by caspase-9 inhibitors, by ERK-inhibitor U0126 as well as by mevalonic acid.
The HMG-CoA reductase inhibitors are widely used in clinics for their cholesterol-lowering properties. Recent experimental and clinical evidence indicates that some of the beneficial effects of statins are because of their pleiotropic effects, such as anti-inflammation and immunomodulation, encouraging further research aimed at evaluating the potential of statins (7–21, 23).
Activated HSCs are one of the major matrix-producing cell types during liver repair and fibrosis. During recovery from liver fibrosis in the rat carbon tetrachloride and bile duct ligation model of fibrosis, there is a reduction in α-SMA-positive cell number. Further findings strongly suggested that apoptosis of activated HSCs may be responsible for mediating cell loss during recovery from fibrosis (3–6). Evolving antifibrotic therapies are therefore targeted at inhibiting activation of HSCs or in inducing their apoptosis.
In a previous in vitro study, the effects of two different HMG-CoA inhibitors (simvastatin and lovastatin) on rat HSCs were evaluated. The results show that these statins are able to reverse the activation and to inhibit the synthesis of collagen type I and reduce the synthesis of collagen types III and IV. These effects were partially reversible with PDGF and mevalonate (24). We believe that these effects are due to apoptosis induced by statins.
In this study we have shown that atorvastatin, one of the statins of the newest generation, is capable of blocking activated HSCs in the G2 phase of the cellular cycle and of inducing their apoptosis in vitro. The apoptosis induced by atorvastatin is dose dependent in activated HSCs. The fraction of cells undergoing apoptosis increased from a control value of 19% apoptosis (spontaneous apoptosis) proportionally with increasing doses of atorvastatin to 60% at the serum concentration used in clinic (10−7–10−8 mol/L) and to 92% apoptosis at a dose of 10−3 mol/L atorvastatin. In quiescent HSCs, the fraction of cells undergoing apoptosis at the serum concentration used in clinics, does not differ significantly from the control. However, at the dosage used in clinic the concentration of atorvastatin in the tissue remains unknown; nevertheless no toxic effect and no changes at the mitochondrial level could be detected in quiescent HSCs even at higher dosage (10−3 and 10−5 mol/L).
These data suggest that atorvastatin, by up-regulating the apoptosis of activated HSCs, could decrease the extent of fibrosis in chronic liver injury while having no effects on quiescent HSCs. Induction and execution of apoptosis programmes are generally believed to be mediated through a hierarchy of caspase activation. Caspases are synthesized as catalytically inactive pro-enzymes and need to be activated by proteolytic cleavage at internal aspartate residues (41, 42). Two pathways of caspase activation during apoptosis have been described. The first one is mediated by death receptors, such as CD95 or tumour necrosis factor (TNF) receptors, controlled by caspases-8/10, which in turn activate downstream effector caspases such as caspase-3 and -7. Spontaneous apoptosis occurring in activated HSCs involves an increased expression of CD95/CD95L (Fas and Fas-ligand) (3).
We evaluated the expression of those proteins in control and in atorvastatin-treated cultures. Although a strong increase in the expression of CD95 and a significant increase in the expression of CD95L were noted in activated HSCs compared with the control cells, no significant changes between the control cells and the cells treated with atorvastatin were detectable, showing that the CD95/CD95L pathway is not involved in the apoptosis induced by atorvastatin. In the second pathway, diverse apoptotic signals converge at the mitochondrial level, inducing the release of cytochrome C from the mitochondria to the cytosol (the cytochrome C is being released into the cytoplasm following the loss of the mitochondrial transmembrane potential) (43, 44). Once in the cytosol, cytochrome C binds to its cytosolic partner apoptotic protease activating factor-1 (APAF-1) and induces the oligomerization of APAF-1–cytochrome C complex in a dATP/ATP-dependent manner (45, 46). This multimeric complex, named ‘apoptosome’, is sufficient to recruit the initiator caspase, procaspase-9, to the complex and induces procaspase-9 autoactivation (47). The activated caspase-9 is released from the apoptosome and subsequently initiates a caspase cascade involving effector caspases such as caspase-3, -6 and -7 (48, 49).
Once active, these caspases cleave various cellular targets, ultimately leading to cell death. It is known that both the death receptor and the mitochondrial pathway can be interconnected via the activation of the pro-apoptotic Bcl-2 family member, Bid, through caspase-8, possibly serving as an amplification loop (50). The mitochondrial pathway of caspase activation is largely determined by the Bcl-2 family of anti- and pro-apoptotic regulators. These proteins respond to cues from various forms of intracellular stress, such as DNA damage or cytokine deprivation, and interact with opposing family members to determine whether or not the caspase proteolytic cascade should be unleashed (51). Some Bcl-2 family members that are located on the mitochondrial membrane can alter the permeability of the mitochondrial membrane and trigger the activation of caspases. Bid is distinguished from other pro-apoptotic Bcl-2 family members in that it plays a critical role in both receptor- and granule-mediated apoptosis because Bid is a substrate for both active caspase-8 and granzyme B. Active caspase-8 cleaves Bid at aspartic acid residue 60, while granzyme B preferentially cleaves Bid as aspartic acid residue 76. The cleavage product as processed by either protease, termed t-Bid, then translocates to the mitochondria through mechanisms that include selective binding to cardiolipin, a mitochondrial membrane lipid. Once at the mitochondria, t-Bid induces the oligomerization of Bax and Bak, leading to mitochondrial membrane permeability and release of the apoptosome, which activates caspase 9.
We have previously shown that the ERK-dependent activation of the Bcl-2 family can have a pro-apoptotic effect on HSCs (52).
The mitogen-activated protein (MAP) kinases are a family of second-messenger kinases that are essential for transferring signals from the cell surface to the nucleus.
These kinases are a family of serine–threonine protein kinases that are activated in response to a variety of extracellular stimuli. ERK, c-Jun NH1-terminal kinase (JNK) and p38 MAP kinase constitute three major subfamilies of MAP kinases that appear to mediate cellular responses, including proliferation, differentiation and apoptosis. (53)
Extracellular signal-regulated protein kinase plays a major role in cell proliferation and differentiation, as well as in survival mediated by various growth factors (54).
On the other hand, JNK and p38 MAP kinase are activated by various inflammatory cytokines and environmental stressors, and play an important role in the signal cascades leading to the induction of cytokines and chemical mediators (55).
Cellular ERK activation either inhibits or enhances apoptosis in some cells (56, 57). Even in the same cell, the role of ERK activation is likely to differ depending on the cellular insult (56).
Several studies revealed a pathway of apoptosis induced by an ERK-dependent increased activation of the downstream kinases JNK and p38 MAP kinase. On the other hand, other studies have shown that ERK-induced apoptosis may occur with or without activation of JNK (58, 59).
In the present study we have shown the effect of atorvastatin in leading to apoptosis in activated HSCs, and we gained insight into mechanisms of apoptotic response, previously undetected in HSCs.
When we evaluated the expression of some of the pro- and anti-apoptotic members of the Bcl-2 family, in quiescent and activated HSCs, in control and in atorvastatin-treated cultures, the results showed an increased expression of Bax and a decreased expression of Bcl-2 and Bcl-xl (pro-apoptotic ratio) in activated HSCs compared with the quiescent HSCs. Between the control cells and the cells treated with atorvastatin, we could detect an activation of t-Bid in activated HSCs treated over 6 h with atorvastatin, but the activity of caspase-8 remained unchanged, showing that there is no link between the death receptor and the mitochondrial pathway. We followed the MAP kinase and the caspase activation pathways; atorvastatin induces an increased phosphorylation of ERK(1/2), but not its basal expression level. By adding the ERK-inhibitor U0126 and also by adding mevalonic acid to activated HSCs, we could fully block the apoptosis induced by atorvastatin. The increased phosphorylation of ERK(1/2) is not followed by activation of JNK.
In this study we have shown that the pro-apoptotic effect of atorvastatin on HSCs does not involve death receptors such as Fas, while it seems to be mediated by the release of cytochrome C from the mitochondria, induced by an ERK-dependent activation of t-Bid, followed by an increase in the activity of caspase-9 and -3 (Fig. 8).
Fig. 8.

Diagram summarizing the pathway of apoptosis induced by atorvastatin in hepatic stellate cells (HSCs). Atorvastatin induces an increased phosphorylation of extracellular signal-regulated protein kinase (ERK1/2) which is followed by activation and cleavage of Bid into truncated Bid (t-Bid). t-Bid translocates to the mitochondria leading to changes in the mitochondrial membrane permeability (see Fig. 7), which initiates activation of procaspase-9 to caspase-9. The activated caspase-9 is released from the apoptosome and subsequently initiates a caspase cascade involving the effector caspases such as caspase-3. Once active, this caspase cleaves various cellular targets, ultimately leading to DNA damage and apoptosis.
In cells treated with atorvastatin, we could detect apoptotic changes at the mitochondrial level 6 h after beginning the treatment, while the detection of apoptosis with other methods (annexin V-FITC, TUNEL) was first possible after 20 h.
Apoptosis induced by atorvastatin in activated HSCs in culture is fully inhibited by caspase-9 inhibitors, ERK-inhibitor and mevalonate, showing that this effect is specific.
We also evaluated the expression of the transcription factors NF-κB and p53. Activation of NF-κB in cultured HSCs is required for expression of specific genes associated with the activated phenotype and is anti-apoptotic for rat HSCs, while the normal function of p53 is to effect cell cycle arrest at the G1 and G2 phases in response to DNA damage to allow DNA repair and, if repair is not successful, p53 initiates programmed cell death to prevent the propagation of genetic defects to successive generations of cells. Involvement of those proteins in apoptosis regulation in HSCs could be shown for the signals of transforming growth factor (TGF)-α and TNF-α (60). Both transcription factors are increasingly expressed in activated HSCs compared with quiescent cells, but no differences were detectable in atorvastatin-treated cells compared with the control, showing that the increased ERK1/2 phosphorylation and the apoptotic pathway initiated by atorvastatin are not regulated by those transcription factors.
Considering the importance of apoptosis occurring in activated HSCs, these in vitro data suggest that atorvastatin could act not only as an anti-inflammatory agent but also as an antifibrotic agent. On the other hand, other fibrogenic cells (like the resident myofibroblasts and fibroblasts) are involved in liver fibrogenesis. Preliminary observations suggest that liver myofibroblasts show a different response to atorvastatin administration with respect to apoptosis when compared with activated HSCs.
These results suggest atorvastatin as a candidate for further in vitro as well as in vivo study in antifibrotic strategies.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft SFB 402, project C6.
References
- 1.Harada Y, Iwai M, Kakusui M, et al. Activated hepatic stellate cells participate in liver fibrosis in a patient with transfusional iron overload. J Gastroenterol. 1998;33:751–4. doi: 10.1007/s005350050168. [DOI] [PubMed] [Google Scholar]
- 2.Mak KM, Leo MA, Lieber CS. Alcoholic liver injury in baboons: transformation of lipocytes to transitional cells. Gastroenterology. 1984;87:188–200. [PubMed] [Google Scholar]
- 3.Saile B, Knittel T, Matthes N, Scott P, Ramadori G. CD95/CD95L-mediated apoptosis of hepatic stellate cells. Am J Pathol. 1997;151:1265–72. [PMC free article] [PubMed] [Google Scholar]
- 4.Saile B, Matthes N, Knittel T, Ramadori G. TGF-α and TNF-α inhibit both apoptosis and proliferation of activated rat hepatic stellate cells. Hepatology. 1999;30:196–202. doi: 10.1002/hep.510300144. [DOI] [PubMed] [Google Scholar]
- 5.Issa R, Williams E, Trim N, et al. Apoptosis of hepatic stellate cells: involvment in resolution of biliary fibrosis and regulation by soluble growth factors. Gut. 2001;48:548–57. doi: 10.1136/gut.48.4.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iredale JP, Benyon RC, Pickering J, et al. Mechanisms of spontaneous resolution of rat liver injury following release of the mechanical stress. Virchows Arch. 2003;442:372–80. doi: 10.1007/s00428-003-0773-7. [DOI] [PubMed] [Google Scholar]
- 7.Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the scandinavian simvastatin survival study (4S) Lancet. 1994;344:1383–89. [PubMed] [Google Scholar]
- 8.Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333:1301–7. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- 9.Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events trial investigators. N Engl J Med. 1996;335:1001–9. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- 10.Collins R, Peto R, Armitage J. The MRC/BHF heart protection study: preliminary results. Int J Clin Pract. 2002;53:53–6. [PubMed] [Google Scholar]
- 11.Vasa M, Fichtlscherer S, Adler K, et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103:2885–90. doi: 10.1161/hc2401.092816. [DOI] [PubMed] [Google Scholar]
- 12.Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes. The MIRACLE study: a randomized controlled trial. JAMA. 2001;285:1711–8. doi: 10.1001/jama.285.13.1711. [DOI] [PubMed] [Google Scholar]
- 13.Crouse JR, III, Byington RP, Furberg CD. HMG-CoA reductase inhibitor therapy and stroke risk reduction: an analysis of clinical trials data. Atherosclerosis. 1998;138:11–24. doi: 10.1016/s0021-9150(98)00014-8. Review. Erratum in: Atherosclerosis 1998 September; 140(1): 193–4. [DOI] [PubMed] [Google Scholar]
- 14.Kannel WB, Castelli WP, Gordon T, McNamara PM. Serum cholesterol, lipoproteins, and the risk of coronary heart disease. The framingham study. Ann Intern Med. 1971;74:1–12. doi: 10.7326/0003-4819-74-1-1. [DOI] [PubMed] [Google Scholar]
- 15.Multiple risk factor intervention trial research group. Multiple risk factor changes and mortality results. JAMA. 1982;248:1465–77. [PubMed] [Google Scholar]
- 16.Brown BG, Zhao XQ. Importance of endothelial function in mediating the benefits of lipid lowering therapy. Am J Cardiol. 1998;82:49–52T. doi: 10.1016/s0002-9149(98)00726-7. [DOI] [PubMed] [Google Scholar]
- 17.Essig M, Nguyen G, Prie D, Escoubet B, Sraer JD, Friedlander G. 3-hydroxy-3- methylglutaryl-CoA reductase inhibitors increase fibrinolytic activity in rat aortic endothelial cells. Role of geranylgeranylation and Rho proteins. Circ Res. 1998;83:683–90. doi: 10.1161/01.res.83.7.683. [DOI] [PubMed] [Google Scholar]
- 18.Endres M, Laufs U, Huang Z, et al. Stroke protection by 3-hydroxy-3-methylglutaryl(HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1998;95:8880–5. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sparrow CP, Burton CA, Hernandez M, et al. Simvastatin has anti-inflammatory and antiatherosclerotic activities independent of plasma cholesterol lowering. Arterioscler Thromb Vasc Biol. 2001;21:115–21. doi: 10.1161/01.atv.21.1.115. [DOI] [PubMed] [Google Scholar]
- 20.Crisby M, Nordin-Fredriksson G, Shah PK, Yano J, Zhu J, Nilsson J. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases and cell death in human carotid plaques: implications for plaque stabilization. Circulation. 2001;103:276–83. doi: 10.1161/01.cir.103.7.926. [DOI] [PubMed] [Google Scholar]
- 21.Kwan B, Mulhaupt F, Myit S, Mach F. Statins as a newly recognised type of immunomodulator. Nat Med. 2000;6:1399–402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- 22.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 23.Liao JK. Isoprenoids as mediators of the biological effects of statins. J Clin Invest. 2002;110:285–8. doi: 10.1172/JCI16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roumbouts K, Wielant A, Kisanga E, Hellemans K, Schuppan D, Geerts A. Effects of HMG-CoA reductase inhibitors on proliferation and ECM protein synthesis by rat hepatic stellate cells. J Hepatol. 2003;38:564–72. doi: 10.1016/s0168-8278(03)00051-5. [DOI] [PubMed] [Google Scholar]
- 25.de Leeuw AM, McCarthy SP, Geerts A, Knook DL. Purified rat liver fat-storing cells in culture divide and contain collagen. Hepatology. 1984;4:392–403. doi: 10.1002/hep.1840040307. [DOI] [PubMed] [Google Scholar]
- 26.Knook DL, Blansjaar N, Sleyster EC. Isolation and characterization of Kupffer and endothelial cells from the rat liver. Exp Cell Res. 1977;109:317–29. doi: 10.1016/0014-4827(77)90011-8. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto M, Maehara Y, Sakaguchi Y, Kusumoto T, Ichiyoshi Y, Sugimachi K. Transforming growth factor-beta 1 induces apoptosis in gastric cancer cells through a p53-independent pathway. Cancer. 1996;77:1628–33. doi: 10.1002/(SICI)1097-0142(19960415)77:8<1628::AID-CNCR31>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 28.Tsutsumi M, Takada A, Takase S. Characterization of desmin-positive rat liver sinusoidal cells. Hepatology. 1987;7:277–84. doi: 10.1002/hep.1840070212. [DOI] [PubMed] [Google Scholar]
- 29.Neubauer K, Knittel T, Aurisch S, Fellmer P, Ramadori G. Glial fibrillary acidic protein – a cell type specific marker for Ito cells in vivo and in vitro. J Hepatol. 1996;24:719–30. doi: 10.1016/s0168-8278(96)80269-8. [DOI] [PubMed] [Google Scholar]
- 30.Dijkstra CD, Dopp EA, Joling P, Kraal G. The heterogeneity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in the rat recognized by monoclonal antibodies ED1, ED2 and ED3. Immunology. 1985;54:589–99. [PMC free article] [PubMed] [Google Scholar]
- 31.Neubauer K, Knittel T, Armbrust T, Ramadori G. Accumulation and cellular localization of fibrinogen/fibrin during short-term and longterm rat liver injury. Gastroenterology. 1995;108:1124–35. doi: 10.1016/0016-5085(95)90211-2. [DOI] [PubMed] [Google Scholar]
- 32.Ramadori G, Veit T, Schwogler S, et al. Expression of the gene of the alpha-smooth muscle-actin isoform in rat liver and in rat fat-storing (ITO) cells. Virchows Arch B Cell Pathol Incl Mol Pathol. 1990;59:349–57. doi: 10.1007/BF02899424. [DOI] [PubMed] [Google Scholar]
- 33.Knittel T, Kobold D, Saile B, et al. Rat liver myofibroblasts and hepatic stellate cells: different cell populations of the fibroblast lineage with fibrogenic potential. Gastroenterology. 1999;117:1205–21. doi: 10.1016/s0016-5085(99)70407-5. [DOI] [PubMed] [Google Scholar]
- 34.Saile B, Matthes N, Neubauer K, et al. Rat liver myofibroblasts and hepatic stellate cells differ in CD95-mediated apoptosis and response to TNF-alpha. Am J Physiol Gastrointest Liver Physiol. 2002;283:G435–44. doi: 10.1152/ajpgi.00441.2001. [DOI] [PubMed] [Google Scholar]
- 35.Darzynkiewicz Z, Williamson B, Carswell EA, Old LJ. Cell cycle – specific effects of tumor necrosis factor. Cancer Res. 1984;44:83–90. [PubMed] [Google Scholar]
- 36.Andree HA, Reutelingsperger CP, Hauptmann R, Hemker HC, Hermens WT, Willems GM. Binding of vascular anticoagulant alpha (VAC alpha) to planar phospholipid bilayers. J Biol Chem. 1990;265:4923–8. [PubMed] [Google Scholar]
- 37.Fadok VA, Savill JS, Haslett C, et al. Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J Immunol. 1992;149:4029–35. [PubMed] [Google Scholar]
- 38.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 39.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cilla DD, Jr, Gibson DM, Whitfield LR, Sedman AJ. Pharmacodynamic effects and pharmacokinetics of atorvastatin after administration to normocholesterolemic subjects in the morning and evening. J Clin Pharmacol. 1996;36:604–9. doi: 10.1002/j.1552-4604.1996.tb04224.x. [DOI] [PubMed] [Google Scholar]
- 41.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–42. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 43.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–12. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 44.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–9. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 45.Li P, Nijhawan D, Budihardjo I, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 46.Hu Y, Benedict MA, Ding L, Nunez G. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. EMBO J. 1999;18:3586–95. doi: 10.1093/emboj/18.13.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zou H, Li Y, Liu X, Wang X. An APAF-1. cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biochem Chem. 1999;274:11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 48.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–57. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- 49.Pan G, O'Rourke K, Dixit VM. Caspase-9, Bcl-XL, and Apaf-1 form a ternary complex. J Biochem Chem. 1998;273:5841–5. doi: 10.1074/jbc.273.10.5841. [DOI] [PubMed] [Google Scholar]
- 50.Gross A, Yin XM, Wang K, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biochem Chem. 1999;274:1156–63. doi: 10.1074/jbc.274.2.1156. [DOI] [PubMed] [Google Scholar]
- 51.Hermann JL, Bruckheimer E, McDonnell TJ. Cell death signal trasduction and BCL-2 function. Biochem Soc Trans. 1999;24:1059–65. doi: 10.1042/bst0241059. [DOI] [PubMed] [Google Scholar]
- 52.Saile B, DiRocco P, Dudas J. IGF-I induces DNA synthesis and apoptosis in rat liver hepatic stellate cells (HSC) but DNA synthesis and proliferation in rat liver myofibroblasts (rMF) Lab Invest. 2004;84:1037–49. doi: 10.1038/labinvest.3700116. [DOI] [PubMed] [Google Scholar]
- 53.Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–6. [PubMed] [Google Scholar]
- 54.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–6. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 55.Tuyt LM, Dokter WH, Birkenkamp K. Extracellular-regulated kinase 1/2, Jun N-terminal kinase, and c-Jun are involved in NF-kappa B-dependent IL-6 expression in human monocytes. J Immunol. 1999;162:4893–902. [PubMed] [Google Scholar]
- 56.Iles KE, Dickinson DA, Wigley AF. HNE increases HO-1 through activation of the ERK pathway in pulmonary epithelial cells. Free Radic Biol Med. 2005;39:355–64. doi: 10.1016/j.freeradbiomed.2005.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ruffels J, Griffin M, Dickenson JM. Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human SH-SY5Y neuroblastoma cells: role of ERK1/2 in H2O2-induced cell death. Eur J Pharmacol. 2004;483:63–173. doi: 10.1016/j.ejphar.2003.10.032. [DOI] [PubMed] [Google Scholar]
- 58.Su J-L, Lin MT, Hong CC. Resveratrol induces FasL-related apoptosis through Cdc42 activation of ASK1/JNK-dependent signalling pathway in human leukaemia HL-60 cells. Carcinogenesis. 2005;26:1–10. doi: 10.1093/carcin/bgh220. [DOI] [PubMed] [Google Scholar]
- 59.Soh Y, Jeong KS, Lee IJ. Selective activation of the c-Jun N-terminal protein kinase pathway during 4-hydroxynonenal-induced apoptosis of PC12. Cells Mol Pharmacol. 2000;58:535–41. doi: 10.1124/mol.58.3.535. [DOI] [PubMed] [Google Scholar]
- 60.Saile B, Matthes N, El Armouche H, Neubauer K, Ramadori G. The Bcl, NFkB and p53/p21WAF1 system are involved in spontaneous apoptosis and in the anti-apoptotic effect of TGF-alpha and TNF-alpha on activated hepatic stellate cells. Eur J Cell Biol. 2001;80:554–61. doi: 10.1078/0171-9335-00182. [DOI] [PubMed] [Google Scholar]