

Abstract
Leucocyte adhesion deficiency (LAD) is an autosomal-recessive genetic disease that is characterized clinically by severe bacterial infections and caused by mutations in the CD18 gene that codes for the β2 integrin subunit. A patient with a severe LAD phenotype was studied and the molecular basis of the disease was identified as a single homozygous defect in a Herpes virus saimiri (HVS)-transformed T-cell line. The defect identified involves a deletion of 171 bp in the cDNA that encodes part of the proteic extracellular domain. This genetic abnormality was further studied at the genomic DNA level and found to consist of a deletion of 169 bp (from − 37 of intron 4 to + 132 of exon 5), which abolishes the normal splicing and results in the total skipping of exon 5. The 171-bp shortened ‘in-frame’ mRNA not only resulted in the absence of CD18 expression on the cell surface but also in its absence in the cytoplasm of HVS T-cell lines. Functionally, the LAD-derived HVS T-cell lines showed a severe, selective T-cell activation impairment in the CD2 (but not in the CD3) pathway. This defect was not reversible when exogenous interleukin-2 (IL-2) was added, suggesting that there is also a functional interaction of the lymphocyte function-associated antigen-1 (LFA-1) protein in the CD2 signal transduction pathway in human T cells, as has been previously reported in mice and in the human Papillon–Lefèvre syndrome. Thus, HVS transformation is not only a suitable model for T-cell immunodeficiency studies and characterization, but is also a good system for investigating the immune system in pathological conditions. It may also be used in the future in cellular models for in vitro gene-therapy trials.
Introduction
Leucocyte adhesion deficiency (LAD; MIM: Mendelian Inheritance in Man, number 116920) is an autosomal recessive hereditary disease caused by defects in the CD18 gene,1,2 which codes for the β2 integrin. The β2 integrin subunit combines non-covalently with the αL, αM and αX integrin subunits to form the lymphocyte function-associated antigen-1 (LFA-1) (αLβ2, CD11a/CD18), Mac-1 (αMβ2, CD11b/CD18) and p150, 95 (αXβ2, CD11c/CD18) antigens, respectively.3 As these three molecules are expressed exclusively on leucocytes, they are also referred to as the leucocyte integrins.4,5 In vitro and in vivo studies have demonstrated that the CD11/CD18 complex is the major determinant of firm adhesion and transendothelial migration of neutrophils.6
The CD18 gene spans ≈ 40 kb and is organized into 16 exons. All the exons have been sequenced and the exon/intron boundaries of the gene have been previously reported.7 The CD18 gene has been located at chromosome 218,9 band q22.3.10,11
The usual consequence of a defect in the CD18 gene is a low or ‘null’ expression of the three leucocyte integrins. The expression level in different patients varies from 0 to ≈ 10% of normal values and the patients’ cells show corresponding degrees of impairment in their adhesion properties.12 Clinically, LAD patients suffer from recurrent bacterial infections, and those with severe phenotype (< 1% expression of normal) often do not survive into adulthood. The common outcome is failure to produce a functional leucocyte integrin as a result of heterogeneous mutations within the common β2 (CD18) gene.13,14
Two clinical phenotypes of LAD (severe and moderate or partial), which correlate with the severity of the disease, have been defined according to the level of β2 integrin expression on patients’ leucocytes.12,13 The diversity of the alterations has also led to the definition of several types of LAD (I–V), the classification of which is based on the size and levels of the CD18 subunit precursor, the CD18 mRNA and the resulting phenotype.13 Analysis of severe and moderate LAD CD18 alleles has identified missense mutations,15–21 aberrant splicing events17,19,20,22 and a 1-bp deletion15,21 within the structural region of the CD18 gene. The molecular basis of the LAD defect has been described for only a few patients3,16,17,19–26(Table 1).
Table 1.
Mutations in the CD18 gene causing leucocyte adhesion deficiency (LAD)
| Missense/nonsense | ||||
| Codon | Exon | Nucleotide | Amino acid | Reference |
| 1 | 2 | ATG-AAG | Met-Lys | 22 |
| 128 | 5 | GAC-AAC | Asp-Asn | 14 |
| 149 | 5 | CTA-CCA | Leu-Pro | 11 |
| 169 | 6 | GGG-AGG | Gly-Arg | 11 |
| 178 | 6 | CCG-CTG | Pro-Leu | 12 |
| 196 | 6 | AAA-ACA | Lys-Thr | 2 |
| 284 | 7 | GGC-AGC | Gly-Ser | 21 |
| 351 | 9 | AAT-AGT | Asn-Ser | 15 |
| 534 | 12 | TGC-TGA | Cys-Stop | 17 |
| 586 | 13 | CGG-TGG | Arg-Trp | 15 |
| 593 | 13 | CGT-TGT | Arg-Cys | 2 |
| Splicing | ||||
| Relative location | Intron | Donor/acceptor | Substitution | Reference |
| − 14 | 6 | as | C-A | 15 |
| + 3 | 9 | ds | G-C | 4 |
| + 1 | 7 | ds | G-A | 14 |
| Deletions | ||||
| Codon | Exon | Size (bp) | Reference | |
| 39 | 3 | 10 | 17 | |
| 418 | 11 | 2 | 20 | |
| 690 | 14 | 1 | 16 | |
| 553 | 13 | 220 | 12 | |
| 110 | 5 | 171 | This work | |
as, acceptor site; ds, donor site.
The establishment of T-cell lines that somehow reproduce the defects observed in peripheral T lymphocytes from patients with primary immunodeficiencies has been of great interest owing to the enormous progress in the diagnosis and characterization of T-cell defects.27,28 Herpes virus saimiri (HVS), a γ-2 herpes virus isolated from the squirrel monkey, was reported to transform CD4+ CD8+ T cells and thymocytes to continuous growth in vitro, 29 but infected cells remained dependent on the addition of exogenous interleukin-2 (IL-2). It has also been reported that HVS-transformed T cells maintain a functional T-cell receptor (TCR), the stimulation of which initiates a biochemical cascade of events (Ca2+ mobilization, pattern of tyrosine phosphorylation) identical to that observed in peripheral blood mononuclear cells (PBMCs).30,31 Furthermore, the analysis of primary T-cell defects might shed new light on the physiology of T lymphocytes, and HVS-transformed T cells is a good in vitro model for analysing T-lymphocyte pathophysiology.
Molecular characterization of the defective CD18 gene from a patient with severe LAD and functional analysis of his lymphocytes (PBMCs and HVS-transformed T cell lines) has been carried out. The results revealed a novel CD18 deletion causing LAD, the total absence of CD18 (not only on the cell surface but also in the cytoplasm) suggested a specific biochemical interaction between the CD18 protein and the CD2 activation pathway in T cells. Thus, HVS-transformed T-cell lines are not only useful for studying the molecular defects underlying immunodeficiencies or as a target for ‘in vitro’ gene therapy trials, but also for further investigation of specific immune system mechanisms as immortalized natural human knockouts.
Materials and methods
Case report
The patient investigated in this report is a deceased 1-year-old male whose father was not known and who had no previous family history of LAD. He had experienced omphalitis and periumbilical cellulitis (caused by Escherichia coli) for 3 weeks, and cerebrospinal fluid (CSF) infection with Staphylococcus simulans. At the age of 3 months, he suffered from Streptococcus viridans septicemia and oral candidiasis. The laboratory findings at 7 months were consistent with a severe LAD diagnosis: leucocytosis (up to 60 000/µl) and thrombocytosis. The patient was classified as having the severe form of the disease, based on the severity of the clinical course and the total absence of CD11a/CD18 in his peripheral lymphocytes (Fig. 1). Moreover, the phagocytic capacity of his neutrophils was reduced (data not shown) while the oxidative burst test was within the normal range. The patient underwent two bone marrow transplantations (BMT) at 7 and 10 months, respectively. After T-cell depletion (with anti-CD3 + anti-CD5 + anti-CD7 monoclonal antibodies [mAbs]) from human leucocyte antigen (HLA) non-identical bone marrow donors, the patient was conditioned with Busulfan and cyclophosphamide [+ Vp16 in the first BMT or horse anti-thymocyte globulin (ATGAM; Pharmacia-Upjohn) in the second]. Two months after the second failed BMT, the patient died from a Fusarium infection.
Figure 1.
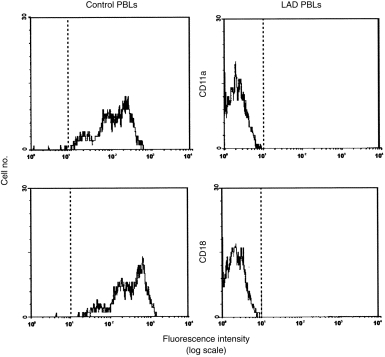
Flow cytometry of peripheral blood lymphocytes (PBLs). Surface expression is shown of CD11a (top) and CD18 (bottom) on lymphocytes in the leucocyte adhesion deficiency (LAD) patient at diagnosis (right panel) and in a healthy control (left panel). Lymphocytes were gated on the basis of CD45 staining and side scatter. The results shown are representative of three independent stainings performed on the same day on lymphocytes from the patient. Dotted vertical lines represent cursor located based on background staining (isotype-matched controls).
HVS-transformed T-cell lines
HVS-transformed T-cell lines were derived from the PBMCs of a patient with severe LAD (LAD-HVS) and a normal control (CT-HVS), as described previously.29 Three months after inoculation with HVS, cell lines were completely established with a stable morphology as well as surface phenotype.32 The presence of the HVS genome was determined by using the polymerase chain reaction (PCR) with specific HVS primers (Table 2, see under HVS genome amplification).33
Table 2.
The different primers used in this work
| CD18 mRNA amplification | ||
| Coding | Non-coding | Size (bp) |
| CD18-1: TATAAGCTTCAGGGCAGACTGGTAGCAAA | CD18–4: CTTAGCAAGCTTGTGCGCCA | 1–1007 |
| CD18-5: AACGAATTCGACTACCCATCGGT | CD18–8: CTAGAATTCTCCTGACGGCCTTGTCTTCA | 970–2410 |
| HVS genome amplification | ||
| Coding | Non-coding | Size (bp) |
| HVS.1: GAGTTTCCAAAATGTACTAAGCTAAC | HVS.2-ACTAATAAAAAGTTCCACACAACTAAC | 89 |
| CD18 genomic DNA amplification | ||
| Coding | Non-coding | Localization (size bp) |
| CD18-1.1: GACATCATGGACCCCACAAGC | CD18–1·2: CTTCAGCACGTGCCTGAAGGC | Exon 4–exon 6 (5400) |
| I4F-1: CTGAAACTCCAGCCTGCCTGG | I5R-2: TACAAGTGTTGGGGAGCCAGG | Intron 4–intron 5 (572) |
| Direct sequencing | ||
| Primer | Localization | |
| CD18-2: | AACAAGCTTCTTGACATTCC | Exon 5 |
| CD18-3: | TCCAAGCTTGATGACCTCAG | Exon 5 |
| I5R-2: | TACAAGTGTTGGGGAGCCAGG | Intron 5 |
| I5R-1: | AGATGGAAGGGGAAGTGGAGG | Intron 5 |
| Competitive PCR analysis | ||
| Coding | Non-coding | Localization (size bp) |
| E4-FM: TAMRA-CTTCACAGGGCCGGGGGATCC | E4-R: CTGGTCGCAGGTAAAGCGTCT | Exon 4 (180) |
| E5-RM: TAMRA-GTCCTGCAGTGCCTGGGCC | I4F-3: GGGGAGAGGGGAGGTCAGGAG | Exon 5–intron 4 (*) |
| E6-FM: TAMRA-CTTCGGGTCCTTCGTGGACAAG | E6-R: GGGCAGGCGGCGACCTGCATCA | Exon 6 (240) |
Amplified size in CT-HVS was 331 bp and in LAD-HVS was 162 bp.
Cytofluorographic analysis
For direct immunofluorescence, 1 × 106 LAD-HVS cells were incubated with the corresponding mAbs: anti-CD3-tricolor (Cy5-phycoerythrin [PE])-labelled (Caltag, San Francisco, CA), anti-CD18 fluorescein isothiocyanate (FITC)-labelled (Dako, Glostrup, Denmark), anti-CD11a (FITC) (Dako), anti-CD11b PE-labelled (Dako), anti-CD11c (FITC) (Dako), anti-CD2 (PE) (Coulter Clone, Hialeah, FL) and an anti-CD4 (PE)/CD8 (FITC) cocktail (Coulter Clone). Cells were washed twice with phosphate-buffered saline (PBS) containing 0·01% NaN3, and three-colour analysis of stained cells was carried out in an EPICS-XL or a FACScan flow cytometer. The precise quantification of CD18 molecules per cell in HVS-transformed cell lines was performed by using indirect immunofluorescence in parallel with calibrated beads (Qifikit; Biocytex, Marseille, France) following the manufacturer’s protocol.32 Quantitative comparisons of fluorescence intensities was also performed by using an appropriate program, which calculated the mean fluorescence, in a linear scale, for each sample (epics cytological software, version 2·01; Coulter).
Intracellular labelling
LAD-HVS and CT-HVS cells were lysed and permeabilized with Fix & Perm (Caltag) using the intracellular labelling procedure,34 after which the cells were labelled for CD18 intracellular protein with anti-CD18 FITC (Dako) mAb. Before permeabilization, both cell lines were incubated with an excess of pure unlabelled anti-CD18 (Dako) mAb to block the surface CD18 antigen.
Amplification of CD18 mRNA
Cytoplasmic RNA was extracted from 1 × 107 HVS-transformed T cells from CT-HVS and from LAD-HVS using the Nonidet P-40 (NP-40) lysis method.35 Reverse transcription (RT) was performed on 5 µg of cytoplasmic RNA using an oligo(dT) primer and the reverse transcriptase kit from Promega (Madison, WI), following the manufacturer’s protocols. PCR amplification of the CD18 mRNA was carried out with 10 µl of the cDNA synthesis reaction in a solution containing 0·2 mm of each deoxynucleotide, 2·5 mm MgCl2, 50 mm KCl, 10 mm Tris-HCl pH 8·3, 1 µm of each oligonucleotide primer (Table 2, see under CD18 mRNA amplification) and 2·5 U of Taq DNA polymerase. PCR amplification was performed as follows: 30 cycles of denaturation (at 95° for 30 seconds), annealing (at 58° for 30 seconds) and extension (at 72° for 1·5 min), followed by a final 10-min extension step at 72°. Amplified fragments were identified by differential migration in a 1% agarose gel and purified using the QIA-quick system (Qiagen, Hilden, Germany). The purified fragments were inserted into the P-GEM-T vector (Promega). Ligation, transformation and identification of recombinant colonies were carried out according to the manufacturer’s recommendations. Double-stranded DNA templates were sequenced using the dideoxy chain-terminator method of Sanger, with dye-labelled dideoxy terminators.36 Each sequence was confirmed by analysis of four or more clones from two different amplifications.
Amplification of CD18 genomic DNA
PCR amplification of genomic DNA was performed using cell lysates of normal and LAD HVS T cells, as described by Saiki et al.37 The 171-bp deletion corresponds precisely to exon 5 in the CD18 transcript. The primers listed in Table 2(see under CD18 genomic DNA amplification) were designed to amplify the intron/exon boundaries for exon 5. Reagents used for high-length fragment amplification were purchased from Boehringer Mannheim (Mannheim, Germany), and the PCR was performed, according to the manufacturer’s specifications, using the hot-start procedure. Amplification using primers CD18-1.1 and CD18-1.2 required 35 cycles comprising a denaturation step at 94° for 10 seconds and an annealing/extension step at 68° for 4·5 min. Primers CD18-1.1 and CD18-1.2 were designed outside exon 5, in exon 4 and exon 6, respectively, and the expected amplification product was ≈ 5400 bp. Amplified fragments were sequenced by direct sequencing of the PCR product using the dideoxy chain terminator method of Sanger. The primers used for direct sequencing are listed in Table 2 (see under Direct sequencing).
Cytogenetic studies
Chromosome preparations and Giemsa-stained metaphases were obtained by using standard methods38,39 from phytohemagglutininin (PHA)-stimulated HVS T lymphocytes.
Competitive PCR
Competitive PCR amplification was performed on genomic DNA obtained from the HVS-transformed T-cell lines derived from LAD-HVS and CT-HVS. Three oligonucleotide primer pairs were used: two were designed to amplify an undeleted exon of the CD18 gene (exons 4 and 6) and the third to amplify the exon that was found to be deleted in the cDNA of the patient (exon 5). The reaction was limited to 20 cycles of PCR, and a limiting amount of Taq polymerase (0·8 U) was added to the PCR mixture (Table 2; see under Competitive PCR analysis; the dye used to end-label PCR primers was TAMRA).40 Each cycle comprised denaturation (at 95° for 15 seconds), annealing (at 58° for 15 seconds) and extension (at 72° for 3 min). Under these conditions, the PCR was quantitative and did not reach a plateau. The DNA was electrophoresed at 1400 V for 14 hr and the data was automatically analysed using the genescan Fluorescent Fragment Analyser (Applied Biosystems, Foster City, CA). The ratios between the two band areas (control/target exons) were calculated and compared between LAD-HVS and CT-HVS.
Functional assays
Proliferative assays in PBMCs
Isolated cells (80 000) were placed in round-bottomed microtitre plates (Nunc, Roskilde, Denmark) in 160 µl of serum-free final culture medium (AIM-V, Gibco, Paisley, Strathclyde, UK) supplemented with 1% penicillin/streptomycin and 1% 20 mm l-glutamine (Whittaker, Walkersville, MD). The following stimuli were used: anti-CD3 (OKT3, Ortho Pharmaceuticals, Raritan, NJ), 12·5 ng/ml and PHA (Difco, Detroit, MI) 1 : 100 final dilution. Wells were pulsed individually with 1 µCi of [3H]thymidine after 3 days of culture and its uptake was measured in a liquid scintillation counter (Beckman, Brea, CA).
Proliferative assays in HVS-T cells
Cells were rested in IL-2-free medium for 1 week before assaying their proliferation. Cells (4 × 104) were resuspended in AIM-V medium supplemented with 1% 20 mm l-glutamine and incubated in the plates. The following stimuli, or their combinations, were used: IOT3b mAb (1 µg/ml, 2 µg/ml, 4 µg/ml; anti-CD3; Immunotech, Marseille), anti-CD2 (1.1 + 2.1) (CLB) 1 : 40 final dilution; (increasing concentrations of CD2 antibody combination were tested without any effect), PHA 1 : 100 final dilution, concanavalin A (Con A; Calbiochem, La Jolla, CA) and human recombinant IL-2 (hrIL-2; Boehringer Mannheim). After 24 hr of culture, cells were pulsed with [3H]thymidine for another 16 hr, harvested onto glass-fibre filters and analysed by direct β-counting.
Results
Null surface expression of leucocyte integrins on LAD cells
At diagnosis, flow cytometry analysis of the CD11a/CD18 integrin complex showed a null expression of this molecule in the PBMCs of the patient (Fig. 1). No other phenotypic alterations were detected with normal T, B and natural killer (NK) cell proportions (60%, 28% and 12%, respectively). Flow cytometry studies confirmed that control HVS-transformed T cells expressed the CD11/CD18 complex, while LAD-HVS cells were totally deficient in surface expression of the CD11a, b, c/CD18 complex (Fig. 2). Both T-cell lines were CD3+ CD4+ CD8− and showed the characteristic surface phenotype of memory-activated T cells (HLA-DR+ CD38+ CD45R0+ CD45RA−).
Figure 2.
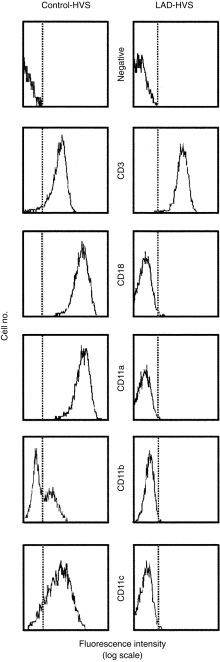
Flow cytometry of Herpes virus saimiri (HVS)-transformed T lymphocytes. Cell-surface cytofluorographic analysis of HVS-transformed T lymphocytes from the leucocyte adhesion deficiency (LAD) patient (right) and a healthy control (left) is shown. Dotted vertical lines represent cursor located based on background staining (isotype-matched controls, histograms labelled as ‘negative’). Cell-surface staining was determined of CD11a, b, c and CD18 integrins, with CD3 included as a positive labelling control.
Quantitative analysis of the exact number of CD18 molecules per cell with an anti-CD18 mAb revealed a null expression of CD18 in the LAD-HVS cell line compared with 97 000 molecules per cell in the CT-HVS cell line (data not shown).
Analysis of intracellular expression of CD18 protein
To evaluate whether or not the CD18 protein was synthesized, the presence of CD18 in the cytoplasm of the LAD cell line (LAD-HVS) and the control cell line (CT-HVS) was studied. CD18 surface expression was blocked with cold anti-CD18 mAb and then the cell membranes were permeabilized and the CD18 cytoplasmic protein was stained. Again, null expression of CD18 in the LAD-HVS cell line was obtained when it was compared to 100% staining in the CT-HVS cell line. CD3 staining was obtained in 100% of both LAD and CT-HVS cell lines, as a positive control (Fig. 3).
Figure 3.
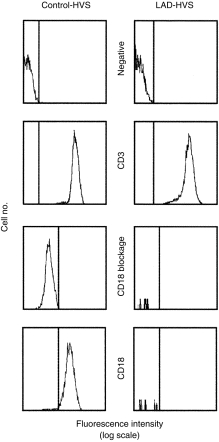
Intracellular cytofluorographic analysis of Herpes virus saimiri (HVS)-transformed T lymphocytes from the leucocyte adhesion deficiency (LAD) patient and a healthy control. Dotted vertical lines represent cursor located based on background staining (isotype-matched controls, labelled as ‘negative’). CD3 staining was used as a positive labelling control (labelled as ‘CD3’). CD18 surface expression was previously blocked with pure unlabelled anti-CD18 monoclonal antibody (mAb), as described in the Materials and methods, and then stained with fluorescein isothiocyanate (FITC)-conjugated anti-CD18 mAb with (labelled as ‘CD18’) or without (labelled as ‘CD18 blockage’) surface membrane permeabilization.
CD18 mRNA of LAD cells showed a 171-nucleotide deletion
CD18 mRNAs from the patient and the control were amplified by PCR. The presence of transcripts in the LAD HVS cell line and the total absence of detectable CD18 protein, both on the cell surface and in the cytoplasm, indicated the presence of deletions, insertions or nucleotide substitutions in the CD18 mRNA that precluded the synthesis of a stable protein. To analyse the complete mRNA, RT and amplification of the CD18 cDNA were carried out using the oligonucleotide primers listed in Table 2 (see under CD18 mRNA amplification) with the strategy described in the Materials and methods. The CD18 transcript of the LAD-HVS cell line was shorter than that obtained with the CT-HVS cell line. DNA sequencing of the amplified cDNA demonstrated a 171-nucleotide deletion corresponding to the total absence of exon 5 (from 329 to 500 bp). No other amplification product corresponding to the normal size of the CD18 mRNA was obtained from the patient’s cell line; thus another mutation that affected the mRNA synthesis, or a homozygous deletion, were suspected.
Analysis of LAD CD18 genomic DNA reveals a homozygous deletion
To determine whether the deletion was present at the genomic level or was a splicing artefact, the CD18 genomic DNA from the control cell line was amplified from intron 4 to intron 5 by PCR (using primers CD18-1.1 and CD18-1.2) and subsequently sequenced (using primers CD18-2 and CD18-3). The intronic sequence obtained (previously unknown; data not shown) allowed the synthesis of a pair of internal primers (I4F-1 and I5R-2) to amplify the genomic DNA surrounding exon 5 of the patient (Fig. 4). The amplification product was directly sequenced using primers I4F-3 and I5R-1 and the sequence revealed a deletion of 169 bp, which included the 3′ end of the intron 4 and the 5′ end of exon 5 (from − 37 intron 4 to + 132 exon 5) (Fig. 5). This confirmed the RT–PCR results and demonstrated that the deletion was present in the patient’s genome. Therefore, the deletion at the genomic level abolished the intron 4 acceptor splicing site that resulted in the total skipping of the residual exon 5 of the patient. Again, the lack of an amplification fragment of normal size was observed in the patient; thus a genomic homozygous deletion was suspected.
Figure 4.
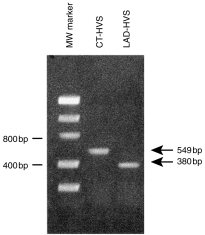
Products of the polymerase chain reaction (PCR) using primers flanking exon 5. Partial CD18 genomic DNA amplification was performed by PCR using the primers and conditions described in the Materials and methods. Unique bands of normal (549 bp) and 169 nucleotide-deleted (380 bp) sequence were observed in the control cell line (CT-HVS) and the patient (LAD-HVS), respectively. Left lane, molecular-weight (Mw) standards. CT, control; HVS, Herpes virus saimiri; LAD, leucocyte adhesion deficiency.
Figure 5.
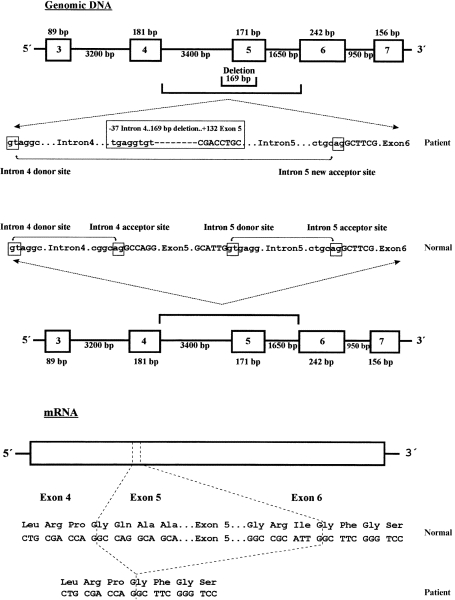
Schematic representation of the homozygous mutation in the leucocyte adhesion deficiency (LAD) patient. The 169-bp genomic deletion is depicted in the upper part of the figure. Exons are shown by thick boxes and introns by thick horizontal lines. Intron sequences are in lowercase letters and exon sequences in uppercase letters. Acceptor and donor splicing sites are also boxed and normal or pathological splicing is depicted by lines with small arrows. The 171 bp ‘in-frame’ mRNA deletion caused by the loss of the intron 4 acceptor splicing site and total exon 5 skipping, as well as the putative translated protein, are depicted in the lower part of the figure.
Major cytogenetic alterations in the patient’s chromosomes were rejected because a normal karyotype in the LAD-HVS cell line was obtained (data not shown). No genetic material from the parents was available, but consanguinity of the parents was suspected and the described results, both at the mRNA and genomic DNA level, strongly suggested a homozygous mutation in the CD18 gene. To definitively characterize the genetic defect, a competitive PCR amplification was designed and developed. In a fluorescent-labelled PCR with limiting conditions (see the Materials and methods), the exon 5 to exon 4 (or exon 6) amplification ratios were determined in the patient and in healthy controls. The results obtained (not shown) showed that the exon 5-shortened amplification product of the patient corresponded to two copies with exon5/exon4–6 ratios identical to those obtained in the healthy controls.
The CD2 activation pathway is severely impaired in LAD-HVS cells
PBMCs isolated from the patient were challenged in vitro with OKT3 (anti-CD3 mAb) and PHA with results comparable to those obtained from a healthy control (data not shown). To further investigate the functional immune status of LAD-HVS T-cell line, cells were rested and then stimulated with different mitogens or their combinations. As expected, HVS-transformed cells responded normally to anti-CD3 mAb and PHA. The results obtained are shown in Table 3 and revealed a normal proliferative response of LAD cells to all mitogens, but that of CD2 was severely impaired, with a fivefold reduction when compared to controls. This poor response of the patient’s T cells to CD2 was not corrected by the addition of exogenous IL-2.
Table 3.
Response index* of Herpes virus saimiri (HVS)-transfected T-cell lines from the patient with leucocyte adhesion deficiency (LAD) and the controls
| Stimuli | LAD-HVS† | Controls‡ |
|---|---|---|
| IL-2 | 8·4 | 9·3 ± 2 |
| PHA | 5·7 | 3·5 ± 1 |
| PHA + IL-2 | 5·9 | 7·4 ± 2 |
| Con A | 4·6 | 8·1 ± 3 |
| Con A + IL-2 | 4·9 | 7·8 ± 3 |
| CD2 | 2·9 | 14·5 ± 3 |
| CD2 + IL-2 | 1·1 | 10·5 ± 3 |
| CD3 | 3·4 | 2·6 ± 1 |
| CD3 + IL-2 | 4·0 | 5·4 ± 2 |
Con A, concanavalin A; IL-2, interleukin-2; PHA, phytohaemagglutinin.
Expressed as net relative increase of each stimulus to the minimum stimulus (background).
The results shown are representative of three independent experiments performed on the patient.
The results are expressed as mean ± standard deviation of three different HVS T-cell lines from normal donors.
Discussion
A novel 169-nucleotide homozygous genomic deletion causes a 171-nucleotide mRNA in-frame deletion and total lack of CD18 protein expression
In this work, the molecular basis of LAD was characterized in a patient with a severe clinical LAD phenotype. A novel genetic alteration in the CD18 gene, consisting of a homozygous genomic deletion, was found following RT–PCR with subsequent genomic DNA amplification and sequencing of the affected region, and finally competitive PCR analysis to study the gene dosage in the patient. The mutation, a genomic deletion of 169-nucleotides comprised the last 37 nucleotides of intron 4 and the first 132 nucleotides of exon 5. The deletion included intron 4 acceptor splicing, causing the direct joining of exon 4 to exon 6 during mRNA splicing and the total lack of the residual exon 5, which is observed as a 171-nucleotide deletion in the sequenced cDNA. This ‘in-frame’ deletion did not result in a premature stop codon, but rather a 57-residue shortened protein.
The deleted region is crucial for CD11/CD18 integrin assembly and expression
The deleted 57 amino acids are located in the extracellular domain of the CD18 protein, belong to a highly conserved 60 amino acid region and are critical for heterodimer formation in the integrin superfamily.7 Thus, the total lack of this region would explain not only the total absence of α/β heterodimers on the cell surface but also the absence of any stable protein in the cell cytoplasm. Biosynthesis studies on CD11 and CD18 proteins have shown that association of the two subunits precedes carbohydrate processing, which occurs in the Golgi apparatus.3 Thus, if the CD18 protein of the patient was unable to associate with the CD11 subunit in the rough endoplasmic reticulum, as observed by the lack of intracellular staining, it would probably be degraded. Other CD18 gene defects causing LAD have been shown to result in production of a CD18 protein precursor that failed to mature to membrane form and these CD18 precursors were rapidly degraded.16
Stimulation impairment in LAD cells highlights the functional association of CD2 and LFA-1 molecules on human T cells
LAD-HVS showed mainly normal responses to most of the stimuli assayed in the in vitro functional evaluation. DNA synthesis for each stimulus was normalized as the net ratio of counts per minute (c.p.m.) obtained with a given stimulus relative to the minimum stimulus (background DNA synthesis); this strategy allowed us to standardize the results obtained from the different assays. The data obtained showed a selective anergy in the CD2 activation pathway of LAD cells. In order to study this, both the proportion of positive cells (%) and the mean cell surface density of the CD2 marker were studied in LAD-HVS and in CT-HVS T lymphocytes: no comparative difference was found (data not shown). Moreover, other mitogenic responses were similar to the values obtained from the normal control (CD3, Con A, PHA). The impaired response to anti-CD2 mAbs was not reversed following the addition of exogenous IL-2. The lack of CD11a/CD18 at the T-cell surface seemed to be responsible for this CD2 activation impairment.
Previous studies have reported the molecular association between CD2 and LFA-1 in mouse T lymphocytes41 and the present work confirms this type of association in humans. The significance of this association is still unknown, but CD2 has been found to be associated with other surface molecules, including CD45 and TCRζ and ε chains.42–44 The CD2/LFA-1 interrelation observed suggests that CD2 (an adhesion molecule) can form complexes not only with molecules involved in signalling through the TCR, but also with other adhesion molecules, namely LFA-1. As recognition of antigen by the TCR results in increased LFA-1-mediated adhesion between an effector T cell and its target,45 the possibility exists that the association of CD2 with LFA-1 might play a role in co-ordinating the adhesion events that occur, in humans and mice, between the cells upon recognition of an antigen by the TCR.41
An alternative possibility is that the CD2/LFA-1 complex provides LFA-1 with the ability to transduce a more diverse range of intracellular signals than when unassociated. Occupancy of CD11a/CD18 with its complementary ligands or with specific mAb has been found to modulate neutrophil oxidative bursts, and to deliver mitogenic or co-mitogenic signals during B- or T-cell activation.46 In addition, recognition of the specific epitope defined by the anti-HVSB6 mAb on LFA-1 can modulate early signal transduction via the CD2 pathway on peripheral human T cells, as determined by measurement of Ca2+ flux and phosphoinositol (PI) hydrolysis.47
Furthermore, the possibility that CD2 and LFA-1 adhesion molecule synthesis28 are conjointly regulated in T lymphocytes was previously put forward by us in a different human disease: the Papillon–Lefèvre Syndrome (PLS) patients.48 The evidence that LAD shares some clinical symptoms (i.e. prepuberal periodontitis) with other diseases, such as PLS, is now somehow explained by the similarities of laboratory findings affecting expression of adhesion molecule in both diseases (in PLS, a low-density expression of major adhesion molecules, such as CD2, CD18, CD11a, CD29 and CD45RO, was observed). Finally, it is necessary to study a greater number of patients with LAD to further confirm our present findings.
The HVS T-cell line is a good model for LAD research
HVS-transformed T-cell lines in the LAD patient not only maintained the peripheral CD18 expression defect but also allowed us to further investigate the immune consequences of the lack of CD18 on T lymphocytes. Moreover, HVS cell lines could be used in the near future as a target for in vitro gene therapy experiments, in the same way as EBV lymphoblastoid cells have been used in other gene therapy trials.49
Acknowledgments
We are indebted to Pilar Gutiérrez, Paloma Muñoz, Paloma de Pablos and Maria Jose Recio for their technical help. This work has been partially supported by Comunidad de Madrid grants (6/70/97 and 8.3/14/98) and by a Ministerio de Educación grant (PM-95-57 and PM-96-21). The nucleotide sequence of the patient’s CD18 gene transcript was submitted to GenBank under accession no.: L78790. The genomic nucleotide sequences from introns 4 and 5 of the patient and the control were submitted to GenBank under accession nos: AF016895 and AF016894, respectively.
References
- 1.Fischer A, Lisowska-grospierre B, Anderson DC, Springer TA. Leukocyte adhesion deficiency: molecular basis and functional consequences. Immunodefic Rev. 1988;1:39. [PubMed] [Google Scholar]
- 2.Arnaout MA. Leukocyte adhesion molecules deficiency: its structural basis, pathophysiology and implications for modulating the inflammatory response. Immunol Rev. 1990;114:145. doi: 10.1111/j.1600-065x.1990.tb00564.x. [DOI] [PubMed] [Google Scholar]
- 3.Sánchez-madrid F, Nagy JA, Robbins E, Simon P, Springer TA. A human leukocyte differentiation antigen family with distinct alpha-subunits and a common beta-subunit: the lymphocyte function-associated antigen (LFA-1), the C3bi complement receptor (OKM1/Mac-1), and the p150, 95 molecule. J Exp Med. 1983;158:1785. doi: 10.1084/jem.158.6.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kishimoto TK, Larson RS, Corbí AL, Dustin ML, Staunton DE, Springer TA. The leukocyte integrins. Adv Immunol. 1989;46:149. doi: 10.1016/s0065-2776(08)60653-7. [DOI] [PubMed] [Google Scholar]
- 5.Larson RS, Springer TA. Structure and function of leukocyte integrins. Immunol Rev. 1990;114:181. doi: 10.1111/j.1600-065x.1990.tb00565.x. [DOI] [PubMed] [Google Scholar]
- 6.Harlan JM. Leukocyte adhesion deficiency syndrome: insights into the molecular basis of leukocyte emigration. Clin Immunol Immunopathol. 1993;67:S16. doi: 10.1006/clin.1993.1079. [DOI] [PubMed] [Google Scholar]
- 7.Weitzman JB, Wells CE, Wright AH, Clark PA, Law SK. The gene organisation of the human β2 integrin subunit (CD18) FEBS Lett. 1991;294:97. doi: 10.1016/0014-5793(91)81351-8. [DOI] [PubMed] [Google Scholar]
- 8.Marlin SD, Morton CC, Anderson DC, Springer TA. LFA-1 immunodeficiency disease. Definition of the genetic defect and chromosomal mapping of alpha and beta subunits of the lymphocyte function-associated antigen 1 (LFA-1) by complementation in hybrid cells. J Exp Med. 1986;164:855. doi: 10.1084/jem.164.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corbí AL, Larson RS, Kishimoto TK, Springer TA, Morton CC. Chromosomal location of the genes encoding the leukocyte adhesion receptors LFA-1, Mac-1 and p150, 95. Identification of a gene cluster involved in cell adhesion. J Exp Med. 1988;167:1597. doi: 10.1084/jem.167.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Law SK, Taylor GM. Restriction fragment length polymorphism of the gene of the human leukocyte integrin β-subunit (CD18) Immunogenetics. 1991;34:341. doi: 10.1007/BF00211998. [DOI] [PubMed] [Google Scholar]
- 11.Solomon E, Palmer RW, Hing S, Law SK. Regional localization of CD18, the beta-subunit of the cell surface adhesion molecule LFA-1, on human chromosome 21 by in situ hybridization. Ann Hum Genet. 1988;52:123. doi: 10.1111/j.1469-1809.1988.tb01087.x. [DOI] [PubMed] [Google Scholar]
- 12.Anderson DC, Schmalsteig FC, Finegold MJ, et al. The severe and moderate phenotypes of heritable Mac-1, LFA-1 deficiency: their quantitative definition and relation to leukocyte dysfunction and clinical features. J Infect Dis. 1985;152:668. doi: 10.1093/infdis/152.4.668. [DOI] [PubMed] [Google Scholar]
- 13.Kishimoto TK, Hollander N, Roberts RM, Anderson DC, Springer TA. Heterogeneous mutations in the beta subunit common to the LFA-1, Mac-1, and p150, 95 glycoproteins cause leukocyte adhesion deficiency. Cell. 1987;50:193. doi: 10.1016/0092-8674(87)90215-7. [DOI] [PubMed] [Google Scholar]
- 14.Dimanche-boitrel MT, Guyot A, De Saint-basile G, Fischer A, Griscelli C, Lisowska-grospierre B. Heterogeneity in the molecular defect leading to the leukocyte adhesion deficiency. Eur J Immunol. 1988;18:1575. doi: 10.1002/eji.1830181016. [DOI] [PubMed] [Google Scholar]
- 15.Arnaout MA, Dana N, Gupta SK, Tenen DG, Fathallah DM. Point mutations impairing cell surface expression of the common beta subunit (CD18) in a patient with leukocyte adhesion molecule (Leu-Cam) deficiency. J Clin Invest. 1990;85:977. doi: 10.1172/JCI114529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wardlaw AJ, Hibbs ML, Springer TA. Distinct mutations in two patients with leukocyte adhesion deficiency and their functional correlates. J Exp Med. 1990;172:335. doi: 10.1084/jem.172.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Back AL, Kwok WW, Hickstein DD. Identification of two molecular defects in a child with leukocyte adherence deficiency. J Biol Chem. 1992;267:5482. [PubMed] [Google Scholar]
- 18.Corbí AL, Vara A, Ursa A, García-rodríguez MC, Fontan G, Sanchez-madrid F. Molecular basis for a severe case of leukocyte adhesion deficiency. Eur J Immunol. 1992;22:1877. doi: 10.1002/eji.1830220730. [DOI] [PubMed] [Google Scholar]
- 19.Matsuura S, Kishi F, Tsukahara M, et al. Leukocyte adhesion deficiency: identification of novel mutations in two Japanese patients with a severe form. Biochem Biophys Res Commun. 1992;184:1460. doi: 10.1016/s0006-291x(05)80047-6. [DOI] [PubMed] [Google Scholar]
- 20.Nelson C, Rabb H, Arnaout MA. Genetic cause of leukocyte adhesion molecule deficiency. Abnormal splicing and a missense mutation in a conserved region of CD18 impair cell surface expression of beta 2 integrins. J Biol Chem. 1992;267:3351. [PubMed] [Google Scholar]
- 21.Sligh JE, Jr, Hurwitz MY, Zhu CM, Anderson DC, Beaudet AL. An initiation codon mutation in CD18 in association with the moderate phenotype of leukocyte adhesion deficiency. J Biol Chem. 1992;267:714. [PubMed] [Google Scholar]
- 22.Kishimoto TK, O'connor K, Springer TA. Leukocyte adhesion deficiency. Aberrant splicing of a conserved integrin sequence causes a moderate deficiency phenotype. J Biol Chem. 1989;264:3588. [PubMed] [Google Scholar]
- 23.López-rodríguez C, Nueda A, Grospierre B, et al. Characterization of two new CD18 alleles causing severe leukocyte adhesion deficiency. Eur J Immunol. 1993;23:2792. doi: 10.1002/eji.1830231111. [DOI] [PubMed] [Google Scholar]
- 24.Wright AH, Douglass WA, Taylor GM, et al. Molecular characterization of leukocyte adhesion deficiency in six patients. Eur J Immunol. 1995;25:717. doi: 10.1002/eji.1830250313. [DOI] [PubMed] [Google Scholar]
- 25.Back AL, Kerkering M, Baker D, Bauer TR, Embree LJ, Hickstein DD. A point mutation associated with leukocyte adhesion deficiency type 1 of moderate severity. Biochem Biophys Res Commun. 1993;193:912. doi: 10.1006/bbrc.1993.1712. [DOI] [PubMed] [Google Scholar]
- 26.Sligh JE, Jr, Anderson DC, Beaudet AL. A mutation in the initiation codon of the CD18 gene in a patient with the moderate phenotype of leukocyte adhesion deficiency. Am J Hum Genet. 1989;45:A219. [PubMed] [Google Scholar]
- 27.Arnaiz-villena A, Timón M, Rodríguez-gallego C, et al. Human T-cell activation deficiencies. Immunol Today. 1992;13:259. doi: 10.1016/0167-5699(92)90007-t. [DOI] [PubMed] [Google Scholar]
- 28.Regueiro JR, Rodríguez-gallego C, Arnaiz-villena A. Human T Lymphocyte Activation Deficiencies. Austin: R. G. Landes Co., CRC Press; 1994. [Google Scholar]
- 29.Biesinger B, Müller-fleckenstein I, Simmer B, et al. Stable growth transformation of human T lymphocytes by Herpes virus saimiri. Proc Natl Acad Sci USA. 1992;89:3116. doi: 10.1073/pnas.89.7.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bröker BM, Tsygankov AY, Müller-fleckenstein I, et al. Immortalization of human T cell clones by Herpes virus saimiri. J Immunol. 1993;151:1184. [PubMed] [Google Scholar]
- 31.Weber F, Meinl E, Drexler K, et al. Transformation of human T cell clones by herpesvirus saimiri: intact antigen recognition by autonomously growing myelin basic protein-specific T cells. Proc Natl Acad Sci USA. 1993;90:11049. doi: 10.1073/pnas.90.23.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodríguez-gallego C, Corell A, Pacheco A, et al. Herpes virus saimiri transformation of T cells in CD3γ immunodeficiency: phenotypic and functional characterization. J Immunol Methods. 1996;198:177. doi: 10.1016/s0022-1759(96)00156-1. [DOI] [PubMed] [Google Scholar]
- 33.Biesinger B, Trimble J, Desrosiers RC, Fleckenstein B. The divergence between two oncogenic Herpesvirus saimiri strains in a genomic region related to the transforming phenotype. Virology. 1990;176:505. doi: 10.1016/0042-6822(90)90020-r. [DOI] [PubMed] [Google Scholar]
- 34.Tiirikainen M. Evaluation of red blood cell lysing solutions for the detection of intracellular antigens by flow cytometry. Cytometry. 1995;20:341. doi: 10.1002/cyto.990200410. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook T, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 36.Arnaiz-villena A, Timón M, Corell A, Pérez-aciego P, Martín-villa JM, Regueiro JR. Primary immunodeficiency caused by mutations in the gene encoding the CD3-γ subunit of the T-lymphocyte receptor. N Engl J Med. 1992;327:529. doi: 10.1056/NEJM199208203270805. [DOI] [PubMed] [Google Scholar]
- 37.Saiki RK, Bugawan TL, Horn GT, Mullis KB, Erlich HA. Analysis of enzymatically amplified beta-globin and HLA-DQ alpha DNA with allele-specific oligonucleotide probes. Nature. 1986;324:163. doi: 10.1038/324163a0. [DOI] [PubMed] [Google Scholar]
- 38.Boeyum A. Separation of white blood cells. Nature. 1964;204:793. doi: 10.1038/204793a0. [DOI] [PubMed] [Google Scholar]
- 39.Seabright M. A rapid banding technique for human cytogenetics. Lancet. 1971;2:971. doi: 10.1016/s0140-6736(71)90287-x. [DOI] [PubMed] [Google Scholar]
- 40.Fassati A, Tedeschi S, Bordoni A, et al. Rapid direct diagnosis of deletions carriers of Duchenne and Becker muscular dystrophies. Lancet. 1994;344:302. doi: 10.1016/s0140-6736(94)91340-4. [DOI] [PubMed] [Google Scholar]
- 41.Altin JG, Pagler EB, Parish CR. Evidence for cell surface association of CD2 and LFA-1 (CD11a/CD18) on T lymphocytes. Eur J Immunol. 1994;24:450. doi: 10.1002/eji.1830240228. [DOI] [PubMed] [Google Scholar]
- 42.Schraven B, Wild M, Kirchgessner H, et al. Alterations of CD2 T association with T cell receptor signalling molecules in ‘CD2 unresponsive’ human T lymphocytes. Eur J Immunol. 1993;23:119. doi: 10.1002/eji.1830230119. [DOI] [PubMed] [Google Scholar]
- 43.Schraven B, Samstag Y, Altevogt P, Meuer SC. Association of CD2 and CD45 on human T lymphocytes. Nature. 1990;345:71. doi: 10.1038/345071a0. [DOI] [PubMed] [Google Scholar]
- 44.Brown MH, Centrell DA, Brattsand G, Crumpton MJ, Gullberg M. The CD2 antigen associates with the T-cell antigen receptor CD3 antigen complex on the surface of human T lymphocytes. Nature. 1989;339:551. doi: 10.1038/339551a0. [DOI] [PubMed] [Google Scholar]
- 45.Figdor CG, van Kooyk Y, Kyewski B. On the mode of action of LFA-1. Immunol Today. 1990;11:277. doi: 10.1016/0167-5699(90)90112-m. [DOI] [PubMed] [Google Scholar]
- 46.Sen J, Bossu P, Burakoff SJ, Abbas AK. T cell surface molecules regulating noncognate B lymphocyte activation. Role of CD2 and LFA-1. J Immunol. 1992;148:1037. [PubMed] [Google Scholar]
- 47.Yamada A, Kaneyuki T, Torimoto Y, Daley JF, Prado CM, Yokoyama MM. Signaling from LFA-1 contributes signal transduction through CD2 alternative pathway in T cell activation. Cell Immunol. 1992;142:145. doi: 10.1016/0008-8749(92)90276-u. [DOI] [PubMed] [Google Scholar]
- 48.Góngora R, Corell A, Regueiro JR, et al. Peripheral blood reduction of memory (CD29+, CD45R0+, and ‘bright’ CD2+ and LFA-1+) T lymphocytes in Papillon–Lefèvre Syndrome. Hum Immunol. 1994;41:185. doi: 10.1016/0198-8859(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 49.Porter CD, Parkar MH, Collins MK, Kinnon C. X-linked chronic granulomatous disease: correction of NADPH oxidase defect by retrovirus-mediated expression of gp91-phox. Blood. 1993;82:2196. [PubMed] [Google Scholar]