

Octaketide synthase from A. arborescens has been overexpressed in E. coli, purified and crystallized. Diffraction data have been collected to 2.6 Å.
Keywords: octaketide synthase, polyketide synthase, pentaketide chromone synthase
Abstract
Octaketide synthase (OKS) from Aloe arborescens is a plant-specific type III polyketide synthase that produces SEK4 and SEK4b from eight molecules of malonyl-CoA. Recombinant OKS expressed in Escherichia coli was crystallized by the hanging-drop vapour-diffusion method. The crystals belonged to space group I422, with unit-cell parameters a = b = 110.2, c = 281.4 Å, α = β = γ = 90.0°. Diffraction data were collected to 2.6 Å resolution using synchrotron radiation at BL24XU of SPring-8.
1. Introduction
Octaketide synthase (OKS; Abe, Oguro et al., 2005 ▶) and pentaketide chromone synthase (PCS; Abe, Utsumi et al., 2005 ▶) are novel plant-specific chalcone synthase superfamily type III polyketide synthases (PKSs) obtained from the medicinal plant Aloe arborescens. OKS catalyzes the iterative condensation of eight molecules of malonyl-CoA to produce SEK4 and SEK4b (Fig. 1 ▶ a), the longest polyketides generated by the structurally simple type III PKS and the shunt products of the type II PKS for actinorhodin (act from Streptomyces coelicolor; Fu et al., 1994 ▶). PCS produces 5,7-dihydroxy-2-methylchromone from five molecules of malonyl-CoA (Fig. 1 ▶ b). OKS and PCS share 91% amino-acid sequence identity (368 of 403 residues) and maintain a conserved Cys-His-Asn catalytic triad. The most characteristic feature is that the chalcone synthase active-site residue 207 (A. arborescens OKS numbering) is uniquely replaced by Gly in OKS and Met in PCS, respectively. Site-directed mutagenesis has revealed that the chemically inert single residue 207 lining the active-site cavity determines the polyketide-chain length and the product specificity; the octaketide-producing OKS is functionally transformed into a pentaketide-producing enzyme by the replacement of Gly207 with the more bulky Met (Abe, Oguro et al., 2005 ▶). Interestingly, the pentaketide product is a regioisomer of the 5,7-dihydroxy-2-methylchromone produced by PCS, which is formed by a C-6/C-1 Claisen-type cyclization (Fig. 1 ▶ b). In contrast, with the OKS G207M mutant a C-4/C-9 aldol-type cyclization yields 2,7-dihydroxy-5-methylchromone (Fig. 1 ▶ c). This suggests cryptic subtle structural differences in active-site geometry between the two enzymes. In previous work, we have reported the X-ray crystal structures of A. arborescens PCS, including the pentaketide-producing wild-type enzyme and the octaketide-producing M207G mutant enzyme (Morita et al., 2007 ▶). To further elucidate the structure–function relationship between PCS and OKS, we carried out a crystallographic analysis of A. arborescens OKS.
Figure 1.

Proposed pathways for the formation of (a) SEK4 and SEK4b from eight molecules of malonyl-CoA by OKS and the PCS M207G mutant, (b) 5,7-dihydroxy-2-methylchromone from five molecules of malonyl-CoA by PCS and (c) 2,7-dihydroxy-5-methylchromone from five molecules of malonyl-CoA by OKS G207M.
2. Experimental
2.1. Expression and purification
Recombinant OKS was expressed in Escherichia coli BL21(DE3)pLysS as a fusion protein with a hexahistidine tag at the C-terminus, as reported previously (Abe, Oguro et al., 2005 ▶). All of the following procedures were performed at 277 K. The cells were harvested by centrifugation at 5000g and resuspended in 50 mM Tris–HCl buffer pH 8.0 containing 0.2 M NaCl and 5% glycerol (buffer A). The cells were disrupted by sonication and were centrifuged at 12 000g for 30 min. The supernatant was loaded onto a Talon metal-affinity chromatography column (Clontech) equilibrated with buffer A. After washing the resin with 50 mM HEPES–NaOH buffer pH 7.0 containing 0.2 M NaCl and 5% glycerol (buffer B), the recombinant OKS was eluted with buffer B containing 300 mM imidazole. The protein solution was diluted fivefold with 50 mM HEPES–NaOH buffer pH 7.0 containing 5% glycerol and 2 mM DTT (buffer C) and was then applied onto a Resource-Q column (GE Healthcare). The column was washed with buffer C containing 50 mM NaCl and the protein was subsequently eluted using a linear gradient of 50–200 mM NaCl. The protein solution was further purified to homogeneity by chromatography on Superdex 200HR (10/300GL; GE Healthcare) and was concentrated to 10 mg ml−1 in 20 mM HEPES–NaOH pH 7.0 buffer containing 100 mM NaCl and 2 mM DTT.
A dynamic light-scattering (DLS) analysis was carried out using a DynaProMSXTC molecular-sizing instrument (Protein Solutions Inc.). After centrifugation with a 0.22 µm Ultrafree-MC filter (Millipore) to remove particulate material from the protein solution, the solution was used to monitor the solution properties of the purified protein. Data were acquired from 50 scattering measurements at 278 K; five sets of data were analyzed using the DYNAMICS software package (Protein Solutions Inc.) and averaged.
2.2. Crystallization and X-ray data collection
Initial crystallization attempts were carried out at 293 and 278 K using the sitting-drop vapour-diffusion method with a 96-condition crystallization screen originally designed by Mitsubishi Chemical Corporation. Crystals were observed in many crystallization conditions and one of the more promising crystallization conditions was further optimized. Cocrystallization attempts with CoA-SH, a common byproduct released from the substrates during the iterative condensation reactions of type III PKSs, were then carried out. Finally, diffraction-quality crystals were obtained at 278 K using 100 mM Tris–HCl buffer pH 8.5 containing 12%(w/v) PEG 8000, 150 mM calcium acetate and 2 mM CoA-SH with the hanging-drop vapour-diffusion method. The optimized crystallization drops were prepared by mixing 0.5 µl protein solution and an equal volume of reservoir solution and were equilibrated against 500 µl reservoir solution.
The crystals were transferred to reservoir solution containing 18%(v/v) glycerol as a cryoprotectant, picked up in a nylon loop after 20 s and then flash-cooled at 100 K in a nitrogen-gas stream. X-ray diffraction data sets were collected from a single crystal at SPring-8 beamline BL24XU using a Rigaku R-AXIS V imaging-plate area detector. The wavelength of the synchrotron radiation was 0.82656 Å and the distance between the crystal and the detector was 400 mm. A total of 180 frames were recorded with 1° oscillation and 48 s exposure time. Data were indexed, integrated and scaled with the HKL-2000 program package (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
Recombinant OKS was heterologously expressed in E. coli as a fusion protein with a hexahistidine tag at the C-terminus. The typical yield of protein was about 0.7 mg per litre of culture. The crystals appeared after a week and grew to approximate dimensions of 0.3 × 0.1 × 0.1 mm (Fig. 2 ▶). A complete data set was collected to 2.6 Å resolution. From the diffraction data collection, the space group was determined to be I422, with unit-cell parameters a = b = 110.2, c = 281.4 Å, α = β = γ = 90.0°. Detailed data-processing statistics are shown in Table 1 ▶. As previously confirmed by SDS–PAGE and gel-filtration analyses, the recombinant OKS, a 45 kDa protein, forms a homodimer, as do the other known type III PKSs (Abe, Oguro et al., 2005 ▶). DLS analysis after gel filtration revealed a monomodal distribution, with a polydispersity value of 17.1% and a molecular-weight estimate of 83 kDa, which is in good agreement with the results of the gel-filtration analysis. With two monomers in the asymmetric unit, the Matthews volume (V M; Matthews, 1968 ▶) was calculated to be 2.4 Å3 Da−1 and the estimated solvent content was 46.2%, which is in the range normally observed for protein crystals. A self-rotation function analysis using the CNS program (Brünger et al., 1998 ▶) indicated that the twofold symmetry axes are parallel to the crystallographic axes. Further structure determination using the molecular-replacement method, with the crystal structure of the A. arborescens PCS M207 mutant (PDB code 2d52; Morita et al., 2007 ▶) as a search model, is in progress.
Figure 2.

Crystals of A. arborescens OKS grown by the hanging-drop method. The dimensions of the largest crystal were approximately 0.3 × 0.1 × 0.1 mm.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Space group | I422 |
| Unit-cell parameters | |
| a = b (Å) | 110.2 |
| c (Å) | 281.4 |
| α = β = γ (°) | 90.0 |
| Resolution (Å) | 30.0–2.6 (2.69–2.60) |
| Unique reflections | 26897 |
| Redundancy | 12.1 (9.4) |
| Completeness (%) | 99.5 (97.9) |
| 〈I/σ(I)〉 | 42.9 (7.6) |
| Rsym† (%) | 9.3 (33.6) |
R
sym = 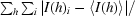
 , where I(h) is the intensity of reflection h,
, where I(h) is the intensity of reflection h,  is the sum over all reflections and
is the sum over all reflections and 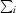 is the sum over i measurements of reflection h.
is the sum over i measurements of reflection h.
Acknowledgments
This work was supported in part by a grant from the National Project on Protein Structural and Functional Analyses.
References
- Abe, I., Oguro, S., Utsumi, Y., Sano, Y. & Noguchi, H. (2005). J. Am. Chem. Soc.127, 12709–12716. [DOI] [PubMed]
- Abe, I., Utsumi, Y., Oguro, S., Morita, H., Sano, Y. & Noguchi, H. (2005). J. Am. Chem. Soc.127, 1362–1363. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Fu, H., Hopwood, D. A. & Khosla, C. (1994). Chem. Biol.1, 205–210. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Morita, H., Kondo, S., Oguro, S., Noguchi, H., Sugio, S., Abe, I. & Kohno, T. (2007). Chem. Biol.14, 359–369. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]