

Abstract
A mitochondrial sulphonylurea-sensitive, ATP-sensitive K+ channel (mitoKATP) that is selectively inhibited by 5-hydroxydecanoate (5-HD) and activated by diazoxide has been implicated in ischaemic preconditioning. Here we re-evaluate the evidence for the existence of this mitoKATP by measuring changes in light scattering (A520) in parallel with direct determination of mitochondrial matrix volumes using 3H2O and [14C]sucrose. Incubation of rat liver and heart mitochondria in KCl medium containing Mg2+ and inorganic phosphate caused a decrease in light scattering over 5 min, which was accompanied by a small (15–30 %) increase in matrix volume. The presence of ATP or ADP in the buffer from the start greatly inhibited the decline in A520, whilst addition after a period of incubation (1–5 min) induced a rapid increase in A520, especially in heart mitochondria. Neither response was accompanied by a change in matrix volume, as measured isotopically. However, the effects of ATP and ADP on A520 were abolished by carboxyatractyloside and bongkrekic acid, inhibitors of the adenine nucleotide translocase (ANT) that lock the transporter in two discrete conformations and cause distinct changes in A520 in their own right. These data suggest that rather than matrix volume changes, the effects of ATP and ADP on A520 reflect changes in mitochondrial shape induced by conformational changes in the ANT. Furthermore, we were unable to demonstrate either a decrease in A520 or increase in matrix volume with a range of ATP-sensitive K+ channel openers such as diazoxide. Nor did glibencamide or 5-HD cause any reduction of matrix volume, whereas the K+ ionophore valinomycin (0.2 nm), produced a 10–20 % increase in matrix volume that was readily detectable by both techniques. Our data argue against the existence of a sulphonylurea-inhibitable mitoKATP channel.
It has been proposed that mitochondrial sulphonylurea-sensitive, ATP-sensitive K+ channels (mitoKATP) may be involved in mediating the effects of ischaemic preconditioning (IPC) of the heart. Much of the evidence for this hypothesis has come from the ability of diazoxide and 5-hydroxydecanoate (5-HD) to mimic and antagonise IPC, since these reagents are regarded as relatively specific openers and blockers of the mitoKATP channel, respectively (Szewczyk & Marban, 1999; Ghosh et al. 2000; Grover & Garlid, 2000). In addition, O'Rourke, Marban and colleagues have demonstrated that diazoxide can oxidise mitochondrial flavoproteins in cardiac myocytes, and argue that this reflects opening of a mitochondrial ATP-sensitive K+ (KATP) channel with resultant mitochondrial depolarisation and stimulation of the respiratory chain (Liu et al. 1998, 1999; Sato et al. 1998, 2000). The effect was blocked by 5-HD. However, other workers have failed to detect these effects (Lawrence et al. 2001), and there are an increasing number of reports in which it is suggested that diazoxide is not as specific for mitoKATP channels as some have claimed. It was demonstrated more than 30 years ago that diazoxide inhibits succinate dehydrogenase activity with the result that the citric acid cycle is blocked and mitochondrial flavoproteins become oxidised (Schäfer et al. 1971). These data have been confirmed more recently by others (Grimmsman & Rustenbeck, 1998; Kowaltowski et al. 2001; Hanley et al. 2002; Lim et al. 2002). In addition, diazoxide has been shown to open the plasma membrane KATP channel at low concentrations when ADP is present (as it will be in the cell; D'Hahan et al. 1999). Furthermore, in mice lacking the plasma membrane K+ channel Kir6.2, IPC was ineffective in reducing infarct size (Suzuki et al. 2002). Finally, diazoxide has been reported to increase the mitochondrial production of reactive oxygen species (ROS) by an unknown mechanism (Pain et al. 2000; Forbes et al. 2001; Liu & O'Rourke, 2001), and ROS have been implicated in IPC (Baines et al. 1997; Vanden Hoek et al. 1998). The use of 5-HD as a specific mitoKATP channel inhibitor is also open to question, since it is a racemic mix of d and l isoforms of a substituted fatty acid that can be activated to its coenzyme A derivative and then act as either a substrate or inhibitor of fatty acid β-oxidation (Hanley et al. 2002; Lim et al. 2002).
It is generally accepted that opening of mitoKATP channels will cause an increase in matrix volume and that this in turn will activate the respiratory chain, providing more ATP to support the recovering heart (Halestrap, 1989,1994; Grover & Garlid, 2000; O'Rourke, 2000). In extensive earlier studies from this laboratory we demonstrated that hormones that elevate mitochondrial Ca2+ in the liver increase the mitochondrial matrix volume by enhancing K+ influx. However, we found no evidence that a sulphonylurea-sensitive K+ channel inhibited by extramitochondrial ATP was responsible, but rather demonstrated a mechanism that was sensitive to matrix [ATP] and involved the adenine nucleotide translocase (ANT, Halestrap, 1989,1994). The first published data that provided direct evidence for the existence of mitoKATP channels came from patch clamping of mitochondria (Inoue et al. 1991). However, there was little evidence to prove that it was actually the inner mitochondrial membrane that was patched rather than the outer mitochondrial membrane or contaminating endoplasmic reticulum and plasma membrane. Indeed, it is known that preparations of isolated mitochondria are extensively contaminated with plasma membranes unless further purified by density gradient centrifugation (Halestrap, 1987; Whipps et al. 1987). Subsequently, characterisation of the mitoKATP channel has utilised light-scattering techniques with isolated mitochondria (Garlid et al. 1996; Yarovyarovoy et al. 1997; Jaburek et al. 1998; Kowaltowski et al. 2001) and reconstitution of detergent solubilised mitochondrial membranes into proteoliposomes (Paucek et al. 1992, 1996; Garlid et al. 1996; Yarovyarovoy et al. 1997; Zhang et al. 2001). The reconstitution experiments were performed with solubilised membranes from isolated mitochondria, and thus it is possible that some contamination with KATP channels from the plasma membrane may have occurred. No data were presented to discount such contamination, although comparison of the dose-response curves of reconstituted plasma membrane and mitochondrial KATP channels for a range of openers might be taken as evidence that this is not the case (Garlid et al. 1996). The use of light scattering to measure changes in matrix volume and thus K+ transport is also questionable since it is well established that ATP and ADP can induce conformational changes in the mitochondria that lead to an increase in light scattering without any change in matrix volume (Klingenberg et al. 1971; Stoner & Sirak, 1973a,b; Halestrap & Davidson, 1990; Doran & Halestrap, 2000).
With the emergence of the putative mitoKATP channel as a potential target of IPC, we decided to re-evaluate the evidence for mitoKATP channels by monitoring their activation and inhibition in isolated mitochondria following addition of putative effectors. This was achieved by measuring changes in matrix volume using a combination of light scattering and isotopic measurements. Despite extensive attempts, we were unable to find any convincing evidence for the existence of a mitoKATP channel. This is consistent with the absence of any sequences in the human genomic database that are likely candidates for a mitochondrial sulphonylurea receptor.
METHODS
Materials
Diazoxide and glibencamide were from Sigma (Gillingham, Dorset, UK), cromakalin from Tocris (Bristol, UK), pinacidil and the nicorandil analogue, N-[2-(acetoxy)ethyl]-3-pyridinecarboxamide from ICN Biomedicals (Basingstoke, UK). 3H2O and [14C]sucrose were from Amersham Biosciences (Little Chalfont, Buckinghamshire, UK).
Methods
Isolation of liver and heart mitochondria
Male Wistar rats (250–275 g) were killed by cervical dislocation, and hearts and livers rapidly removed and placed in ice-cold sucrose buffer (mm): sucrose 300, Tris-HCl 10, EGTA 2; pH 7.4). Each heart was homogenised in 3.5 ml sucrose buffer using a Polytron homogeniser (Fisher, Loughborough, UK) at setting 3 for 5 s. Homogenates from three hearts were diluted to 40 ml with sucrose buffer containing 5 mg ml−1 bovine serum albumin (BSA) and the homogenate centrifuged for 2 min at 2000 g and 4 °C to sediment cell debris. The supernatant was then centrifuged at 10 000 g for 5 min (4 °C) to sediment a crude mitochondrial pellet, which was then resuspended in 12 ml of ice-cold sucrose buffer containing 19 % (w/v) Percoll. Following centrifugation (14 000 g) for 10 min at 4 °C, the mitochondria formed a loose pellet that was retained, whilst membrane contaminants were removed with the supernatant, which was discarded (Halestrap, 1987). The mitochondria were resuspended in 40 ml sucrose buffer and centrifuged (4 °C) for 5 min at 10 000 g. The final mitochondrial pellet was resuspended at about 30 mg protein ml−1 and kept on ice until use (< 3 h). The yield of mitochondria was routinely about 6 mg protein per heart. For liver mitochondria, the procedure was similar except that each liver (8–10 g) was divided into two and each half homogenised using a Dounce Potter homogeniser in 40 ml of sucrose buffer containing 5 mg ml−1 BSA. Cell debris was removed by centrifugation at 600 g for 10 min and the resulting supernatant subjected to the same high-speed centrifugations as employed for heart mitochondria. Mitochondria were stored on ice in sucrose buffer at 60 mg protein ml−1. The yield of liver mitochondria was about 60 mg protein per liver.
Combined light scattering and radioisotope measurements of changes in mitochondrial matrix volume
Absolute determination of the matrix volume of mitochondria requires measurement of the matrix water content and this is most usually performed by incubating mitochondria with 3H2O and analysing the [3H] content of the mitochondrial pellet after centrifugation. In order to correct for the 3H2O trapped between mitochondria and within the intermembrane space, [14C]sucrose is also included in the medium, since this enters the intermembrane space but not the matrix. The [14C] and [3H] content of both the mitochondrial pellet and a sample of the supernatant can then be used to calculate the matrix volume (Halestrap, 1989). For monitoring changes in mitochondrial volume with time, light scattering can be used, since an increase in matrix volume is accompanied by a decrease in light scattering. This is measured as a decrease in A520, since 520 nm is an isosbestic point for the mitochondrial cytochromes and thus insensitive to changes in redox state. The absolute values for A520 are dependent on several factors in addition to the matrix volume, including the concentration of the mitochondria and the optical geometry of the spectrophotometer. In addition, light scattering is sensitive to changes in the intermembrane space and the shape of the mitochondria. As such it is a technique that is better suited for monitoring the rate of change of matrix volume, rather than determination of absolute values (Halestrap, 1989).
The incubation medium routinely contained (mm): KCl 125, Mops 20, Tris 10, EGTA 0.5, KH2PO4 2.5, MgCl2 2.5, succinate 5, oligomycin 1 μm and rotenone 0.2 μm, pH 7.2, and was gassed with 100 % O2. For some experiments this was supplemented with 1 mm ATP + 10 mm KHCO3 and a gas phase of O2:CO2, 95 %:5 % employed to mimic more closely the intracellular conditions. Equilibration of 5 % CO2 in the gas phase with 10 mm KHCO3 in the buffer maintains a pH of 7.2 at 25 °C. Mitochondria were incubated in the required buffer at 25 °C and 1 and 2 mg protein ml−1 for heart and liver mitochondria, respectively. Incubations were performed in 4 ml cuvettes contained in the temperature-controlled housing of a split-beam spectrophotometer that was constantly gassed with 100 % O2 or O2:CO2, 95 %:5 % as appropriate. An overhead stirrer was employed to maintain oxygenation of the mitochondria. Where changes in volume and light scattering (A520) with time were to be determined, mitochondrial suspension was injected only into the sample cuvette, whilst the reference cuvette was replaced with a neutral filter to balance the signal. For measuring the effect of added agents at a fixed time, mitochondria were incubated in both cuvettes, and after equilibration for 1 min the required effector was added only to the sample cuvette as required. At the end of the incubation, usually a further 4 min, 3H2O (370 Bq) and [14C]sucrose (74 Bq) were added to each cuvette and data recording terminated. Four 0.8 ml samples were then rapidly transferred to 1.5 ml microcentrifuge tubes and centrifuged at 14 000 g for 1 min. The supernatant was removed whilst the mitochondrial pellets were lysed with 500 μl water. Both pellet and supernatant samples were deproteinised with perchloric acid (2 % w/v final) and 500 μl of pellet extract and 100 μl of the supernatant sample was added to 10 ml scintillation fluid (Emulsifier Safe, PerkinElmer Life Sciences, Zaventem, Belgium) plus water to give a total aqueous volume of 1 ml. 3H and 14C were determined by scintillation counting using a Packard TriCarb 2100-TR scintillation counter. In order to resolve small differences (< 10 %) in matrix volumes, all samples were subjected to three cycles of counting, and great care was taken to avoid evaporation of 3H2O. The procedure used for calculating matrix volume from the 3H and 14C dpm was that described previously (Halestrap & McGivan, 1979; Halestrap, 1989). The protein content of each mitochondrial pellet was determined by dissolving the perchloric acid precipitated protein in 3 ml biuret reagent and measuring the A540 using bovine serum albumin BSA as a standard. Volumes are expressed as μl (mg mitochondrial protein)−1.
Statistical analysis
Data are expressed as means ±s.e.m., with the statistical significance of each effector being analysed using Student's paired t test.
RESULTS
The effects of ATP and ADP on the time course of mitochondrial swelling and light scattering in KCl medium
Several studies have shown that mitochondria isolated in sucrose medium and then incubated in KCl medium under energised conditions show a decrease in light scattering that is taken to indicate matrix swelling. However, in none of these studies was matrix volume actually measured directly with isotopes. In Fig. 1 we present typical data of such light scattering experiments for both heart (A) and liver (B) mitochondria and companion data for the changes in matrix volume measured with 3H2O and [14C]sucrose. Although we could detect the decrease in light scattering (A520) over the first 5 min observed by others, the increase in matrix volume measured isotopically was small and variable (10–30 %). This is consistent with previous data from this laboratory (Halestrap & Dunlop, 1986; Halestrap, 1987). The presence of 0.2 mm ATP or ADP in the medium (in the presence of oligomycin) caused a major reduction in the rate and extent of the decrease in A520 with heart mitochondria, in agreement with data from other laboratories; a similar but smaller effect was seen in liver mitochondria. These data might be interpreted as ATP and ADP inhibiting the entry of K+ into mitochondria, slowing the swelling and decrease in light scattering. However, as shown in Fig. 1, when the matrix volumes were measured directly, it was found that the effects of ATP and ADP on the change in A520 were not accompanied by any change in matrix volume measured with 3H2O and [14C]sucrose.
Figure 1. The effects of ATP and ADP on the light scattering response and measured matrix volume of heart (A) and liver (B) mitochondria.
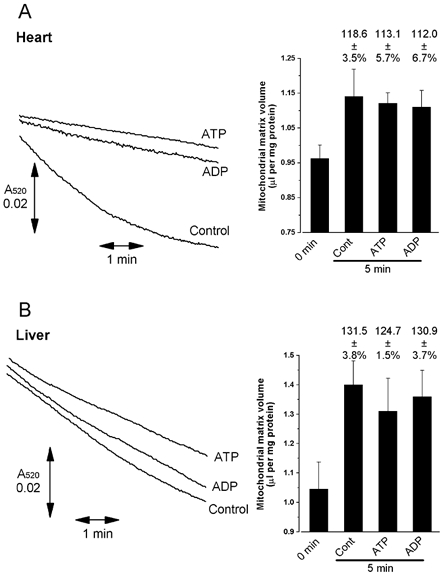
Heart mitochondria were added through an injection port into 3.5 ml standard KCl buffer contained within the sample cuvette of the spectrophotometer and incubated with continuous stirring at 25 °C under a gas phase of 100 % O2, as outlined in Methods. This enabled changes in light scattering to be recorded immediately following complete mixing of the mitochondrial suspension with the buffer (within 5 s of addition of the mitochondria). Where indicated by the labelling to the right of the trace, 0.2 mm ATP or ADP were present in the medium from the start. After 5 min, 3H2O and [14C]sucrose were added to the cuvette and matrix volumes determined as described under Methods. For determination of matrix volumes at time zero, mitochondria were added to ice-cold buffer containing isotopes, and volumes were determined immediately. Data from three separate experiments on different batches of mitochondria are summarised in the accompanying bar graphs. Error bars represent s.e.m. and the numbers above each bar the mean (±s.e.m.) volume after incubation expressed as a percentage of the time zero value. Cont = control.
Changes in light scattering induced by ATP and ADP reflect conformational changes of the ANT rather than changes in the matrix volume
In Fig. 2A we demonstrate that the presence of 0.2 mm ATP or ADP not only inhibited the initial decrease in light scattering of heart mitochondria (traces i-iv compared to traces v-viii), but also rapidly reversed it when added after a period of incubation in their absence (traces v-xii). This rapid increase in A520 was greater if mitochondria had been preincubated for 5 min (traces v and viiii) than for 1 min (traces ix-xii). The speed and magnitude of this increase in light scattering was reminiscent of previous data obtained in this laboratory (Halestrap, 1987) and elsewhere (Klingenberg et al. 1971) demonstrating that conformational changes of the ANT are accompanied by changes in the shape of heart mitochondria that may be detected by light scattering, but not in their matrix volume. Such changes in the ANT conformation were induced by the addition of the two inhibitors of the ANT, carboxyatractyloside (CAT) and bongkrekic acid (BKA), that trap the transporter in the ‘c’ and ‘m’ conformations, respectively (Klingenberg, 1980). We have used these inhibitors to confirm that the effects of ATP and ADP on light scattering also reflect such conformational changes of the ANT. These data are presented in Fig. 2A and B.
Figure 2. The effects of adenine nucleotide translocase ligands on the changes in light scattering of heart mitochondria induced by ATP and ADP.
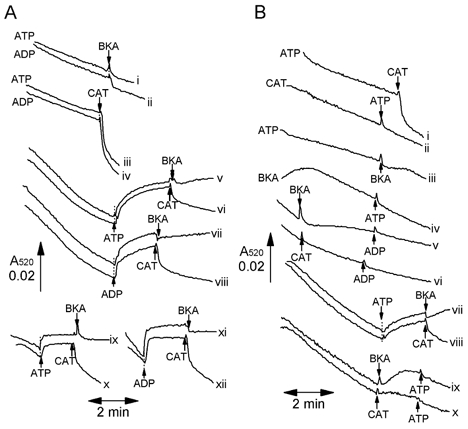
Heart mitochondria were added through an injection port into incubation buffer contained within the sample cuvette of the spectrophotometer as described in Fig. 1, and additions of ATP (0.2 mm), ADP (0.2 mm), bongkrekic acid (BKA, 10 μm) or carboxyatractyloside (CAT, 10 μm) made as indicated by the arrows. In some incubations (indicated by the label to the left of the trace) these effectors were present in the buffer before addition of mitochondria. A and B, representative traces from two separate experiments with different mitochondrial preparations. All traces shown are typical of at least three separate experiments.
In Fig. 2A we demonstrate that in the presence of either ATP or ADP, CAT (traces iii and iv) but not BKA (traces i and ii) caused a rapid decrease in light scattering. This effect was greater if the ATP and ADP were present from the start of the incubation (traces i-iv) than if it were added after preincubation for 1 min (traces ix-xii) or 5 min (traces v-viii). In the absence of ATP or ADP, addition of BKA induced an increase in A520 in its own right that prevented subsequent addition of ATP or ADP from causing an increase in A520 (Fig. 2B traces iii-v, vii, ix). In contrast, CAT added in the absence of ATP slightly reduced the rate of decrease in light scattering, but unlike BKA did not reverse it. However, it shared with BKA the ability to prevent subsequent addition of ATP or ADP from inducing an increase in A520 (Fig. 2B traces ii, vi, x). These data strongly support our contention that changes in light scattering induced by ATP and ADP reflect changes in the ANT conformation rather than matrix volume, although similar observations by Beavis et al. (1993) were interpreted as reflecting an interaction of the ANT with the mitoKATP channel.
In order to ascertain which explanation is correct, measurements of matrix volume with 3H2O and [14C]sucrose were made in parallel with the light scattering measurements, and these data are reported in Fig. 3. Note that in these experiments, the spectrophotometer was used in ‘split-beam’ mode with the mitochondrial suspension being present in both sample and reference cuvettes, and additions of the required reagent being made only to the sample cuvette. Under these conditions the changes observed in A520 represent the difference in light scattering induced by the reagent relative to the control incubation without added reagent. In the experiments reported in Fig. 3, neither the increase in A520 induced by addition of ATP or ADP, nor the subsequent decrease in A520 upon addition of CAT, were accompanied by any detectable change in matrix volume. In contrast, the addition of the K+ ionophore valinomycin to produce a change in light scattering of similar magnitude to that induced by CAT did give the anticipated increase in matrix volume. Increases of 110.6 ± 2.27 (P < 0.02) and 123.4 ± 1.93 (P < 0.01) were determined at 0.2 and 0.5 nm (0.2 and 0.5 pmol (mg protein)−1), respectively, consistent with previous data from this laboratory (Halestrap et al. 1986; Halestrap, 1987). Thus we can be confident that if the changes in light scattering induced by ATP and ADP were caused by a change in matrix volume, we should have detected it. In the same series of experiments we included the addition of glibencamide, a sulphonylurea that is reported to inhibit the mitoKATP with a K1/2 of about 2 μm at the Mg2+ concentration (2.5 mm) used here (Paucek et al. 1992; Szewczyk et al. 1997). A very slight increase in A520 was observed at 50 μm, but this was not accompanied by a detectable decrease in matrix volume. No significant effect on A520 was observed at 2 or 10 μm glibencamide (data not shown). The effects of other putative mitoKATP channel openers or blockers will be considered in detail below.
Figure 3. Changes in light scattering of heart mitochondria induced by ATP and CAT are not accompanied by changes in isotopically measured matrix volume.
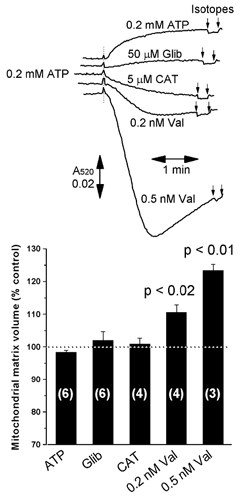
Heart mitochondria were incubated under the same conditions as described for Figs 1 and 2, but using both sample and reference cuvettes of the spectrophotometer as described under Methods. After 1 min incubation in the cuvette the indicated reagent was added to the mitochondrial suspension in the sample cuvette only, and after a further 2 min 3H2O and [14C]sucrose were added to both cuvettes, and matrix volumes were determined as described under Methods. In the bar graph, the changes in matrix volume induced by the reagent added are expressed as the percentage of the volumes in the absence of reagent (reference cuvette). Data are presented as the means ±s.e.m. (error bars) of the number of experiments shown, each performed with a separate mitochondrial preparation. When CAT (10 μm) was added, 0.2 mm ATP was present in both the sample and reference cuvettes from the start of the incubation. The mean control matrix volume (reference cuvette) for the total of six mitochondrial preparations represented was 1.26 ± 0.10 μl (mg protein)−1. Glib, glibenclamide; Val, valinomycin.
Changes in light scattering induced by ATP are enhanced by phosphate and Mg2+
The experiments reported above were all carried out in the presence of 2.5 mm inorganic phosphate (Pi) and 2.5 mm Mg2+ since previous work from Garlid's laboratory had shown that the effects of ATP could only be observed under such conditions (Paucek et al. 1992; Garlid et al. 1996; Jaburek et al. 1998). In Fig. 4A we confirm that both Pi and Mg2+ are required to produce the maximal increase in light scattering induced by ATP. However, in Fig. 4B we demonstrate that in the absence of Pi and Mg2+ (trace i) the decrease in light scattering during 5 min incubation is much less than in the presence of these agents (trace iii), but similar to that observed in the presence of ATP, Pi and Mg2+ (trace ii). Thus our data suggest that Mg2+ and Pi induce a decrease in light scattering that is inhibited by the presence of ATP, but that this is not associated with any change in matrix volume (Fig. 3). Once again these data imply that changes in A520 are detecting alterations in mitochondrial shape rather than matrix volume.
Figure 4. Effects of phosphate and Mg2+ ions on the increase in light scattering of heart mitochondria induced by ATP.
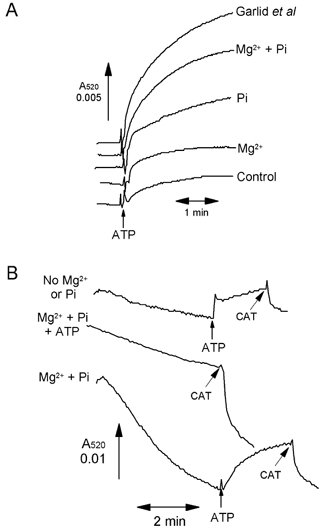
A, experiments were performed as in Fig. 3, with mitochondria present in both sample and reference cuvettes, but the medium was varied as indicated. Where shown, ATP (0.2 mm) was added to the sample cuvette only. The control medium lacked the 2.5 mm MgCl2 (Mg2+) and 2.5 mm potassium phosphate (Pi) usually present, and these reagents were added back as indicated. In addition, a parallel experiment was performed using the buffer described by Garlid and colleagues (Kowaltowski et al. 2001). B, mitochondria were present only in the sample cuvette as for Figs 1 and 2. Incubation was performed in the absence and presence of 0.2 mm ATP, 2.5 mm MgCl2 and 2.5 mm phosphate, as shown by the label to the left of the trace, and further additions of CAT (10 μm) or 0.2 mm ATP were made as indicated by the arrows.
The effects of KATP openers and blockers on the matrix volume of isolated mitochondria
Studies from Garlid's laboratory using a range of KATP channel openers and blockers has led to the proposal that diazoxide and 5-hydroxydecanoate have a much higher affinity for the mitoKATP channel than for the sarcolemmal KATP channel (Garlid et al. 1996). In view of the effects of these agents to mimic and oppose IPC, respectively, these agents have been used extensively to implicate mitoKATP channels in IPC (Szewczyk & Marban, 1999; Ghosh et al. 2000; Grover & Garlid, 2000). However, no studies have been reported on the effects of these agents on the matrix volume of isolated mitochondria determined directly with 3H2O and [14C]sucrose. In Fig. 5 we present data from such experiments employing a range of KATP channel openers and blockers with mitochondria incubated under conditions designed to mimic the physiological setting. For this purpose we used the basic KCl medium supplemented with 1 mm ATP and 10 mm bicarbonate, and gassed with O2:CO2 (19:1) to maintain a pH of 7.2. Changes in matrix volume were followed in real time using light scattering and quantitative measurements made in parallel using 3H2O and [14C]sucrose. Our experiments were routinely performed at 25 °C to ensure that the integrity of the mitochondria was maintained during the incubation. However, we confirmed that similar light-scattering results were obtained at 37 °C and some of these data are shown for comparison. We also confirmed that identical results were obtained in the absence of bicarbonate (data not shown).
Figure 5. ATP-sensitive K+ (KATP) channel openers and blockers are without effect on heart or liver mitochondrial matrix volume.
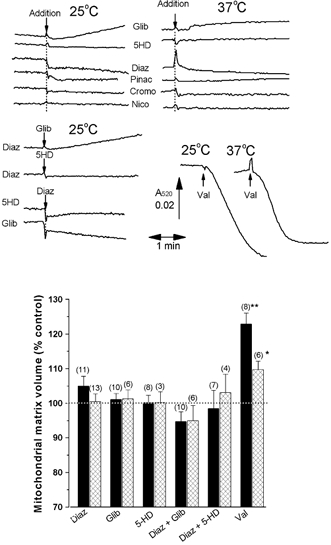
The experimental protocol was the same as described in Fig. 3, but the buffer used was supplemented with 10 mm KHCO3 and 1 mm ATP, and the gas phase 95 % O2:5 % CO2 to mimic physiological conditions, as explained under Methods. Where noted, to the left of the trace of light scattering, the medium was supplemented with 50 μm glibencamide (Glib), 5-hydroxydecanoate (5-HD) or diazoxide (Diaz). Where indicated, additions were made to the sample cuvette of 0.2 nm valinomycin (Val) or 50 μm glibencamide (Glib), 5-HD, diazoxide (Diaz), pinacidil (Pinac), cromokalin (Cromo) and the nicorandil analogue N-[2-(acetoxy)ethyl]-3-pyridinecarboxamide (Nico). At the end of each run 3H2O and [14C]sucrose were added to both cuvettes and matrix volumes were determined as described under Methods. In the bar graph beneath, the change in matrix volume induced by the reagent added is expressed as the percentage of the volume in the absence of reagent (reference cuvette), presented as the means ±s.e.m. (error bars) of the number of experiments shown, each performed with a separate mitochondrial preparation. Data are given for both heart (black bars) and liver (cross-hatched bars) mitochondria. Traces of light scattering are only shown for heart mitochondria, but essentially identical results were found for liver mitochondria. The effects of valinomycin were statistically significant by Student's t test (*P < 0.02; **P < 0.01).
The data of Fig. 5 confirm that neither 5-HD (50 μm) nor diazoxide (50 μm) added alone or together caused any change in the matrix volume of heart or liver mitochondria, whether measured isotopically or with light scattering. These concentrations are well above the K1/2 values reported by others (Garlid et al. 1996; Szewczyk et al. 1996; Bajgar et al. 2001). Glibencamide (50 μm) was also without effect. In order to confirm that small changes in matrix volume could be detected under these conditions, we demonstrated that addition of 0.2 nm valinomycin, a K+ ionophore, gave the expected decrease in light scattering (Fig. 5A). This was accompanied by percentage increases in matrix volume measured isotopically 122.9 ± 3.21 (n = 4; P < 0.01) and 109.7 ± 2.50 (n = 6; P < 0.02) for heart and liver mitochondria, respectively. The smaller increase in volume for liver mitochondria is a consequence of the different mitochondrial protein concentrations used (2 mg ml−1 for liver and 1 mg ml−1 for heart mitochondria, equivalent to 0.1 and 0.2 pmol valinomycin (mg protein)−1, respectively). The increases in matrix volume induced by such low valinomycin concentrations are similar to those we have measured previously (Quinlan et al. 1983; Quinlan & Halestrap, 1986; Halestrap, 1987). Cromakalin and pinacidil have also been reported to open the mitoKATP channel (Garlid et al. 1996; Szewczyk et al. 1996; Bajgar et al. 2001), but at 50 μm we found these agents were also without effect on the light scattering of heart mitochondria. Nor did the nicorandil analogue, N-[2-(acetoxy)ethyl]-3-pyridinecarboxamide, another KATP channel opener, have any effect on light scattering.
DISCUSSION
The data we have presented in this paper reveal that the changes in mitochondrial light scattering induced by ATP do not reflect changes in matrix volume (measured isotopically) and thus are unlikely to represent inhibition of mitochondrial K+ entry. Rather, the effects of CAT and BKA implicate changes in the conformation of the ANT that are known to cause changes in mitochondrial morphology, revealed as changes in light scattering (Klingenberg et al. 1971; Stoner & Sirak, 1973a,b; Halestrap & Davidson, 1990). These effects are much more profound in heart than liver mitochondria (Fig. 1 and Klingenberg et al. 1971), probably because the latter have only about 25 % of the ANT content of heart mitochondria (Klingenberg, 1980). It is known that interactions between the inner and outer membrane can influence light scattering independently of matrix volume (Beavis et al. 1985), perhaps through a process involving the formation and breakage of contact sites that are rich in the ANT (Doran & Halestrap, 2000; Vyssokikh et al. 2001). Indeed, even the initial decrease in light scattering upon addition of mitochondria to the KCl medium is not accompanied by a large increase in matrix volume (Fig. 1), something we have shown previously (Halestrap & Dunlop, 1986; Halestrap, 1987).
Our data confirm that of Garlid and colleagues (Paucek et al. 1992) that the effects of adenine nucleotides on light scattering are most readily detected in the presence of Mg2+ and Pi (Fig. 4). These workers have suggested that it is ATP (or ADP) complexed with Mg2+ that is the active ligand for inhibiting the mitoKATP channel and that phosphate acts as a charge-compensating anion, providing net uptake of potassium phosphate sufficient to induce osmotic swelling. However, our data show that the presence of adenine nucleotides from the start of the incubation prevents the decrease in light scattering induced by Mg2+ and Pi and partially reverses the effect if added after a period of incubation. The effect of ATP and ADP are reversed by CAT but not by BKA (Fig. 2A), which itself causes an increase in A520 (Fig. 2B). Thus it would seem probable that all of these effectors are working through an interaction with the ANT. One explanation for the observed results would be that, as isolated in sucrose medium (low ionic strength), the ANT is present largely in the ‘m’ conformation with ATP or ADP bound to the external substrate binding site. Upon incubation in the KCl medium (high ionic strength), the adenine nucleotides will be displaced, especially in the presence of phosphate, which can bind weakly to the ANT in competition with adenine nucleotides (Asimakis & Conti, 1985; Wilson & Asimakis, 1987). Displacement of ATP and ADP will be further enhanced by Mg2+, which will readily form MgATP and MgADP complexes with bound adenine nucleotides, displacing them from the ANT (Klingenberg, 1980). Mg2+ may have additional effects on the conformation of the ANT since it is known that divalent cations such as Mg2+ and Ca2+ can interact with it (Boos, 1982).
Thus, although we agree with other workers that ATP and ADP do cause changes in light scattering of mitochondria consistent with inhibition of a mitoKATP channel, our data suggest that the explanation of these changes in light scattering is unrelated to such channel activity. Furthermore, we were unable to demonstrate any effects of putative mitoKATP channel openers and blockers, such as diazoxide and 5-HD, on light scattering under conditions in which 0.2 nm valinomycin produced reproducible increases in matrix volume and decreases in A520 (Fig. 4 and Fig. 6). We have attempted to replicate exactly the various protocols described for the published data of Garlid and colleagues (Garlid et al. 1996; Jaburek et al. 1998; Bajgar et al. 2001), including differences in the respiratory substrate and buffer composition, but have still been unable to observe any appreciable change in light scattering induced by any of these agents. We are unable to offer an explanation for why our data differ from those of Garlid and colleagues. Whatever the reason for the difference, our isotopic measurements of matrix volume lead us to conclude that none of these mitoKATP openers or blockers actually affect mitochondrial K+, entry and thus cast doubt on the existence of a mitoKATP channel.
We conclude that there is no strong evidence for the existence of a mitoKATP channel related to the sulphonylurea-sensitive channels of the plasma membrane. That is not to dismiss the presence of mitochondrial K+ channels, for there is extensive evidence for the existence of electrogenic K+ uptake pathways working in combination with a K+/H+ antiporter to regulate the matrix volume (Halestrap, 1989; Brierley et al. 1994; Bernardi, 1999; Garlid & Paucek, 2001). Indeed, changes in matrix volume, most probably mediated by increased matrix [Ca2+], may play a vital role in the regulation of mitochondrial function in the heart and liver (Halestrap, 1989,1994; Kowaltowski et al. 2001; Xu et al. 2002). However, our data provide further evidence that caution should be applied when using agents such as 5-HD and diazoxide as pharmacological tools to probe the role of the putative mitoKATP channel in IPC. Rather, other effects of these agents may account for their ability to mimic and oppose the effects of IPC. For diazoxide, alternative mechanisms include opening of the plasma membrane KATP channel (D'Hahan et al. 1999; Suzuki et al. 2002) or inhibition of mitochondrial respiration (Schäfer et al. 1971; Grimmsman & Rustenbeck, 1998; Kowaltowski et al. 2001; Hanley et al. 2002; Lim et al. 2002) with concomitant production of ROS, which are known to be critical in mediating IPC (Baines et al. 1997; Vanden Hoek et al. 1998; Pain et al. 2000; Forbes et al. 2001), perhaps through activation of protein kinase pathways (Kawamura et al. 1998; Wang & Ashraf, 1999; Baines et al. 1999; Wang et al. 1999). For 5-HD, alternative mechanisms include its activation to 5-HD-CoA, which in turn may affect fatty acid oxidation and other aspects of mitochondrial metabolism (Hanley et al. 2002; Lim et al. 2002).
Acknowledgments
This work was supported by a project grant from the British Heart Foundation.
REFERENCES
- Asimakis GK, Conti VR. Phosphate-induced efflux of adenine nucleotides from heart mitochondria. Am J Physiol. 1985;249:H1009–1016. doi: 10.1152/ajpheart.1985.249.5.H1009. [DOI] [PubMed] [Google Scholar]
- Baines CP, Cohen MV, Downey JM. Signal transduction in ischemic preconditioning: The role of kinases and mitochondrial K-ATP channels. J Cardiovasc Electrophysiol. 1999;10:741–754. doi: 10.1111/j.1540-8167.1999.tb00251.x. [DOI] [PubMed] [Google Scholar]
- Baines CP, Goto M, Downey JM. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J Mol Cell Cardiol. 1997;29:207–216. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- Bajgar R, Seetharaman S, Kowaltowski AJ, Garlid KD, Paucek P. Identification and properties of a novel intracellular (mitochondrial) ATP-sensitive potassium channel in brain. J Biol Chem. 2001;276:33369–33374. doi: 10.1074/jbc.M103320200. [DOI] [PubMed] [Google Scholar]
- Beavis AD, Brannan RD, Garlid KD. Swelling and contraction of the mitochondrial matrix. 1. A structural interpretation of the relationship between light scattering and mitochondrial volume. J Biol Chem. 1985;260:1324–13433. [PubMed] [Google Scholar]
- Beavis AD, Lu Y, Garlid KD. On the regulation of K+ uniport in intact mitochondria by adenine nucleotides and nucleotide analogs. J Biol Chem. 1993;268:997–1004. [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Boos KS. ADP and ATP transport in mitochondriA Evidence for a metal-ion involved in transport catalysis by use of metallochromic indicators. Biochim Biophys Acta. 1982;693:68–74. doi: 10.1016/0005-2736(82)90471-0. [DOI] [PubMed] [Google Scholar]
- Brierley GP, Baysal K, Jung DW. Cation transport systems in mitochondria: Na+ and K+ uniports and exchangers. J Bioenerg Biomembr. 1994;26:519–526. doi: 10.1007/BF00762736. [DOI] [PubMed] [Google Scholar]
- D'Hahan N, Moreau C, Prost AL, Jacquet H, Alekseev AE, Terzic A, Vivaudou M. Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Proc Natl Acad Sci U S A. 1999;96:12162–12167. doi: 10.1073/pnas.96.21.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran E, Halestrap AP. Cytochrome c release from isolated rat liver mitochondria can occur independently of outer-membrane rupture: possible role of contact sites. Biochem J. 2000;348:343–350. [PMC free article] [PubMed] [Google Scholar]
- Forbes RA, Steenbergen C, Murphy E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ Res. 2001;88:802–809. doi: 10.1161/hh0801.089342. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P. The mitochondrial potassium cycle. IUBMB Life. 2001;52:153–158. doi: 10.1080/15216540152845948. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarovyarovoy V, Sun XC, Schindler PA. The mitochondrial K-ATP channel as a receptor for potassium channel openers. J Biol Chem. 1996;271:8796–8799. doi: 10.1074/jbc.271.15.8796. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Standen NB, Galinanes M. Evidence for mitochondrial K-ATP channels as effectors of human myocardial preconditioning. Cardiovasc Res. 2000;45:934–940. doi: 10.1016/s0008-6363(99)00407-1. [DOI] [PubMed] [Google Scholar]
- Grimmsman T, Rustenbeck I. Direct effects of diazoxide on mitochondria in pancreatic B-cells and on isolated liver mitochondria. Br J Pharmacol. 1998;123:781–788. doi: 10.1038/sj.bjp.0701663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover GJ, Garlid KD. ATP-sensitive potassium channels: A review of their cardioprotective pharmacology. J Mol Cell Cardiol. 2000;32:677–695. doi: 10.1006/jmcc.2000.1111. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. The regulation of the oxidation of fatty acids and other substrates in rat heart mitochondria by changes in matrix volume induced by osmotic strength, valinomycin and Ca2+ Biochem J. 1987;244:159–164. doi: 10.1042/bj2440159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP. The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro, and its role in the control of mitochondrial metabolism. Biochim Biophys ActA. 1989;973:355–382. doi: 10.1016/s0005-2728(89)80378-0. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. Regulation of mitochondrial metabolism through changes in matrix volume. Biochem Soc Trans. 1994;22:522–529. doi: 10.1042/bst0220522. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Davidson AM. Inhibition of Ca2+-induced large amplitude swelling of liver and heart mitochondria by Cyclosporin A is probably caused by the inhibitor binding to mitochondrial matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Dunlop JL. Intramitochondrial regulation of fatty acid β-oxidation occurs between flavoprotein and ubiquinone. A role for changes in matrix volume. Biochem J. 1986;239:559–565. doi: 10.1042/bj2390559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, McGivan JD. Measurement of membrane transport phenomena. In: Kornberg HL, Metcalfe JC, Northcote DH, Pogson CI, Tipton KF, editors. Techniques in Metabolic Research. B206. Amsterdam: Elsevier/North Holland; 1979. pp. 1–23. [Google Scholar]
- Halestrap AP, Quinlan PT, Whipps DE, Armston AE. Regulation of the mitochondrial matrix volume in vivo and in vitro. The role of calcium. Biochem J. 1986;236:779–787. doi: 10.1042/bj2360779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanley PJ, Mickel M, Loffler M, Brandt U, Daut J. KATP channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol. 2002;542:735–741. doi: 10.1113/jphysiol.2002.023960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue I, Nagase H, Kishi K, Higuti T. ATP-Sensitive K+ channel in the mitochondrial inner membrane. Nature. 1991;352:244–247. doi: 10.1038/352244a0. [DOI] [PubMed] [Google Scholar]
- Jaburek M, Yarovyarovoy V, Paucek P, Garlid KD. State-dependent inhibition of the mitochondrial K-ATP channel by glyburide and 5-hydroxydecanoate. J Biol Chem. 1998;273:13578–13582. [PubMed] [Google Scholar]
- Kawamura S, Yoshida K, Miura T, Mizukami Y, Matsuzaki M. Ischemic preconditioning translocates PKC-delta and -epsilon, which mediate functional protection in isolated rat heart. Am J Physiol. 1998;275:H2266–2271. doi: 10.1152/ajpheart.1998.275.6.H2266. [DOI] [PubMed] [Google Scholar]
- Klingenberg M. The ADP-ATP translocation in mitochondria, a membrane potential controlled transport. J Membr Biol. 1980;56:97–105. doi: 10.1007/BF01875961. [DOI] [PubMed] [Google Scholar]
- Klingenberg M, Grebe K, Schere B. Opposite effects of bongkrekic acid and atractyloside on the adenine nucleotides induced mitochondrial volume changes and on the efflux of adenine nucleotides. FEBS Lett. 1971;16:253–256. doi: 10.1016/0014-5793(71)80363-0. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Seetharaman S, Paucek P, Garlid KD. Bioenergetic consequences of opening the ATP-sensitive K+ channel of heart mitochondria. Am J Physiol Heart Circ Physiol. 2001;280:H649–657. doi: 10.1152/ajpheart.2001.280.2.H649. [DOI] [PubMed] [Google Scholar]
- Lawrence CL, Billups B, Rodrigo GC, Standen NB. The K-ATP channel opener diazoxide protects cardiac myocytes during metabolic inhibition without causing mitochondrial depolarization or flavoprotein oxidation. Br J Pharmacol. 2001;134:535–542. doi: 10.1038/sj.bjp.0704289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KHH, Javadov SJ, Das M, Clarke SJ, Suleiman S-M, Halestrap AP. The effects of ischaemic preconditioning, diazoxide and 5-hydroxydecanoate on rat heart mitochondrial volume and respiration. J Physiol. 2002;545:961–974. doi: 10.1113/jphysiol.2002.031484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YG, O'Rourke B. Opening of mitochondrial K-ATP channels triggers cardioprotection are reactive oxygen species involved? Circ Res. 2001;88:750–752. doi: 10.1161/hh0801.090537. [DOI] [PubMed] [Google Scholar]
- Liu YG, Sato T, O'Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: Novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- Liu YG, Sato T, Seharaseyon J, Szewczyk A, O'Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: viable candidate effectors of ischemic preconditioning. Ann N Y Acad Sci. 1999;874:27–37. doi: 10.1111/j.1749-6632.1999.tb09222.x. [DOI] [PubMed] [Google Scholar]
- O'Rourke B. Myocardial K-ATP channels in preconditioning. Circ Res. 2000;87:845–855. doi: 10.1161/01.res.87.10.845. [DOI] [PubMed] [Google Scholar]
- Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS, Heusch G, Cohen MV, Downey JM. Opening of mitochondrial K-ATP channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–466. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- Paucek P, Mironova G, Mahdi F, Beavis AD, Woldegiorgis G, Garlid KD. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+-channel from rat liver and beef heart mitochondria. J Biol Chem. 1992;267:26062–26069. [PubMed] [Google Scholar]
- Paucek P, Yarovyarovoy V, Sun XC, Garlid KD. Inhibition of the mitochondrial K-ATP channel by long-chain Acyl-CoA esters and activation by guanine nucleotides. J Biol Chem. 1996;271:32084–32088. doi: 10.1074/jbc.271.50.32084. [DOI] [PubMed] [Google Scholar]
- Quinlan PT, Halestrap AP. The mechanism of the hormonal activation of respiration in isolated rat heptocytes and its importance in the regulation of gluconeogenesis. Biochem J. 1986;236:789–800. doi: 10.1042/bj2360789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan PT, Thomas AP, Armston AE, Halestrap AP. Measurement of the intramitochondrial volume in hepatocytes without cell disruption and its elevation by hormones and valinomycin. Biochem J. 1983;214:395–404. doi: 10.1042/bj2140395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, O'Rourke B, Marban E. Modulation of mitochondrial ATP-dependent K+ channels by protein kinase C. Circ Res. 1998;83:110–114. doi: 10.1161/01.res.83.1.110. [DOI] [PubMed] [Google Scholar]
- Sato T, Sasaki N, Seharaseyon J, O'Rourke B, Marban E. Selective pharmacological agents implicate mitochondrial but not sarcolemmal K-ATP channels in ischemic cardioprotection. Circulation. 2000;101:2418–2423. doi: 10.1161/01.cir.101.20.2418. [DOI] [PubMed] [Google Scholar]
- Schäfer G, Portenhauser R, Trolp R. Inhibition of mitochondrial metabolism by the diabetogenic thiadiazine diazoxide. I. Action on succinate dehydrogenase and TCA-cycle oxidations. Biochem Pharmacol. 1971;20:1271–1280. doi: 10.1016/0006-2952(71)90358-3. [DOI] [PubMed] [Google Scholar]
- Stoner CD, Sirak HD. Adenine nucleotide-induced contraction of the inner mitochondrial membrane. I. General characterization. J Cell Biol. 1973a;56:51–64. doi: 10.1083/jcb.56.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner CD, Sirak HD. Adenine nucleotide-induced contraction on the inner mitochondrial membrane. II. Effect of bongkrekic acid. J Cell Biol. 1973b;56:65–73. doi: 10.1083/jcb.56.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Sasaki N, Miki T, Sakamoto N, Ohmoto Sekine Y, Tamagawa M, Seino S, Marban E, Nakaya H. Role of sarcolemmal K-ATP channels in cardioprotection against ischemia/reperfusion injury in mice. J Clin Invest. 2002;109:509–516. doi: 10.1172/JCI14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk A, Czyz A, Wojcik G, Wojtczak L, Nalecz MJ. ATP-regulated K+ channel in mitochondria: Pharmacology and function. J Bioenerg Biomembr. 1996;28:147–152. doi: 10.1007/BF02110645. [DOI] [PubMed] [Google Scholar]
- Szewczyk A, Marban E. Mitochondria: a new target for K+ channel openers? Trends Pharmacol Sci. 1999;20:157–161. doi: 10.1016/s0165-6147(99)01301-2. [DOI] [PubMed] [Google Scholar]
- Vanden Hoek TL, Becker LB, Shao ZH, Li CQ, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998;273:18092–18098. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- Vyssokikh MY, Katz A, Rueck A, Wuensch C, Dorner A, Zorov DB, Brdiczka D. Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. Biochem J. 2001;358:349–358. doi: 10.1042/0264-6021:3580349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YG, Ashraf M. Role of protein kinase C in mitochondrial K-ATP channel-mediated protection against Ca2+ overload injury in rat myocardium. Circ Res. 1999;84:1156–1165. doi: 10.1161/01.res.84.10.1156. [DOI] [PubMed] [Google Scholar]
- Wang YG, Hirai K, Ashraf M. Activation of mitochondrial ATP-sensitive K+ channel for cardiac protection against ischemic injury is dependent on protein kinase C activity. Circ Res. 1999;85:731–741. doi: 10.1161/01.res.85.8.731. [DOI] [PubMed] [Google Scholar]
- Whipps DE, Armston AE, Pryor HJ, Halestrap AP. Effects of glucagon and Ca2+ on the metabolism of phosphatidylinositol-4-phosphate and phosphatidylinositol-4, 5-bisphosphate in isolated rat hepatocytes and plasma membranes. Biochem J. 1987;241:835–845. doi: 10.1042/bj2410835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DE, Asimakis GK. Phosphate-induced efflux of adenine nucleotides from rat-heart mitochondria: evaluation of the roles of the phosphate/hydroxyl exchanger and the dicarboxylate carrier. Biochim Biophys Acta. 1987;893:470–479. doi: 10.1016/0005-2728(87)90098-3. [DOI] [PubMed] [Google Scholar]
- Xu W, Liu Y, Wang S, McDonald T, Van Eyk JE, Sidor A, O'Rourke B. Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science. 2002;298:1029–1033. doi: 10.1126/science.1074360. [DOI] [PubMed] [Google Scholar]
- Yarovyarovoy V, Paucek P, Jaburek M, Garlid KD. The nucleotide regulatory sites on the mitochondrial K-ATP channel face the cytosol. Biochim Biophys Acta. 1997;1321:128–136. doi: 10.1016/s0005-2728(97)00051-0. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Chen YF, Campbell WB, Zou AP, Gross GJ, Li PL. Characteristics and superoxide-induced activation of reconstituted myocardial mitochondrial ATP-sensitive potassium channels. Circ Res. 2001;89:1177–1183. doi: 10.1161/hh2401.101752. [DOI] [PubMed] [Google Scholar]