

Abstract
We have reported that intravenous administration of hypocretin (Hcrt or orexin) reverses the symptoms of narcolepsy in genetically narcoleptic dogs. We have also reported that the onset of symptoms in canine genetic narcolepsy is accompanied by degenerative changes in forebrain regions, particularly the septal nucleus and amygdala. In the present in vivo microdialysis study we have investigated the effect of intravenous administration of Hcrt-1 (orexin-A) to anaesthetized rats on glutamate and GABA release in the amygdala, a region with moderate Hcrt innervation, and in the cerebellar cortex, a region with sparse or no Hcrt innervation. We found that intravenous Hcrt administration caused a marked (> 60 %) and sustained (> 50 min) increase in glutamate release within the amygdala, but no change in release in the cerebellar cortex. We did not detect a significant change in GABA release. When calcium-free artificial cerebrospinal fluid was used as the microdialysis perfusate, Hcrt-1 no longer produced an increase in glutamate release. Hcrt may act via the calcium-dependent regulation of glutamate release in certain nuclei of the central nervous system.
The somata of hypocretin (Hcrt, also called orexin) neurons are localized to the perifornical and lateral hypothalamic regions. These neurons project widely but unevenly throughout the brain (Peyron et al. 1998; Nambu et al. 1999; van den Pol, 1999; Fung et al. 2001). Hcrt neurons have also been described in the rat enteric nervous system (Kirchgessner & Liu, 1999). Certain brain regions such as the cerebellar cortex are virtually devoid of Hcrt projections and receptors, whereas forebrain limbic regions including the amygdala and some brainstem nuclei are heavily innervated and have high levels of Hcrt receptor mRNA (Peyron et al. 1998; Trivedi et al. 1998; Nambu et al. 1999; Hervieu et al. 2001; Marcus et al. 2001). Human narcolepsy has been shown to be linked to a loss of 85–95 % of Hcrt cells (Thannickal et al. 2000) and to undetectable Hcrt mRNA (Peyron et al. 2000). Narcoleptic dogs show degenerative changes in the hypothalamus, amygdala and other forebrain regions, with peak levels of axonal loss at the time of symptom onset (Siegel et al. 1999). We have reported recently that the amygdala contains a population of cataplexy-related neurons (Gulyani et al. 2002).
We have also reported that motor activation produces a marked increase in Hcrt release (Kiyashchenko et al. 2002; Wu et al. 2002). Therefore, in the current study we monitored the effects of Hcrt administration in anaesthetized rats to eliminate the possibility of motor mediation, and to prevent other effects that are inhibited by barbiturate anaesthesia (Matsukawa et al. 1993; Lim et al. 1997). Hcrt-1 crosses the blood-brain barrier by diffusion (Kastin & Akerstrom, 1999). We found that systemic administration of Hcrt-1 reverses sleepiness and disrupted night-time sleep, and reduces cataplexy in canine narcolepsy (John et al. 2000). It has been shown in in vitro studies that Hcrt application to hypothalamic slices causes changes in glutamate and GABA release (van den Pol et al. 1998). In the study reported here, we have administered Hcrt-1 intravenously while monitoring glutamate and GABA release with in vivo microdialysis and HPLC in the amygdala and cerebellum, regions of high and low Hcrt innervation and receptor concentration, respectively, to determine whether systemic administration of Hcrt produces changes in glutamate and GABA release in Hcrt-innervated regions. These data have been reported in abstract form (John et al. 2001).
METHODS
This study was conducted on 15 adult male Sprague-Dawley rats (275–350 g) in accordance with National Research Council's ‘Guide for the Care and Use of Laboratory Animals’. All animal use protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the University of California, Los Angeles and the V.A. Greater Los Angeles Health Care System.
Surgical procedures
Rats were anaesthetized with 50 mg kg−1 (i.p.) pentobarbital sodium (Nembutal; Abbott Laboratories, North Chicago, IL, USA) and placed in a stereotaxic frame. Under aseptic conditions, 23 G stainless-steel guide cannulae were implanted above the amygdala (central nucleus; A 6.5, L 4.2, H 2) and cerebellum (preculminate fissure 4; A −1.3, L 1.5, H 6.7; Paxinos & Watson, 1986), 2 mm dorsal to their targets. A supplemental dose of Nembutal (30 mg kg−1) was given 3 h after the first dose. Body temperature was maintained using a water-circulating heating pad (Gaymar Industries, NY, USA). Microdialysis probes (Type A-I-01; Eicom, Kyoto, Japan) with 1 mm long membranes (cut-off MW 50 000) were inserted through the guide cannulae to the targets. The recovery rate of the microdialysis probes for amino acids was between 10 and 14 %. Artificial cerebrospinal fluid (ACSF: Harvard Apparatus, MA, USA) was continuously perfused at 4 μl min−1 through the amygdalar and cerebellar probes starting 2 h prior to the experiment, using an infusion pump (EPS-64, Eicom). Hcrt-1 (orexin-A, Phoenix Pharmaceuticals, Mountain View, CA, USA) dissolved in normal saline was administered (3 μg kg−1 in 200 μl) through the tail vein, using a glass syringe. As a control, saline was administered in the same manner. Hcrt-1 or control injections in randomized order were administered between 13.30 h and 14.30 h. Three rats were used to study the Ca2+ dependency of the transmitter release by omitting Ca2+ from the ACSF. In these studies, the amygdalar probe on one side was perfused with ACSF of the following composition (mm): 147 NaCl, 2.5 KCl, 0.9 MgCl2 and 2.2 CaCl2. The probe on the other side was perfused with Ca2+-free ACSF (Reid et al. 2000). Dialysate was collected for 30 min before and until 90 min after the Hcrt-1/saline injection, at 10 min intervals using an automated and refrigerated fraction collector (Eicom). The collecting polyethylene vials were maintained at 5 °C during collection and then stored at −80 °C until analysis.
Amino acid assay
The concentration of amino acid in the dialysate was detected by HPLC (EP-300, Eicom) with fluorescence detection (Soma S-3350: excitation/emission = 340/440 nm), as described in our prior studies (Nitz & Siegel, 1997; Kodama et al. 1998). The sample was injected using an autoinjector (ESA 540). Pre-column derivatization was performed with o-phthaldialdehyde and 2-mercaptoethanol at 5 °C for 3 min. The derivatives were then separated in a liquid chromatography column (MA-5ODS, 2.1 × 150 mm, Eicom) at 30 °C with 30 % methanol in 0.1 m phosphate buffer (pH 6.0), being degassed by an online degasser (DG-100, Eicom). Quantification was achieved with a PowerChrom analysis system (ADInstruments, Australia) using external amino acid standards (Sigma, USA). Neurotransmitter concentrations were calculated by comparing the HPLC peak of glutamate and GABA in microdialysis samples with the peak area of known concentrations of the same compounds analysed on the same day. The detection limit for glutamate and GABA was 20 fmol. Release of glutamate and GABA was stable 2 h after the implantation of the dialysis probe, as reported earlier (Herbison et al. 1990). Thus, the mean value of glutamate and GABA from three consecutive samples during the next 30 min was taken as the basal output. The values obtained subsequently are expressed as a percentage of this basal value. In some cases (n = 3), Hcrt-1 injection was performed within 60 min of the start of Ca2+-free ACSF perfusion into the amygdala. Three samples were taken immediately prior to the Hcrt-1 injection and used as the baseline for further comparison with postinjection levels.
Histology
Rats were deeply anaesthetized with pentobarbital sodium (60 mg kg−1) and transcardial perfusion was carried out with normal saline followed by 4 % buffered paraformaldehyde. After treatment with 30 % sucrose in 0.1 m phosphate-buffered saline, brains were cut at 40 μm intervals using a cryostat. Cresyl violet staining was carried out to verify placement of dialysis probes.
Data analysis
Changes in neurotransmitter release were analysed by one-way analysis of variance, followed by post-hoc comparisons using Fisher's least significant difference (LSD) test.
RESULTS
Glutamate and GABA release in the amygdala
Intravenous administration of Hcrt-1 produced a significant change in the glutamate level in the central nucleus of the amygdala. The basal level of glutamate in the amygdala was 22.93 ± 1.18 pmol per 20 μl sample. A significant increase in glutamate level was apparent within 10 min of intravenous Hcrt-1 injection. The increase was maintained for more than 50 min (F9,99= 2.35, P < 0.02; P < 0.05, Fisher's LSD; Fig. 1). In the amygdala perfused with Ca2+-free ACSF, a 21 % decrease in basal glutamate release was seen (25.34 ± 1.39 to 20.13 ± 0.84 pmol). This tonic decrease in glutamate release was apparent starting 30 min after the start of Ca2+-free ACSF perfusion and was stable for 30 min before Hcrt-1 injection. The Ca2+-free ACSF perfusion abolished the Hcrt-1-induced increase in glutamate release in the amygdala. There was no increase in glutamate throughout the 90 min postinjection period (F11,22= 1.30, P < 0.28; Fig. 1). However, the contralateral amygdala, which was perfused with normal ACSF, exhibited a significant increase in glutamate (F11,22= 2.45, P < 0.04).
Figure 1. Intravenous hypocretin (Hcrt)-1 administration elevates glutamate release, and this elevation is calcium dependent.
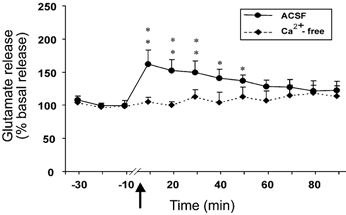
This graph shows the level of glutamate release in the amygdala from 30 min before to 90 min after the intravenous administration of Hcrt-1 (3 μg kg−1). The arrow indicates the point of injection. The continuous line shows data from the artificial cerebrospinal fluid (ACSF)-perfused site (n = 12) and the broken line shows data from the Ca2+-free ACSF-perfused site (n = 3). Note that the Ca2+-free ACSF perfusion completely blocked the effect of Hcrt-1. All values are means ±s.e.m; **P < 0.01, *P < 0.05, compared to the corresponding pretreatment level, Fisher's LSD test.
The basal level of GABA in the amygdala was 1.98 ± 0.25 pmol per 20 μl sample. The level of GABA exhibited a non-significant decrease at 40–60 min after Hcrt-1 injection in the amygdala perfused with Ca2+-containing ACSF (F9,99= 0.68, P < 0.71; Fig. 2). Intravenous Hcrt-1 injection did not alter the level of amygdala GABA release when Ca2+-free perfusion was performed. The tonic level of GABA release decreased by 18 % from the basal level (1.61 ± 0.25 to 1.32 ± 0.20 pmol) following Ca2+-free perfusion in this group, and remained stable for 30 min before Hcrt-1 injection. Intravenous administration of saline instead of Hcrt-1 did not change amino acid levels in the amygdala (Fig. 3).
Figure 2. Intravenous Hcrt-1 administration does not significantly alter GABA release.
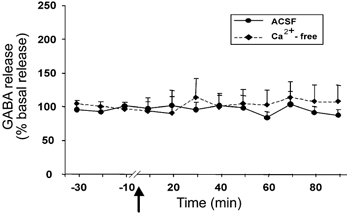
GABA levels in the amygdala from 30 min before to 90 min after intravenous administration of Hcrt-1 (3 μg kg−1). The arrow indicates the point of injection, the continuous line shows data from the ACSF-perfused site (n = 12) and the broken line shows data from the Ca2+-free ACSF-perfused site (n = 3). All values are means ±s.e.m.
Figure 3. Intravenous saline administration does not alter glutamate and GABA levels.
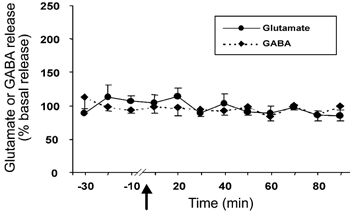
Glutamate and GABA levels in the amygdala from 30 min before to 90 min after intravenous administration of saline. The arrow indicates the point of injection, the continuous line shows glutamate levels (n = 9) and the broken line shows GABA levels (n = 9). All values are means ±s.e.m.
Glutamate and GABA release in the cerebellum
The basal level of glutamate and GABA in the cerebellar cortex was 5.71 ± 0.93 pmol per 20 μl sample and 1.53 ± 0.48 pmol per 20 μl sample, respectively. There was no significant change in the glutamate (n = 7) or GABA (n = 5) level in the cerebellum after intravenous administration of Hcrt-1 (Fig. 4).
Figure 4. Intravenous administration of Hcrt-1 (3 μg kg−1) has no effect on glutamate and GABA levels in the cerebellar cortex, a region that lacks significant Hcrt innervation.
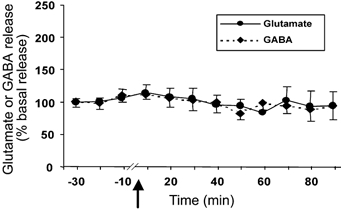
Glutamate and GABA levels in the cerebellar cortex from 30 min before to 90 min after intravenous administration of Hcrt-1 (3 μg kg−1). The arrow indicates the point of injection, the continuous line shows glutamate levels (n = 7) and the broken line shows GABA levels (n = 5). All values are means ±s.e.m.
Cresyl violet staining of brain sections revealed that the microdialysis probes were located in the central nucleus of the amygdala and the cerebellar cortex (Fig. 5).
Figure 5. Cresyl-violet-stained coronal brain section showing location of the microdialysis probes.
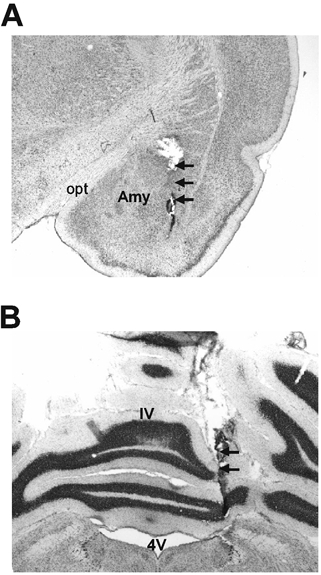
A, the amygdala. B, the cerebellar cortex. The locations of the microdialysis probes are indicated by arrows. Opt: optic tract; Amy: central nucleus of amygdala; 4V: 4th ventricle; IV: preculminate fissure 4.
DISCUSSION
We found that intravenous Hcrt administration causes a marked and sustained increase in glutamate release in the amygdala. Because these studies were conducted in anaesthetized animals, the observed changes in release could not have been mediated by behavioural changes. Ca2+-free ACSF perfusion abolished the Hcrt-1-induced glutamate release, indicating that these changes were dependent on vesicular release. Hcrt-1 has been shown to cross the blood-brain barrier (Kastin & Akerstrom, 1999). Therefore, the observed effect is probably mediated by Hcrt activation of Hcrt-1 and Hcrt-2 receptors in the amygdala (Trivedi et al.1998; Marcus et al. 2001). The lack of Hcrt innervation (Peyron et al. 1998; Nambu et al. 1999) and equivocal evidence for Hcrt receptors in the cerebellar cortex (Trivedi et al.1998; Hervieu et al. 2001; Marcus et al. 2001) is consistent with the lack of change in glutamate and GABA release in this structure.
In the in vivo situation, Hcrt could act by receptor-mediated presynaptic regulation of glutamate release near the collection site or through action at a distant site, affecting glutamate release in the amygdala through a multisynaptic pathway. The in vitro release of glutamate by Hcrt has been shown to be present even when axonal transmission was blocked (van den Pol et al. 1998), suggesting presynaptic regulation. The changes in GABA release that we saw were not significant, although an increase was reported in an in vitro study of hypothalamic tissue. It remains to be seen whether this difference is a function of the regions sampled, the dose of Hcrt to which the tissue was exposed, differences between the slice and in vivo preparations, anaesthesia or other factors. The distribution of Hcrt-1 receptor protein-like immunoreactivity is very dense in the hypothalamus compared to the amygdala (Hervieu et al. 2001) and this could also affect the GABA response to Hcrt administration.
It is possible that an indirect effect through peripheral pathways might trigger glutamate release, although barbiturate anaesthesia has an inhibitory effect on many peripheral pathways (Matsukawa et al. 1993; Lim et al. 1997). Evidence for peripheral Hcrt release and receptors has been contradictory (Gautvik et al. 1996; Sakurai et al. 1998; Kirchgessner & Liu, 1999). The presence of Ca2+-dependent glutamate release in the amygdala, with its high levels of Hcrt receptors, and the absence of release in the cerebellar cortex, a region relatively devoid of Hcrt receptors, can be most parsimoniously explained by the hypothesis that systemically administered Hcrt may be acting directly on Hcrt receptors in the amygdala. This is consistent with a recent study showing increased levels of glutamate in the locus coeruleus after Hcrt administration (Kodama & Kimura, 2002). Recent work by our group has demonstrated that the Hcrt effect on trigeminal motoneurons is mediated by the local release of glutamate (Peever et al. 2003).
It has been found that most human narcolepsy is due to a loss of Hcrt neurons (Thannickal et al. 2000) or of the mRNA of the Hcrt peptide (Peyron et al. 2000). Narcolepsy is characterized by overwhelming sleepiness and sudden losses of muscle tone, known as cataplexy. We have reported previously that systemic administration of Hcrt produces motor activation and reverses the symptoms of narcolepsy in genetically narcoleptic dogs (John et al. 2000). We have hypothesized that these effects are mediated by Hcrt-1 receptors, which are functional in these Hcrt-2-receptor-mutant animals. In a recent relevant abstract (Fujiki et al. 2001), there was no report of any effect of systemic Hcrt administration on cataplexy, although the other indicators of narcoleptic symptomatology monitored by John et al. (2000), including waking and sleep amounts and continuity, were not measured. Other studies of the effects of systemically administered Hcrt on centrally mediated responses have reported both positive (Dyer et al. 1999; Mitsuma et al. 1999; Kodama & Kimura, 2002) and negative results (Takahashi et al. 1999; Chen et al. 2000). The causes of these differences remain to be identified, but it is likely that the dose, time course and method of assessment of effects are critical. The present results suggest that amino acid release is an important mediator of the effects of systemically administered Hcrt.
Prior work has shown that application of Hcrt to Hcrt-1-receptor-rich regions in the brainstem and intracerebroventricularly increases motor activity and alters muscle tone (Hagan et al. 1999; Kiyashchenko et al. 2001; Peever et al. 2003). Hcrt release is maximal during periods of motor activation (Kiyashchenko et al. 2002; Wu et al. 2002). Regulation of amino acid release may play a major part in these and other actions of Hcrt in the central nervous system.
Acknowledgments
This work was supported by the Medical Research Services of the Department of Veterans Affairs and PHS grants NS14610, HL 41370 and HL 60296.
references
- Chen CT, Hwang LL, Chang JK, Dun NJ. Pressor effects of orexins injected intracisternally and to rostral ventrolateral medulla of anesthetized rats. Am J Physiol. 2000;278:R692–697. doi: 10.1152/ajpregu.2000.278.3.R692. [DOI] [PubMed] [Google Scholar]
- Dyer CJ, Touchette KJ, Carroll JA, Allee GL, Matteri RL. Cloning of porcine prepro-orexin cDNA and effects of an intramuscular injection of synthetic porcine orexin-B on feed intake in young pigs. Domest Anim Endocrinol. 1999;16:145–148. doi: 10.1016/s0739-7240(99)00011-9. [DOI] [PubMed] [Google Scholar]
- Fujiki N, Yoshida Y, Ripley B, Mignot E, Nishino S. Effect of systemic and central administration of hypocretin-1 in narcoleptic (Hcrtr 2 mutated) and control dogs. Sleep. 2001;24:A96–97. [Google Scholar]
- Fung SJ, Yamuy J, Sampogna S, Morales FR, Chase MH. Hypocretin (orexin) input to trigeminal and hypoglossal motoneurons in the cat: a double-labeling immunohistochemical study. Brain Res. 2001;903:257–262. doi: 10.1016/s0006-8993(01)02318-6. [DOI] [PubMed] [Google Scholar]
- Gautvik KM, De Lecea L, Gautvik VT, Danielson PE, Tranque P, Dopazo A, Bloom FE, Sutcliffe JG. Overview of the most prevalent hypothalamus-specific mRNAs, as identified by directional tag PCR subtraction. Proc Natl Acad Sci U S A. 1996;93:8733–8738. doi: 10.1073/pnas.93.16.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyani S, Wu M-F, Nienhuis R, John J, Siegel JM. Cataplexy-related neurons in the amygdala of the narcoleptic dog. Neuroscience. 2002;112:355–365. doi: 10.1016/s0306-4522(02)00089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan JJ, Leslie RA, Patel S, Evans ML, Wattam TA, Holmes S, Benham CD, Taylor SG, Routledge C, Hemmati P, Munton RP, Ashmeade TE, Shah AS, Hatcher JP, Hatcher PD, Jones DN, Smith MI, Piper DC, Hunter AJ, Porter RA, Upton N. Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci U S A. 1999;96:10911–10916. doi: 10.1073/pnas.96.19.10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbison AE, Heavens RP, Dyer RG. Endogenous release of gamma-aminobutyric acid from the medial preoptic area measured by microdialysis in the anaesthetised rat. J Neurochem. 1990;55:1617–1623. doi: 10.1111/j.1471-4159.1990.tb04947.x. [DOI] [PubMed] [Google Scholar]
- Hervieu GJ, Cluderay JE, Harrison DC, Roberts JC, Leslie RA. Gene expression and protein distribution of the orexin-1 receptor in the rat brain and spinal cord. Neuroscience. 2001;103:777–797. doi: 10.1016/s0306-4522(01)00033-1. [DOI] [PubMed] [Google Scholar]
- John J, Wu MF, Kodama T, Siegel JM. Hypocretin-1 (orexin-A) produced changes in glutamate and GABA release: an in vivo microdialysis study. Sleep. 2001;24:A20. [Google Scholar]
- John J, Wu MF, Siegel JM. Systemic Administration of hypocretin-1 reduces cataplexy and normalizes sleep and waking durations in narcoleptic dogs. Sleep Res Online. 2000;3:23–28. [PMC free article] [PubMed] [Google Scholar]
- Kastin AJ, Akerstrom V. Orexin A but not orexin B rapidly enters brain from blood by simple diffusion. J Pharmacol Exp Ther. 1999;289:219–223. [PubMed] [Google Scholar]
- Kirchgessner AL, Liu MT. Orexin synthesis and response in the gut. Neuron. 1999;24:941–951. doi: 10.1016/s0896-6273(00)81041-7. [DOI] [PubMed] [Google Scholar]
- Kiyashchenko LI, Mileykovskiy BY, Lai YY, Siegel JM. Increased and decreased muscle tone with orexin (hypocretin) microinjections in the locus coeruleus and pontine inhibitory area. J Neurophysiol. 2001;85:2008–2016. doi: 10.1152/jn.2001.85.5.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyashchenko LI, Mileykovskiy BY, Maidment N, Lam HA, Wu MF, John J, Peever J, Siegel JM. Release of hypocretin (orexin) during waking and sleep states. J Neurosci. 2002;22:5282–5286. doi: 10.1523/JNEUROSCI.22-13-05282.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama T, Kimura M. Arousal effects of orexin-A correlate with GLU release from the locus coeruleus in rats. Peptides. 2002;23:1673–1681. doi: 10.1016/s0196-9781(02)00109-2. [DOI] [PubMed] [Google Scholar]
- Kodama T, Lai YY, Siegel JM. Enhanced glutamate release during REM sleep in the rostromedial medulla as measured by in vivo microdialysis. Brain Res. 1998;780:178–181. [PMC free article] [PubMed] [Google Scholar]
- Lim DY, Kang TJ, Hong SP, Chung CH, Choi CH, Lee SI, Park YW, Kwack JJ, Ki JD, Kim CW, Park CY. Influence of pentobarbital-Na on stimulation-evoked catecholamine secretion in the perfused rat adrenal gland. Korean J Intern Med. 1997;12:163–175. doi: 10.3904/kjim.1997.12.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- Matsukawa K, Ninomiya I, Nishiura N. Effects of anesthesia on cardiac and renal sympathetic nerve activities and plasma catecholamines. Am J Physiol. 1993;265:R792–797. doi: 10.1152/ajpregu.1993.265.4.R792. [DOI] [PubMed] [Google Scholar]
- Mitsuma T, Hirooka Y, Mori Y, Kayama M, Adachi K, Rhue N, Ping J, Nogimori T. Effect of orexin-A on thyrotropin-releasing hormone and thyrotropin secretion in rats. Horm Metab Res. 1999;31:606–609. doi: 10.1055/s-2007-978805. [DOI] [PubMed] [Google Scholar]
- Nambu T, Sakurai T, Mizukami K, Hosoya Y, Yanagisawa M, Goto K. Distribution of orexin neurons in the adult rat brain. Brain Res. 1999;827:243–260. doi: 10.1016/s0006-8993(99)01336-0. [DOI] [PubMed] [Google Scholar]
- Nitz D, Siegel JM. GABA release in the cat locus coeruleus as a function of the sleep/wake state. Neuroscience. 1997;78:795–801. doi: 10.1016/s0306-4522(96)00549-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 2. New York: Blackwell Science Inc; 1986. [Google Scholar]
- Peever JH, Lai YY, Siegel JM. Excitatory effects of hypocretin-1 (orexin-A) in the trigeminal motor nucleus are reversed by NMDA antagonism. J Neurophysiol. 2003 doi: 10.1152/jn.00968.2002. (in the Press) [DOI] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, Nishino S, Mignot E. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, Van Den Pol AN, De Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MS, Fox L, Ho LB, Berger SP. Nicotine stimulation of extracellular glutamate levels in the nucleus accumbens: neuropharmacological characterization. Synapse. 2000;35:129–136. doi: 10.1002/(SICI)1098-2396(200002)35:2<129::AID-SYN5>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, Gulyani S, Ouyang S, Wu MF, Mignot E, Switzer RC, McMurry G, Cornford M. Neuronal degeneration in canine narcolepsy. J Neurosci. 1999;19:248–257. doi: 10.1523/JNEUROSCI.19-01-00248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Okumura T, Yamada H, Kohgo Y. Stimulation of gastric acid secretion by centrally administered orexin-A in conscious rats. Biochem Biophys Res Commun. 1999;254:623–627. doi: 10.1006/bbrc.1998.9994. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi P, Yu H, MacNeil DJ, Van Der Ploeg LH, Guan XM. Distribution of orexin receptor mRNA in the rat brain. FEBS Letters. 1998;438:71–75. doi: 10.1016/s0014-5793(98)01266-6. [DOI] [PubMed] [Google Scholar]
- Van Den Pol AN. Hypothalamic hypocretin (orexin): robust innervation of the spinal cord. J Neurosci. 1999;19:3171–3182. doi: 10.1523/JNEUROSCI.19-08-03171.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Pol AN, Gao XB, Obrietan K, Kilduff TS, Belousov AB. Presynaptic and postsynaptic actions and modulation of neuroendocrine neurons by a new hypothalamic peptide, hypocretin/orexin. J Neurosci. 1998;18:7962–7971. doi: 10.1523/JNEUROSCI.18-19-07962.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, John J, Maidment N, Lam HA, Siegel JM. Hypocretin release in normal and narcoleptic dogs after food and sleep deprivation, eating, and movement. Am J Physiol. 2002;283:R1079–1086. doi: 10.1152/ajpregu.00207.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]