

Abstract
In sedentary individuals, postexercise hypotension following a single bout of aerobic exercise is due to an unexplained peripheral vasodilatation. We tested the hypothesis that α-adrenergic responsiveness in the forearm and leg vasculatures is blunted during postexercise hypotension. We studied 12 men and two women before and 30 min after a 60 min bout of cycling at 60 % V̇O2, peak. In the first five subjects, arterial pressure (brachial artery catheter) and forearm blood flow (plethysmography) were measured and vascular conductance was calculated during intraarterial infusions of the α1-agonist phenylephrine and the α2-agonist clonidine. Exercise reduced mean arterial pressure (89 ± 2 vs. 95 ± 2 mmHg, P < 0.05) and increased forearm vascular conductance 77 ± 33 % (P < 0.05). Despite these changes in baseline vascular conductance, vasoconstrictor responses in the forearm to phenylephrine and clonidine were similar (or enhanced) postexercise vs. preexercise. In the remaining nine subjects, arterial pressure (femoral artery catheter) and leg blood flow (Doppler ultrasound of the femoral artery) were measured and vascular conductance was calculated during intraarterial infusions of phenylephrine and clonidine. Exercise reduced mean arterial pressure (80 ± 2 vs. 89 ± 2 mmHg, P < 0.05) and increased leg vascular conductance 94 ± 16 % (P < 0.05). Despite these changes in baseline vascular conductance, vasoconstrictor responses in the leg to phenylephrine and clonidine were similar (or enhanced) postexercise vs. preexercise. These results suggest that vascular responsiveness to α-adrenergic agonists is maintained during postexercise hypotension in humans. Thus, while postexercise hypotension is associated with increased vascular conductance in the forearm and leg, it does not appear that blunting of α-adrenergic responsiveness is the cause.
After a single bout of dynamic exercise, there are profound changes in the mechanisms that regulate and determine arterial pressure, resulting in a postexercise hypotension that lasts nearly 2 h in sedentary but otherwise healthy individuals (Halliwill et al. 1996b; Halliwill, 2001). Whereas shorter or less vigorous exercise protocols elicit inconsistent changes in arterial pressure in normotensive subjects, postexercise hypotension is consistently elicited after longer (30–60 min) bouts of moderate-intensity (50–60 % peak aerobic capacity (V̇O2, peak)) exercise (Hannum & Kasch, 1981; Wilcox et al. 1982; Kaufman et al. 1987; Somers et al. 1991; Landry et al. 1992; Halliwill et al. 1996a; b Halliwill, 2001). It is generally accepted that postexercise hypotension is due to a persistent rise in peripheral vascular conductance that is not completely offset by increases in cardiac output (Halliwill et al. 1996a, b, 2000; Halliwill, 2001), although there are some exceptions (e.g. endurance-trained men; Senitko et al. 2002). Forearm (Coats et al. 1989; Cléroux et al. 1992a, b; Isea et al. 1994; Piepoli et al. 1994; Halliwill et al. 2000) and calf (Hara & Floras, 1992; Halliwill et al. 1996a; Halliwill et al. 2000) vascular conductances are increased in parallel with total peripheral conductance. Although the mechanisms of the vasodilatation underlying postexercise hypotension are poorly understood, the sympathetic nervous system (Floras et al. 1989; Hara & Floras, 1992; Halliwill et al. 1996a; Kulics et al. 1999), baroreflex resetting (Halliwill et al. 1996a), nitric oxide (Patil et al. 1993; Halliwill et al. 2000; Rao et al. 2002), and an unknown vasodilator have all been implicated (Halliwill et al. 2000; Halliwill, 2001).
Previously, Halliwill et al. (1996a) have shown in humans that vascular responsiveness to sympathetic vasoconstrictor outflow is impaired so that vascular resistance is reduced for a given level of sympathetic nerve activity. The nature of this ‘vascular component’ of postexercise hypotension is unknown, but ineffective transduction of sympathetic outflow into vascular resistance could be the result of competing influences at the level of the arterial smooth muscle, such as the release of local vasodilator substances (Halliwill, 2001). Along these lines, extensive work by DiCarlo and co-workers in animal models (isolated aortic strips; Howard et al. 1992) and in conscious rabbits (Howard & DiCarlo, 1992) and rats (Patil et al. 1993; Rao et al. 2002) have demonstrated α1-adrenergic receptor hyporesponsiveness following exercise. However, it is unknown whether humans have reduced α1-adrenergic receptor responsiveness during postexercise hypotension. In addition, recent work suggests that α2-adrenergic receptors contribute substantially to resting vascular tone in the human forearm (Dinenno et al. 2002). There is also evidence that α2-adrenergic receptors are more susceptible to interference from metabolic vasodilatation than α1-adrenergic receptors (Anderson & Faber, 1991; Thomas et al. 1994; Buckwalter et al. 2001). In this context, it is unknown whether exercise produces a lasting α2-adrenergic receptor hyporesponsiveness similar to what has been seen for α1-adrenergic receptors in animal models of postexercise hypotension.
Therefore, the goal of this study was to determine whether hyporesponsiveness of either α1- or α2-adrenergic receptors underlies the vascular component of postexercise hypotension in humans. We tested the hypothesis that α-adrenergic responsiveness in the forearm and leg vasculatures is blunted during postexercise hypotension in humans.
METHODS
This study was approved by the Institutional Review Board of the Mayo Clinic and Foundation. Each subject gave his or her informed written consent prior to participation. All studies were performed according to the Declaration of Helsinki.
Subjects
Fourteen healthy, non-smoking, normotensive subjects participated in this study (12 men, 2 women, ages 18–34 years, height 176 ± 10 cm, weight 78.8 ± 13.6 kg (mean ± S.D.)). None of the subjects were taking medications other than oral contraceptives. All female subjects had a negative serum pregnancy test on a screening day. While a recent study suggests that the phase of oral contraceptive use does not have any major effects on postexercise hypotension (Birch et al. 2002), it is unknown whether the menstrual cycle has major effects on postexercise hypotension. Therefore, female subjects were studied during the early follicular phase (1 to 4 days after the onset of menstruation) or during the placebo phase of oral contraceptive use.
On a screening day, peak aerobic capacity (V̇O2, peak) was determined with a graded maximal cycle ergometer test comprising 1 min workload increments. Specifically, after a 5 min warm-up period of easy cycling (20–30 W), workload increased at 20, 25, or 30 W every minute. Selection of the workload increment was subjective, with the goal of producing exhaustion within 8 to 12 min. All subjects achieved exhaustion within this time range; resulting V̇O2, peak values were within the normal range for this population (35.1 ± 4.7 ml−1 kg−1 min−1 (mean ± S.D.)). This test was used to determine the exercise workloads used in the two protocols that followed.
In addition to determination of V̇O2, peak, forearm volume was determined by water displacement in subjects in Protocol 1. Forearm volume, measured from the antecubital crease to the wrist, was used to adjust drug dosing for brachial artery drug administrations. Subjects in Protocol 2 underwent body composition analysis by dual-energy X-ray absorptiometry (DEXA; Lunar Radiation, Madison, WI, USA) in order to determine right leg fat-free mass, which was used to adjust drug dosing for femoral artery drug administrations.
For both protocols, subjects reported for the study at least 2 h post-prandial and abstained from caffeine for 12 h and from exercise for 24 h prior to the study.
Protocol 1
In the first five subjects, arterial pressure (brachial artery catheter) and forearm blood flow (plethysmography) were measured and vascular conductance was calculated during intraarterial infusions of the α1-agonist phenylephrine and the α2-agonist clonidine, before and 30 min after a single bout of exercise. Exercise consisted of a 60 min period of seated upright cycling at 60 % V̇O2, peak. Exercise of this intensity and duration produces a sustained (≈2 h) postexercise hypotension (Halliwill, 2001). During exercise, subjects were allowed to drink water ad libitum. Throughout the protocol, ambient temperature was controlled between 22 and 24 °C.
Measurements
Prior to exercising, subjects were instrumented in the supine position for heart rate (three-lead electrocardiogram) and arterial pressure. Arterial pressure measurements and drug infusions were performed using a 5 cm, 20-gauge brachial artery catheter placed proximal to the antecubital crease in the non-dominant arm (in all cases, left) using sterile techniques after local anaesthesia (1–2 ml of 1 % lidocaine) (Dietz et al. 1994a, b). The catheter was continuously flushed with heparinized saline (3 ml h−1, 2 units ml−1).
Forearm blood flow
Using standard methods, forearm blood flow was estimated by venous occlusion plethysmography with mercury-in-silastic strain gauges (Greenfield et al. 1963; Dietz et al. 1994a, b). During measurements, an arterial occlusion cuff around the wrist was continuously inflated to suprasystolic pressure (250 mmHg) while a venous occlusion cuff around the upper arm was inflated to 50 mmHg for 7.5 s out of every 15 s, providing one blood flow measurement every 15 s. Blood flow was reported as ml (dl of tissue)−1 min−1. In addition, forearm vascular conductance was calculated as blood flow × 100/mean arterial pressure.
α-Adrenergic responsiveness
In order to assess α-adrenergic responsiveness before and after exercise, we infused an α1-adrenergic agonist (phenylephrine-HCl) and an α2-adrenergic agonist (clonidine) via the brachial artery catheter while forearm blood flow was measured. Dose-response curves to each drug were determined using 2 min infusions of increasing doses. Baseline blood flow for each drug was calculated as the average over a 3 min period immediately prior to drug administration. Blood flow was analysed throughout the drug infusions and the response to each dose was calculated as the average of two consecutive flow measurements representing the peak response to that dose.
The order of drug administration was the same before and after exercise in each individual, but randomized across individuals so that half received phenylephrine first and half received clonidine first. Dose-response curves for phenylephrine were generated using the following doses: 0.03125, 0.125, and 0.5 μg (dl forearm volume)−1 min−1. Since forearm volume averaged 11.0 ± 1.5 dl, the highest dose of phenylephrine corresponded to 5.5 ± 0.8 μg min−1. Dose-response curves for clonidine were generated using the following doses: 0.0375, 0.15, and 0.6 μg (dl forearm volume)−1 min−1. The highest dose of clonidine corresponded to 6.6 ± 0.9 μg min−1. All doses were formulated so that infusion rates were between 1 and 3 ml min−1. A 25 min washout period between the two drug dose profiles allowed vascular tone to return to baseline. Thus, while responses to one agonist were assessed at 30 min postexercise, the other was not assessed until ≈60 min postexercise.
Protocol 2
In the remaining nine subjects, arterial pressure (femoral artery catheter) and femoral blood flow (Doppler ultrasound) were measured and vascular conductance was calculated during intraarterial infusions of phenylephrine and clonidine, before and 30 min after a 60 min bout of cycling at 60 % V̇O2, peak.
Measurements
Prior to exercise, subjects were instrumented in the supine position for heart rate (three-lead electrocardiogram) and arterial pressure. For this protocol, a 10-cm, 18-gauge femoral artery catheter was placed immediately distal to the inguinal ligament in the right leg using sterile techniques after local anaesthesia. The catheter was continuously flushed with heparinized saline (3 ml h−1, 2 units ml−1).
Leg blood flow
Femoral artery blood velocity was measured with a 4 MHz pulsed Doppler ultrasound probe (Model 500M, Multigon Industries, Yonkers, NY, USA) placed below the catheter insertion site and proximal to the femoral bifurcation. The artery was insonated with an angle of 60 deg. A 7 MHz linear array ultrasound probe (Acuson 128XP/10-ART Ultrasound System, Mountain View, CA, USA) was used to obtain femoral artery diameter measurements immediately after each femoral artery blood velocity measurement. Leg blood flow was then derived as the product of femoral mean blood velocity and arterial cross-sectional area. Blood flow was reported as millilitres per minute. In addition, leg vascular conductance was calculated as blood flow/mean arterial pressure.
α-Adrenergic responsiveness
In order to assess α-adrenergic responsiveness before and after exercise, we infused phenylephrine and clonidine via the femoral artery catheter while femoral blood flow was measured. Dose-response curves to each drug were determined using 2 min infusions of increasing doses. Baseline blood flow for each drug was calculated as the average over a 2 min period immediately prior to drug administration. The response to each dose was calculated as the average blood flow over 30 s immediately following each 2 min infusion.
The order of drug administration was the same before and after exercise in each individual, but randomized across individuals so that half received phenylephrine first and half received clonidine first. Dose-response curves for phenylephrine were generated using the following doses: 0.1, 0.4 and 1.6 μg (dl leg fat-free volume)−1 min−1. Since leg fat-free volume averaged 89 ± 12 dl, the highest dose of phenylephrine corresponded to 142 ± 7 μg min−1. Dose-response curves for clonidine were generated using the following doses: 0.2 and 0.8 μg (dl leg fat-free volume)−1 min−1. The highest dose of clonidine corresponded to 71 ± 4 μg min−1. All doses were formulated so that infusion rates were between 2 and 4 ml min−1. A 25 min washout period between the two drug dose profiles allowed vascular tone to return to baseline. Thus, while responses to one agonist were assessed at 30 min postexercise, the other was not assessed until ≈60 min postexercise.
Data analysis
All physiological signals were digitized and stored on computer at 250 Hz, and data were analysed off-line with signal processing software (WinDaq, Dataq Instruments, Akron, OH). Mean arterial pressure was derived from the arterial pressure waveform. Forearm blood flow was determined from the derivative of the forearm plethysmogram.
Statistics
The results were analysed with a repeated-measures two-way ANOVA (drug dose vs.exercise). Significant effects were further tested with Fischer's LSD test, and differences were considered significant when P < 0.05. All values are reported as means ±s.e.m.
RESULTS
Exercise
The goal was to have each subject exercise for 60 min at 60 % V̇O2, peak. In Protocol 1, the average workloads were 95 ± 13 W. Heart rate increased from 63 ± 6 beats min−1 at supine rest to 142 ± 11 beats min−1 during exercise (mean for entire 60 min of exercise, P < 0.05). This represented, on average, 61 ± 5 % of heart rate reserve, and is consistent with the target workload. In Protocol 2, the average workloads were 108 ± 7 W. Heart rate increased from 63 ± 2 at supine rest to 142 ± 7 beats min−1 during exercise (P < 0.05). This represented, on average, 64 ± 3 % of heart rate reserve, and is consistent with the target workload.
Postexercise hypotension
Table 1 shows postexercise versus preexercise haemodynamics for both protocols. As expected, mean arterial pressures were less postexercise compared to preexercise. Heart rate was higher postexercise compared to preexercise. Vascular conductance was increased in the arm (77 ± 33 %) 30 min postexercise compared to preexercise but returned to near preexercise levels (10 ± 21 %) at 60 min postexercise. Vascular conductance was increased in the leg (94 ± 16 %) 30 min postexercise compared to preexercise and remained elevated (60 ± 27 %) at 60 min postexercise.
Table 1.
Haemodynamics
| Preexercise | 30 min Postexercise | 60 min Postexercise | |
|---|---|---|---|
| Procotol 1 | |||
| Heart rate (beas min−1) | 62.5 ± 5.8 | 69.0 ± 5.1* | 66.7 ± 6.4 |
| Mean arterial pressure (mm Hg) | 94.8 ± 2.0 | 89.3 ± 1.6* | 89.3 ± 2.2* |
| Forearm blood flow (ml dl−1 min−1) | 2.59 ± 0.23 | 4.18 ± 0.58* | 2.57 ± 0.36† |
| Forearm vascular conductance (ml dl−1 min−1 (100 mmHg)−1) | 2.74 ± 0.27 | 4.72 ± 0.72* | 2.90 ± 0.45† |
| Procotol 2 | |||
| Heart rate (beats min−1) | 62.9 ± 2.3 | 70.1 ± 4.4* | 67.1 ± 2.1*† |
| Mean arterial pressure (mm Hg) | 88.6 ± 2.1 | 79.9 ± 2.1* | 79.2 ± 2.1* |
| Leg blood flow (ml min−1) | 355 ± 27 | 628 ± 70* | 578 ± 62* |
| Leg vascular conuctance (ml min−1 mmHg−1) | 1.03 ± 0.32 | 7.96 ± 0.95* | 7.40 ± 0.88* |
P < 0.05 vs. preexercise.
P < 0.05 vs. 30 min postexcercise. Values are means ±s.e.m.
Forearm vascular responsiveness
Table 2 shows forearm vascular conductance before and during intraarterial infusions of the α1-adrenergic agonist phenylephrine and the α2-adrenergic agonist clonidine. Figure 1 shows the dose-response relationships for the forearm vasculature to phenylephrine and clonidine. Due to the differences in baseline vascular conductance postexercise versus preexercise, forearm vascular responses are expressed as both the absolute and percentage reduction in forearm vascular conductance at each dose. Infusion of either phenylephrine or clonidine produced dose-dependent decreases in forearm vascular conductance. These vasoconstrictor responses were intact after exercise.
Table 2.
Forearm vascular conductance (Protocol 1)
| Forearm vascular conductance (ml dl−1 min−1 (100 mmHg)−1) | ||
|---|---|---|
| Preexcercise | Postexercise | |
| Phenylephrine | ||
| Baseline | 2.28 ± 0.22 | 3.66 ± 0.63* |
| 0.03125 μg dl−1 min−1 | 1.56 ± 0.10 | 2.32 ± 0.46 |
| 0.125 μg dl−1 min−1 | 1.16 ± 0.14 | 1.83 ± 0.44 |
| 0.5 μg dl−1 min−1 | 1.01 ± 0.11 | 1.09 ± 0.16 |
| Clonidine | ||
| Baseline | 2.45 ± 0.40 | 3.96 ± 0.91* |
| 0.0375 μg dl−1 min−1 | 1.60 ± 0.20 | 1.91 ± 0.14 |
| 0.15 μg dl−1 min−1 | 1.49 ± 0.09 | 1.64 ± 0.12 |
| 0.6 μg dl−1 min−1 | 1.24 ± 0.10 | 1.47 ± 0.15 |
P < 0.05 postexerxise vs. preexercise. Values are means ±s.e.m.
Figure 1. Forearm vascular responses.
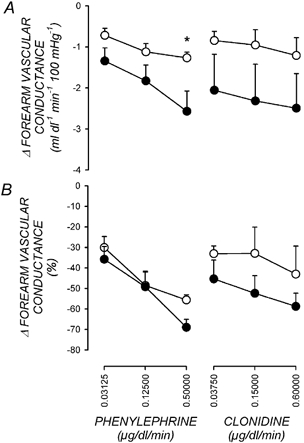
The absolute change in forearm vascular conductance (A) and the percentage change in forearm vascular conductance (B) in response to increasing doses of phenylephrine or clonidine administered prior to exercise (○) and postexercise (•). Values are means +s.e.m., n = 5. *P < 0.05 postexercise versus preexercise at same dose.
Leg vascular responsiveness
Table 3 shows leg vascular conductance before and during intraarterial infusions of the α1-adrenergic agonist phenylephrine and the α2-adrenergic agonist clonidine. Figure 2 shows the dose-response relationships for the leg vasculature to phenylephrine and clonidine. Due to the differences in baseline vascular conductance postexercise versus preexercise, leg vascular responses are expressed as both the absolute and percentage reduction in leg vascular conductance at each dose. Infusion of either phenylephrine or clonidine produced dose-dependent decreases in leg vascular conductance. These vasoconstrictor responses were intact after exercise.
Table 3.
Leg vascular conductance (Protocol 2)
| Leg vascular conductance (ml min−1 mmHg−1) | ||
|---|---|---|
| Preexcercise | Postexercise | |
| Phenylephrine | ||
| Baseline | 4.24 ± 0.40 | 7.96 ± 0.95* |
| 0.1 μg dl−1 min−1 | 2.76 ± 0.36 | 3.70 ± 0.52* |
| 0.4 μg dl−1 min−1 | 1.89 ± 0.30 | 2.94 ± 0.56* |
| 1.6 μg dl−1 min−1 | 1.82 ± 0.47 | 3.02 ± 1.49 |
| Clonidine | ||
| Baseline | 3.89 ± 0.35 | 7.91 ± 1.10* |
| 0.2 μg dl−1 min−1 | 2.00 ± 0.27 | 3.37 ± 0.62 |
| 0.8 μg dl−1 min−1 | 1.61 ± 0.24 | 1.76 ± 0.32 |
P < 0.05 postexerxise vs. preexercise. Values are means ±s.e.m.
Figure 2. Leg vascular responses.
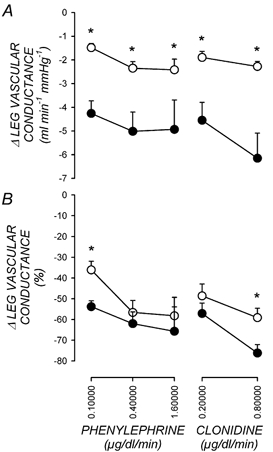
The absolute change in leg vascular conductance (A) and the percentage change in leg vascular conductance (B) in response to increasing doses of phenylephrine or clonidine administered prior to exercise (○) and postexercise (•). Values are means +s.e.m., n = 9. *P < 0.05 postexercise versus preexercise at same dose.
DISCUSSION
The goal of this study was to determine whether hyporesponsiveness of either α1- or α2-adrenergic receptors underlies the vascular component of postexercise hypotension in humans. In contrast to some animal studies, the results suggest that vascular responsiveness to α-adrenergic agonists is maintained during postexercise hypotension in humans. Thus, while postexercise hypotension is associated with increased vascular conductance in both the upper and lower extremities, it does not appear that blunting of α-adrenergic responsiveness is the cause.
Vascular transduction during postexercise hypotension
Previously, Halliwill et al. (1996a) recorded muscle sympathetic nerve activity to the vascular beds of the lower leg while also estimating calf blood flow. From these data, the transduction of sympathetic nerve activity into vascular resistance was quantified non-invasively by augmenting sympathetic outflow via an exercise pressor response. Thus, as muscle sympathetic nerve activity increased, the corresponding rise in calf vascular resistance was observed. The authors demonstrated that the rise in calf vascular resistance for a given rise in muscle sympathetic nerve activity was reduced ≈70 % during postexercise hypotension, and termed this phenomenon the ‘vascular component’ of postexercise hypotension to differentiate it from the ‘neural component’, composed of a resetting of the arterial baroreflex to lower pressures in conjunction with sympathoinhibition (Halliwill et al. 1996a; Halliwill, 2001). The nature of this ‘vascular component’ of postexercise hypotension is unknown, but we had previously considered it a possibility that ineffective transduction of sympathetic outflow into vascular resistance could be the result of competing influences at the level of the arterial smooth muscle, such as the release of local vasodilator substances (Halliwill, 2001). Along similar lines, complicated interactions between nitric oxide and the sympathetic nervous system have been documented by numerous groups (Gonzalez et al. 1992; Pohl & DeWit, 1996; Iida, 1999). Thomas & Victor (1997) have proposed that increased nitric oxide production plays a major role in exercise hyperaemia by causing a ‘functional sympatholysis’ in which sympathetically mediated vasoconstriction is blunted during exercise. Studies by DiCarlo and co-workers (Patil et al. 1993; Rao et al. 2002) in rats are consistent with this notion of sympatholysis, suggesting that it could carry forward into the postexercise period and contribute to postexercise hypotension. However, the findings from the current study indicate that neither α1- nor α2-adrenergic receptor responsiveness is blunted and suggest that this phenomenon is not involved in postexercise hypotension in humans. Furthermore, evidence from Halliwill et al. (2000, 2001) suggests that nitric oxide does not play an important role in postexercise hypotension in humans.
What other mechanisms could explain the vascular component of postexercise hypotension?
Ineffective transduction of sympathetic nerve activity into vasoconstriction could result from lesser neurotransmitter release. For example, circulating opioids may be increased after a single bout of exercise (Schwarz & Kindermann, 1992). Sympathetic nerve terminals possess pre-synaptic inhibitory opioid receptors (Wong-Dusting & Rand, 1989) that may be occupied after exercise, effectively reducing noradrenaline release. Although systemic opioid blockade with naloxone has reversed postexercise hypotension in animals (Shyu & Thorén, 1986; Devine et al. 1991; Sebastian et al. 1995), it has had mixed results in humans (Boone et al. 1992; Hara & Floras, 1992), and the role of opioids remains controversial (Sebastian et al. 1995). Pre-synaptic inhibition can also be caused by noradrenaline via α2-adrenergic receptor activation (Kiowski et al. 1985) or by neuropeptide Y (Lundberg & Stjärne, 1984), which is co-released with noradrenaline during exercise (Pernow et al. 1986). After exercise, neuropeptide Y may remain bound to pre-synaptic receptors, reducing noradrenaline release (Pernow et al. 1986). While a study of noradrenaline kinetics or of the relation between sympathetic nerve activity and noradrenaline release would address these potential changes in vascular regulation, to date such studies have not been done after exercise.
In the present study, we used intraarterial infusions of an α1- and an α2-adrenergic agonist to probe the post-synaptic stage of vascular transduction. We found that the vasculature of both the forearm and the leg exhibits robust vasoconstriction in response to these agonists. While the presence of both α1- and α2-adrenergic receptors has been well documented in the human forearm (Kiowski et al. 1985; Dinenno et al. 2002), we are unaware of any prior studies that have investigated the presence of α2-adrenergic receptors in the leg vasculature of humans. While it would appear from our results that there are functional α2-adrenergic receptors in the leg vasculature of humans, confirmation of this observation would require use of an appropriate α2-adrenergic antagonist. It is unknown to what extent α2-adrenergic receptors contribute to resting vascular tone in the leg, but recent work suggests that α2-adrenergic receptors contribute more to resting vascular tone than α1-adrenergic receptors in the forearm of young healthy men (Dinenno et al. 2002).
The current study suggests that the changes in vascular transduction that have been reported in humans during postexercise hypotension (Halliwill et al. 1996a) may differ from what occurs in some animal models of postexercise hypotension. It would appear that in humans, the change in transduction may be entirely pre-junctional, while in the animal models in which a change in responsiveness has been observed, there exists a significant post-junctional component.
Is it possible that the prior report of impaired vascular transduction in humans after exercise is flawed? When Halliwill et al. (1996a) estimated the transduction of sympathetic nerve activity into vascular resistance in response to isometric handgrip exercise held to fatigue (activation of an exercise pressor reflex), the rise in calf vascular resistance for a given rise in muscle sympathetic nerve activity was blunted during postexercise hypotension. This sympathoexcitatory manoeuvre is associated with increases in circulating adrenaline (Robertson et al. 1979) that could have produced some degree of β-adrenergic receptor-mediated vasodilatation. If so, this vasodilatation may have been superimposed on the α-adrenergic receptor-mediated vasoconstriction, and, if augmented during postexercise hypotension, would have appeared as an impairment of sympathetic vascular transduction. On the other hand, it appears that vascular responses to β-adrenergic agonists are blunted following exercise (Martin et al. 1991; Howard & DiCarlo, 1992). Thus, while we suspect that some degree of β-adrenergic vasodilatation does indeed occur during the exercise pressor reflex, it seems unlikely that this effect is augmented postexercise or could explain the prior observation of blunted vascular transduction during postexercise hypotension in humans (Halliwill et al. 1996a). Taken together, these studies suggest an ongoing impairment in noreadrenaline release after exercise.
Limitations
There are several limitations inherent in the methods we have used to assess vascular responses in this study. First, it must be recognized that the α1- and an α2-adrenergic agonists we employed are imperfect in their selectivity for α1- and α2-adrenergic receptors. For instance, phenylephrine can cause β-adrenergic receptor-mediated vasodilatation when administered in the presence of α-adrenergic blockade (Torp et al. 2001). At present, it is unclear how much impact the non-specificity of phenylephrine had on our results. Along these same lines, we did not perform selective α-adrenergic blockade to confirm the specificity of our α2-adrenergic agonist. However, prior studies suggest that > 80 % of the vasoconstriction caused by clonidine is α2-adrenergic receptor dependent (Jie et al. 1984).
Second, it has been argued that intraarterial infusions of adrenergic agonists will primarily activate luminal receptors and may not replicate the pattern of post-junctional α-adrenergic receptor activation that is produced by activation of sympathetic nerves and subsequent perivascular noradrenaline release (Jie et al. 1987). In this context, it is important to recognize that, despite this potential limitation, an identical methodology of intraarterial infusions of phenylephrine has been used successfully in several studies to demonstrate the lasting α1-adrenergic receptor hyporesponsiveness in animal models of postexercise hypotension (Howard & DiCarlo, 1992; Rao et al. 2002) and to demonstrate functional sympatholysis during exercise in humans (Rosenmeier et al. 2003). Thus, it appears that intraarterial infusions of α-adrenergic agonists are indeed an appropriate method for assessing α-adrenergic vascular responsiveness after exercise.
Third, baseline forearm and leg blood flows were significantly higher postexercise versus preexercise, which complicates interpretation of the data for two important reasons. Since agonist concentrations or infusion rates were not adjusted postexercise, the arterial concentrations of the drugs (i.e. the vasoconstrictor stimulus) should have been lower postexercise versus preexercise. This would not change the conclusion that vasoconstrictor responses were not attenuated postexercise, but does raise the question of whether the responses might have been augmented. Furthermore, the validity of comparing vascular responses when basal blood flows differ is debatable, whether data are expressed as absolute changes or percentage changes. The concern is that subtle differences in vascular responsiveness postexercise versus preexercise may have been obscured by the higher basal blood flows in the postexercise period. While we have not addressed these concerns directly in the present study, similar studies comparing vascular responses across varying basal blood flow levels support the use of percentage change in vascular conductance as a robust measure for such comparisons (Buckwalter et al. 1998; Tschakovsky et al. 2002; Rosenmeier et al. 2003).
Finally, in this study the experimental design was such that the second postexercise drug infusion period was performed at a time when the postexercise effects in the forearm appear to have been waning (see Table 1). As such, the lack of an effect on forearm responses may in part reflect a more rapid return of vascular tone to preexercise levels. While we have previously documented that the vasodilatation that underlies postexercise hypotension is not restricted to the sites of active skeletal muscle but also includes inactive regions as well (such as the arms in this study), we had not previously noted a differential time course for forearm and leg responses. However, one might speculate that the mechanisms and time course of vasodilatation during exercise and postexercise hypotension differ between exercising muscle and resting muscle. Thus, although the current results suggest that α-adrenergic responses are similarly maintained during postexercise hypotension in the forearm and leg vasculatures, this does not imply that the mechanism of the overlying vasodilatation is indeed the same in these regions.
Conclusion
We assessed α1- and α2-adrenergic responsiveness in the vasculature of the forearm and leg during postexercise hypotension. In humans, vascular responsiveness to α-adrenergic agonists is maintained during postexercise hypotension. Thus, while postexercise hypotension is associated with increased vascular conductance, α-adrenergic hyporesponsiveness is not a contributing factor. The cause of the vasodilatation underlying postexercise hypotension in humans remains largely unexplained.
Acknowledgments
We thank Christopher P. Johnson, Shelly K. Roberts, Karen P. Krucker and Pamela Engrav for their technical assistance. We especially thank the subjects who volunteered for this study. This research was supported in part by a grant from the American Heart Association, Northland Affiliate, Scientist Development Grant 30403Z and NIH General Clinical Research Center Grant RR-00585 (to the Mayo Clinic, Rochester, MN, USA).
REFERENCES
- Anderson KM, Faber JE. Differential sensitivity of arteriolar α1- and α2-adrenoreceptor constriction to metabolic inhibition during rat skeletal muscle contraction. Circ Res. 1991;69:174–184. doi: 10.1161/01.res.69.1.174. [DOI] [PubMed] [Google Scholar]
- Birch K, Cable N, George K. Combined oral contraceptives do not influence post-exercise hypotension in women. Exp Physiol. 2002;87:623–632. doi: 10.1113/eph8702400. [DOI] [PubMed] [Google Scholar]
- Boone JB, Jr, Levine M, Flynn MG, Pizza FX, Kubitz ER, Andres FF. Opioid receptor modulation of postexercise hypotension. Med Sci Sports Exer. 1992;24:1108–1113. [PubMed] [Google Scholar]
- Buckwalter JB, Mueller PJ, Clifford PS. α1-Adrenergic-receptor responsiveness in skeletal muscle during dynamic exercise. J Appl Physiol. 1998;85:2277–2283. doi: 10.1152/jappl.1998.85.6.2277. [DOI] [PubMed] [Google Scholar]
- Buckwalter JB, Naik JS, Valic Z, Clifford PS. Exercise attenuates α-adrenergic-receptor responsiveness in skeletal muscle vasculature. J Appl Physiol. 2001;90:172–178. doi: 10.1152/jappl.2001.90.1.172. [DOI] [PubMed] [Google Scholar]
- Cléroux J, Kouamé N, Nadeau A, Coulombe D, Lacourcière Y. Aftereffects of exercise on regional and systemic hemodynamics in hypertension. Hypertension. 1992a;19:183–191. doi: 10.1161/01.hyp.19.2.183. [DOI] [PubMed] [Google Scholar]
- Cléroux J, Kouamé N, Nadeau A, Coulombe D, Lacourcière Y. Baroreflex regulation of forearm vascular resistance after exercise in hypertensive and normotensive humans. Am J Physiol. 1992b;263:H1523–1531. doi: 10.1152/ajpheart.1992.263.5.H1523. [DOI] [PubMed] [Google Scholar]
- Coats AJS, Conway J, Isea JE, Pannarale G, Sleight P, Somers VK. Systemic and forearm vascular resistance changes after upright bicycle exercise in man. J Physiol. 1989;413:289–298. doi: 10.1113/jphysiol.1989.sp017654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MD, Sebastian LA, Monnin KA, Tipton CM. Training and post-exercise hypotension with hypertensive rats (SHR) Physiol. 1991;34:267. [Google Scholar]
- Dietz NM, Rivera JM, Eggener SE, Fix RT, Warner DO, Joyner MJ. Nitric oxide contributes to the rise in forearm blood flow during mental stress in humans. J Physiol. 1994a;480:361–368. doi: 10.1113/jphysiol.1994.sp020366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz NM, Rivera JM, Warner DO, Joyner MJ. Is nitric oxide involved in cutaneous vasodilation during body heating in humans? J Appl Physiol. 1994b;76:2047–2053. doi: 10.1152/jappl.1994.76.5.2047. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Eisenach JH, Dietz NM, Joyner MJ. Postjunctional α-adrenoceptors and basal limb vascular tone in healthy men. J Physiol. 2002;540:1103–1110. doi: 10.1113/jphysiol.2001.015297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floras JS, Sinkey CA, Aylward PE, Seals DR, Thoren PN, Mark AL. Postexercise hypotension and sympathoinhibition in borderline hypertensive men. Hypertension. 1989;14:28–35. doi: 10.1161/01.hyp.14.1.28. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Fernandez A, Martin C, Moncada S, Estrada C. Nitric oxide from endothelium and smooth muscle modulates responses to sympathetic nerve stimulation: implications for endotoxin shock. Biochem Biophys Res Commun. 1992;186:150–156. doi: 10.1016/s0006-291x(05)80787-9. [DOI] [PubMed] [Google Scholar]
- Greenfield ADM, Whitney RJ, Mowbray JF. Methods for the investigation of peripheral blood flow. Br Med Bull. 1963;19:101–109. doi: 10.1093/oxfordjournals.bmb.a070026. [DOI] [PubMed] [Google Scholar]
- Halliwill JR. Mechanisms and clinical implications of post-exercise hypotension in humans. Exerc Sport Sci Rev. 2001;29:65–70. doi: 10.1097/00003677-200104000-00005. [DOI] [PubMed] [Google Scholar]
- Halliwill JR, Minson CT, Joyner MJ. Effect of systemic nitric oxide synthase inhibition on postexercise hypotension in humans. J Appl Physiol. 2000;89:1830–1836. doi: 10.1152/jappl.2000.89.5.1830. [DOI] [PubMed] [Google Scholar]
- Halliwill JR, Taylor JA, Eckberg DL. Impaired sympathetic vascular regulation in humans after acute dynamic exercise. J Physiol. 1996a;495:279–288. doi: 10.1113/jphysiol.1996.sp021592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwill JR, Taylor JA, Hartwig TD, Eckberg DL. Augmented baroreflex heart rate gain after moderate-intensity, dynamic exercise. Am J Physiol. 1996b;270:R420–426. doi: 10.1152/ajpregu.1996.270.2.R420. [DOI] [PubMed] [Google Scholar]
- Hannum SM, Kasch FW. Acute postexercise blood pressure response of hypertensive and normotensive men. Scand J Sports Sci. 1981;3:11–15. [Google Scholar]
- Hara K, Floras JS. Effects of naloxone on hemodynamics and sympathetic activity after exercise. J Appl Physiol. 1992;73:2028–2035. doi: 10.1152/jappl.1992.73.5.2028. [DOI] [PubMed] [Google Scholar]
- Howard MG, DiCarlo SE. Reduced vascular responsiveness after a single bout of dynamic exercise in the conscious rabbit. J Appl Physiol. 1992;73:2662–2667. doi: 10.1152/jappl.1992.73.6.2662. [DOI] [PubMed] [Google Scholar]
- Howard MG, DiCarlo SE, Stallone JN. Acute exercise attenuates phenylephrine-induced contraction of rabbit isolated aortic rings. Med Sci Sports Exerc. 1992;24:1102–1107. [PubMed] [Google Scholar]
- Iida N. Nitric oxide mediates sympathetic vasoconstriction at supraspinal, spinal, and synaptic levels. Am J Physiol. 1999;276:H918–H925. doi: 10.1152/ajpheart.1999.276.3.H918. [DOI] [PubMed] [Google Scholar]
- Isea JE, Piepoli M, Adamopoulos S, Pannarale G, Sleight P, Coats AJS. Time course of haemodynamic changes after maximal exercise. Eur J Clin Invest. 1994;24:824–829. doi: 10.1111/j.1365-2362.1994.tb02026.x. [DOI] [PubMed] [Google Scholar]
- Jie K, van Brummelen P, Vermey P, Timmermans PBMWM, van Zwieten PA. Identification of vascular postsynaptic α1- and α2-adrenoceptors in man. Circ Res. 1984;54:447–452. doi: 10.1161/01.res.54.4.447. [DOI] [PubMed] [Google Scholar]
- Jie K, van Brummelen P, Vermey P, Timmermans PBMWM, van Zwieten PA. Postsynaptic α1- and α2-adrenoceptors in human blood vessels: interactions with exogenous and endogenous catecholamines. Eur J Clin Invest. 1987;17:174–181. doi: 10.1111/j.1365-2362.1987.tb02397.x. [DOI] [PubMed] [Google Scholar]
- Kaufman FL, Hughson RL, Schaman JP. Effect of exercise on recovery blood pressure in normotensive and hypertensive subjects. Med Sci Sport Exerc. 1987;19:17–20. [PubMed] [Google Scholar]
- Kiowski W, Hulthén UL, Ritz R, Bühler FR. Prejunctional α2-adrenoceptors and norepinephrine release in the forearm of normal humans. J Cardiovasc Pharm. 1985;7:S144–S148. doi: 10.1097/00005344-198500076-00024. [DOI] [PubMed] [Google Scholar]
- Kulics JM, Collins HL, Di Carlo SE. Postexercise hypotension is mediated by reductions in sympathetic nerve activity. Am J Physiol. 1999;276:H27–32. doi: 10.1152/ajpheart.1999.276.1.H27. [DOI] [PubMed] [Google Scholar]
- Landry JF, Després JP, Prud'homme D, Lamarche B, Tremblay A, Nadeau A, Bouchard C. A study of some potential correlates of the hypotensive effects of prolonged submaximal exercise in normotensive men. Can J Physiol Pharmacol. 1992;70:53–59. doi: 10.1139/y92-008. [DOI] [PubMed] [Google Scholar]
- Lundberg JM, Stjärne L. Neuropepride Y (NPY) depresses the secretion of 3H-noradrenaline and the contractile response evoked by field stimulation, in rat vas deferens. Acta Physiol Scand. 1984;120:477–479. doi: 10.1111/j.1748-1716.1984.tb07410.x. [DOI] [PubMed] [Google Scholar]
- Martin WH, III, Spina RJ, Korte E, Ogawa T. Effects of chronic and acute exercise on cardiovascular β-adrenergic responses. J Appl Physiol. 1991;71:1523–1528. doi: 10.1152/jappl.1991.71.4.1523. [DOI] [PubMed] [Google Scholar]
- Patil RD, DiCarlo SE, Collins HL. Acute exercise enhances nitric oxide modulation of vascular response to phenylephrine. Am J Physiol. 1993;265:H1184–1188. doi: 10.1152/ajpheart.1993.265.4.H1184. [DOI] [PubMed] [Google Scholar]
- Pernow J, Lundberg JM, Kaijser L, Hjemdahl P, Theodorsson-Norheim E, Martinsson A, Pernow B. Plasma neuropeptide Y-like immunoreactivity and catecholamines during various degrees of sympathetic activation in man. Clin Physiol. 1986;6:561–578. doi: 10.1111/j.1475-097x.1986.tb00789.x. [DOI] [PubMed] [Google Scholar]
- Piepoli M, Isea JE, Pannarale G, Adamopoulos S, Sleight P, Coats AJS. Load dependence of changes in forearm and peripheral vascular resistance after acute leg exercise in man. J Physiol. 1994;478:357–362. doi: 10.1113/jphysiol.1994.sp020256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl U, DeWit C. Interaction of nitric oxide with myogenic and adrenergic vasoconstrictor processes in the control of microcirculatory blood flow. Plugers Arch. 1996;432:R107–110. [PubMed] [Google Scholar]
- Rao SP, Collins HL, DiCarlo SE. Postexercise α-adrenergic receptor hyporesponsiveness in hypertensive rats is due to nitric oxide. Am J Physiol Regul Integr Comp Physiol. 2002;282:R960–968. doi: 10.1152/ajpregu.00490.2001. [DOI] [PubMed] [Google Scholar]
- Robertson D, Johnson GA, Robertson RM, Nies AS, Shand DG, Oates JA. Comparative assessment of stimuli that release neuronal and adrenomedullary catecholamines in man. Circulation. 1979;59:637–643. doi: 10.1161/01.cir.59.4.637. [DOI] [PubMed] [Google Scholar]
- Rosenmeier JB, Dinenno FA, Fritzlar SJ, Joyner MJ. α1-and α2-adrenergic vasoconstriction is blunted in contracting human muscle. J Physiol. 2003;547:971–976. doi: 10.1113/jphysiol.2002.037937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schw arz L, Kindermann W. Changes in β-endorphin levels in response to aerobic and anaerobic exercise. Sports Med. 1992;13:25–36. doi: 10.2165/00007256-199213010-00003. [DOI] [PubMed] [Google Scholar]
- Sebastian LA, Moran MM, Tipton CM. Post-exercise hypotension and intra-cerebro-ventricular (ICV) injections of β-endorphin and naloxone hydrochloride. FASEB J. 1995;9:A361. [Google Scholar]
- Senitko AN, Charkoudian N, Halliwill JR. Influence of endurance exercise training status and gender on postexercise hypotension. J Appl Physiol. 2002;92:2368–2374. doi: 10.1152/japplphysiol.00020.2002. [DOI] [PubMed] [Google Scholar]
- Shyu BC, Thorén P. Circulatory events following spontaneous muscle exercise in normotensive and hypertensive rats. Acta Physiol Scand. 1986;128:515–524. doi: 10.1111/j.1748-1716.1986.tb08007.x. [DOI] [PubMed] [Google Scholar]
- Somers VK, Conway J, Coats A, Isea J, Sleight P. Postexercise hypotension is not sustained in normal and hypertensive humans. Hypertension. 1991;18:211–215. doi: 10.1161/01.hyp.18.2.211. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Hansen J, Victor RG. Inhibition of α2-adrenergic vasoconstriction during contraction of glycolytic, not oxidative, rat himblimb muscle. Am J Physiol. 1994;266:H920–929. doi: 10.1152/ajpheart.1994.266.3.H920. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Victor RG. Nitric oxide mediates contraction-induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J Physiol. 1997;506:817–826. doi: 10.1111/j.1469-7793.1998.817bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torp KD, Tschakovsky ME, Halliwill JR, Minson CT, Joyner MJ. β-receptor agonist activity of phenylephrine in the human forearm. J Appl Physiol. 2001;90:1855–1859. doi: 10.1152/jappl.2001.90.5.1855. [DOI] [PubMed] [Google Scholar]
- Tschakovsky ME, Sujirattanawimol K, Ruble SB, Valic Z, Joyner MJ. Is sympathetic neural vasoconstriction blunted in the vascular bed of exercising human muscle? J Physiol. 2002;541:623–635. doi: 10.1113/jphysiol.2001.014431. Wilcox RG, Bennett T, Brown AM & Macdonald IA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox RG, Bennett T, Brown AM, Macdonald IA. Is exercise good for high blood pressure? Br Med J. 1982;285:767–769. doi: 10.1136/bmj.285.6344.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Dusting H, Rand MJ. Inhibition of sympathetic neurotransmission by the opioid δ-receptor agonist DAMA in the pithed rat. Clin Exp Pharmacol Physiol. 1989;16:821–827. doi: 10.1111/j.1440-1681.1989.tb01521.x. [DOI] [PubMed] [Google Scholar]