

Abstract
We tested the hypothesis that activation of peripheral chemoreceptors with acute isocapnic hypoxia resets arterial baroreflex control of both heart rate and sympathetic vasoconstrictor outflow to higher pressures, resulting in increased heart rate and muscle sympathetic nerve activity without changes in baroreflex sensitivity. We further hypothesized that this resetting would not occur during isocapnic hyperpnoea at the same breathing rate and depth as during isocapnic hypoxia. In 12 healthy, non-smoking, normotensive subjects (6 women, 6 men, 19-36 years), we assessed baroreflex control of heart rate and muscle sympathetic nerve activity using the modified Oxford technique during normoxia, isocapnic hyperpnoea, and isocapnic hypoxia (85 % arterial O2 saturation). While isocapnic hyperpnoea did not alter heart rate, arterial pressure, or sympathetic outflow, hypoxia increased heart rate from 61.9 ± 1.8 to 74.7 ± 2.7 beats min−1 (P < 0.05), increased mean arterial pressure from 97.4 ± 2.0 to 103.9 ± 3.3 mmHg (P < 0.05), and increased sympathetic activity 22 ± 13 % relative to normoxia and 72 ± 21 % (P < 0.05) relative to hyperpnoea alone. The sensitivity for baroreflex control of both heart rate and sympathetic activity was not altered by either hypoxia or hyperpnoea. Thus, it appears that acute activation of peripheral chemoreceptors with isocapnic hypoxia resets baroreflex control of both heart rate and sympathetic activity to higher pressures without changes in baroreflex sensitivity. Furthermore, these effects appear largely independent of breathing rate and tidal volume.
Chemoreflexes and baroreflexes play a major role in control of the cardiovascular system via the profound influences they exert on autonomic outflow. However, the interaction of these powerful reflex systems is not completely understood in humans. The peripheral chemoreceptors located in the carotid and aortic bodies are activated by a fall in O2, and to a lesser degree by a rise in CO2 or acidity (Fitzgerald & Lahiri, 1986). These receptors send signals to the medulla via cranial nerves IX and X, and synapse initially in the nucleus tractus solitarii. The best known effect of peripheral chemoreceptor activation is hyperpnoea, but these pathways also affect the level of activity in the sympathetic outflow tracts located in the intermediolateral cell column of the spinal cord and the parasympathetic efferent tracts of the vagus nerve. In contrast to peripheral chemoreceptors, chemoreceptive areas within the central nervous system (i.e. central chemoreceptors) are not responsive to decreases in O2 until severely hypoxic (e.g. arterial O2 saturations less than 50 %) but are sensitive to changes in CO2 and acidity (Bowes et al. 1981).
Exposure of humans to either altitude or hypoxia produces elevations in heart rate and sympathetic vasoconstrictor nerve activity directed to skeletal muscle vascular beds (Saito et al. 1988; Rowell et al. 1989; Somers et al. 1989; Halliwill & Minson, 2002). Previously, Halliwill & Minson (2002) found that these changes are accompanied by resetting of the arterial baroreflex to higher pressures and higher levels of heart rate and muscle sympathetic nerve activity. This resetting occurs without discernible changes in arterial baroreflex sensitivity. However, it should be noted that these observations were based on exposure of subjects to a hypoxic breathing gas (12 % O2 in N2) which leads to significant hyperpnoea and hypocapnia. Animal studies suggest that primary chemoreflex responses are often modified, or over-ridden, by the effects of increased rate and depth of breathing on autonomic outflow and cardiovascular function (Rutherford & Vatner, 1978). Thus, the prior observation of arterial baroreflex resetting during hypoxia may not be attributable to chemoreflex activation per se but could result from concomitant hyperpnoea and hypocapnia. On the other hand, one series of studies suggests that increased ventilation or changes in breathing patterns in humans modify the pattern of sympathetic nerve activity within breaths but do not change the overall level of sympathetic outflow (Seals et al. 1990, 1993). This would suggest that the sympathoexcitation produced by hypoxia in humans is a direct effect of peripheral chemoreflex activation. Unfortunately, many human studies on peripheral chemoreflex control of cardiovascular function have ignored the potentially confounding changes in ventilation and CO2. The few studies that controlled for these factors have focused on control of heart rate (Bhattacharva et al. 1973; Eckberg et al. 1982). Therefore, observations made at altitude or under conditions of hypocapnic hypoxia may not reflect the direct effects of chemoreflex activation and may differ from responses produced by isocapnic hypoxia or hypercapnic hypoxia (e.g. during sleep apnoea).
Therefore, the goal of the current study was to provide insight into cardiovascular regulation during activation of peripheral chemoreceptors. The purpose of this protocol was to assess the effect of activation of peripheral chemoreceptors with isocapnic hypoxia on baroreflex control of heart rate and muscle sympathetic nerve activity. Isocapnic hypoxia was used to specifically target activation of peripheral chemoreceptors without the confounding effect of co-activating central chemoreceptors. A secondary purpose was to differentiate between the direct effects of chemoreflex activation and secondary effects related to changes in ventilation rate and depth. We tested the hypothesis that activation of peripheral chemoreceptors with acute isocapnic hypoxia resets arterial baroreflex control of both heart rate and sympathetic vasoconstrictor outflow to higher pressures, resulting in increased heart rate and muscle sympathetic nerve activity without changes in baroreflex sensitivity. We further hypothesized that this resetting would not occur during isocapnic hyperpnoea at the same breathing rate and depth as during isocapnic hypoxia.
METHODS
This study received approval from an Institutional Review Board and each subject gave his or her informed written consent prior to participation. All studies were performed according to the Declaration of Helsinki. We assessed baroreflex control of heart rate and muscle sympathetic nerve activity using the modified Oxford technique during normoxia, isocapnic hyperpnoea and isocapnic hypoxia.
Subjects
Twelve healthy, non-smoking, normotensive subjects (6 women, 6 men) between the ages of 19 and 36 years participated in this study (height 172 ± 9 cm, weight 73.0 ± 13.1 kg, body mass index 24.4 ± 2.7 kg m−2 (means ± S.D.)). None of the subjects were taking medications other than oral contraceptives, and none had been at altitude (> 1500 m) for at least 5 months. Haemoglobin concentrations ranged from 11.7 to 15.8 mg dl−1. All female subjects had a negative serum pregnancy test within 12 h prior to participation. Because of the effects of the menstrual cycle on cardiovascular regulation (Minson et al. 2000a,b), all female subjects were studied during the early follicular phase (1-4 days after the onset of menstruation) or during the placebo phase of oral contraceptives to control for this potential influence.
Familiarization visit
Subjects were brought to the lab several days prior to the protocol day to become familiar with the instrumentation and for determination of their individual ventilatory response during a 5 min period of isocapnic hypoxia (85 % arterial O2 saturation). Details regarding the induction of isocapnic hypoxia are provided below. Following these measurements, each subject was instructed and allowed to practice controlled breathing at a prescribed tidal volume and breathing frequency (isocapnic hyperpnoea) that was based on their individual ventilatory response during the preceding bout of isocapnic hypoxia.
Protocol visit
Throughout the protocol, subjects were instrumented in the supine position for measurement of heart rate (electrocardiogram), beat-by-beat arterial pressure via finger photoplethysmography (Finapres blood pressure monitor, Model 2300, Ohmeda, Englewood, CO, USA), and arterial O2 saturation via pulse oximetry of the ear lobe (Cardiocap/5, Datex-Ohmeda, Madison, WI, USA). An intravenous catheter was placed in an antecubital vein for administration of vasoactive substances for the purpose of assessing baroreflex responses. In 10 of the subjects (5 women, 5 men), we recorded muscle sympathetic nerve activity from the fibular (peroneal) nerve via microneurography. In the remaining two subjects, nerve recordings were either inadequate or were not stable during one or more of the study conditions.
After instrumentation, subjects were monitored during a 20 min rest period in which tidal CO2 was measured via a nasal cannula (Cardiocap/5, Datex-Ohmeda, Madison, WI, USA). Mean end-tidal CO2 over the last 5 min was defined as isocapnia for the remainder of the protocol. Subjects then underwent three measurement periods separated by 30 min rest periods. Each measurement period corresponded to one of the following conditions: normoxia with uncontrolled breathing, isocapnic hyperpnoea with controlled breathing, and isocapnic hypoxia with controlled breathing. We continuously recorded heart rate, arterial pressure, arterial O2 saturation, end-tidal PCO2, ventilation, and sympathetic activity during each measurement period. We assessed baroreflex control of heart rate and muscle sympathetic nerve activity after recording a 5 min period of stable pressure and heart rate for each condition. Our prior work has shown that repeated baroreflex trials separated by a 20 min rest period are reproducible (Rudas et al. 1999; Minson et al. 2000a,b). Since we had previously determined each subject's ventilatory response to hypoxia, we were able to randomize the order of conditions between normoxia, isocapnic hyperpnoea and isocapnic hypoxia.
Experimental procedures
Normoxia, isocapnic hyperpnoea and isocapnic hypoxia
In order to induce these three conditions and isolate the effects of hypoxia, we employed a self-regulating partial-rebreathe system developed by Banzett et al. (2000) to maintain constant alveolar fresh-air ventilation independent of changes in breathing frequency or tidal volume as we have done previously (Weisbrod et al. 2001; Dinenno et al. 2003). This system allowed us to clamp end-tidal CO2 levels despite large changes in minute ventilation during hypoxia and hyperpnoea. Using this technique, the level of O2 provided in the inspiratory gas was manipulated by mixing N2 with air via a medical gas blender. For isocapnic hypoxia, the level of O2 was titrated down to achieve an arterial O2 saturation of 85 % as assessed by pulse oximetry of the earlobe. Subjects breathed through a scuba mouthpiece with a nose-clip to prevent any nasal breathing. Gas concentrations were monitored at the mouthpiece (Cardiocap/5, Datex-Ohmeda, Madison, WI, USA). Ventilation was measured via a pneumotachograph (model VMM-2a, Interface Associates, Laguna Niguel, CA, USA). In addition, controlled breathing at a prescribed tidal volume and breathing frequency was used with and without superimposed hypoxia to produce conditions of isocapnic hypoxia and isocapnic hyperpnoea. This was achieved by providing both an auditory and visual cue for breathing frequency and displaying real-time inspiratory tidal volume with predetermined target tidal volumes for each subject. Identical breathing patterns were used for both isocapnic hyperpnoea and isocapnic hypoxia. During the normoxia condition, visual and auditory cues were not provided and the subject's breathing patterns were not controlled.
Muscle sympathetic nerve activity
Muscle sympathetic nerve activity was recorded via microneurography (Sundlöf & Wallin, 1977). Multiunit postganglionic muscle sympathetic nerve activity was recorded from the fibular (peroneal) nerve posterior to the fibular head with a tungsten microelectrode. The recorded signal was amplified 100 000-fold and band-pass filtered (700-2000 Hz), rectified and integrated (resistance-capacitance integrator circuit, time constant 0.1 s) with a custom-built amplifier system for analysis of muscle sympathetic nerve activity.
Baroreflex control of heart rate and sympathetic outflow
Baroreflex responses were assessed by measuring heart rate and muscle sympathetic nerve activity during arterial pressure changes induced by nitroprusside and phenylephrine as developed by Ebert & Cowley (1992) and validated by Rudas et al. (1999). During all three conditions, 100 μg sodium nitroprusside was given intravenously as a bolus, followed 1 min later by 150 μg phenylephrine HCl. This protocol decreases arterial pressure ≈15 mmHg below baseline levels and then increases it ≈15 mmHg above baseline levels, over a short time course. We have previously shown that this pressure stimulus is unaltered by systemic hypoxia (Halliwill & Minson, 2002).
Data analysis
Data were digitized at 250 Hz with signal processing software (WinDaq, Dataq Instruments, Akron, OH, USA) and analysed off-line. Each muscle sympathetic nerve activity recording was normalized by assigning the largest sympathetic burst under resting conditions an amplitude of 1000 (Halliwill, 2000). All other bursts for that recording were calibrated against that value. The zero nerve activity level was determined from the mean voltage during a period of neural silence between sympathetic bursts. A period in which bursts were absent for > 5 s was found in each tracing and used for this purpose.
Baroreflex control of sympathetic outflow was determined from the relation between muscle sympathetic nerve activity and diastolic pressure during vasoactive drug boluses (Ebert & Cowley, 1992; Rudas et al. 1999). The slope of this relation is used as an index of reflex sensitivity. The operating point for the relation in terms of resting arterial pressure and nerve activity was determined as the average values over the 5-min period immediately preceding the nitroprusside bolus. Diastolic pressure was used because muscle sympathetic nerve activity correlates closely with diastolic pressure but not with systolic pressure (Sundlöf & Wallin, 1977; Rudas et al. 1999). The methods used to analyse these data have been described extensively (Halliwill, 2000; Halliwill & Minson, 2002).
Baroreflex control of heart rate was determined from the relation between heart rate and systolic pressure during vasoactive drug boluses. The slope of this relation is used as an index of reflex sensitivity. The operating point for the relation in terms of resting arterial pressure and heart rate was determined as the average values over the 5 min period immediately preceding the nitroprusside bolus. Systolic pressure was used because heart rate correlates closely with systolic pressure but not with diastolic pressure (Sundlöf & Wallin, 1977; Rudas et al. 1999). In order to perform a linear regression between heart rate and pressure, values for heart rate were first pooled over 2 mmHg pressure ranges as described previously (Ebert & Cowley, 1992; Rudas et al. 1999; Halliwill & Minson, 2002). The analogous regression between R-R interval and systolic pressure was also determined.
Statistics
Because there were no discernible differences between men and women, data from the two groups were combined for statistical analysis. Measurements across the three conditions were compared via a one-way repeated measures analysis of variance. Regressions for baroreflex responses were compared across the three conditions via a one-way repeated measures analysis of variance of the individual slopes for each regression. Likewise, in order to quantify parallel shifts in the baroreflex pressure-effector relations, predicted effector responses (e.g. heart rate) were calculated from the individual regressions at an arbitrary pressure for each individual across all conditions and compared via a one-way repeated analysis of variance. The arbitrary pressure was selected as each individual's resting pressure under normoxic conditions. When appropriate, post-hoc comparisons were performed using Tukey's test. Differences were considered significant when P < 0.05. All values are reported as means ± s.e.m. unless otherwise indicated.
RESULTS
Controlled breathing during isocapnic hyperpnoea and isocapnic hypoxia
Table 1 shows ventilation during all three conditions, normoxia, isocapnic hyperpnoea and isocapnic hypoxia. As planned, arterial O2 saturation levels were markedly lower during isocapnic hypoxia than during either normoxia or isocapnic hyperpnoea. Breathing patterns were tightly controlled by the subjects so that the rate and depth of ventilation were similar during isocapnic hyperpnoea and isocapnic hypoxia. Both conditions showed greater tidal volume and minute ventilation compared to normoxic uncontrolled breathing. As planned, end-tidal CO2 did not differ across conditions.
Table 1.
Ventilation
| Normoxia | Isocapnic hyperpnoea | Isocapnic hypoxia | |
|---|---|---|---|
| Arterial O2 saturation (%) | 98.8 ± 0.2 | 99.1 ± 0.2 | 85.7 ± 0.3*† |
| Minute ventilation (1 min−1) | 8.3 ± 1.2 | 18.4 ± 2.4* | 18.9 ± 2.7* |
| Breating frequency (breaths min−1) | 12.9 ± 1.5 | 14.0 ± 1.4 | 14.1 ± 1.5 |
| Tidal volume (ml) | 667 ± 63 | 1444 ± 240* | 1474 ± 252* |
| End-tidal Pco2 (mmHg) | 37.6 ± 1.5 | 38.4 ± 1.1 | 39.1 ± 1.1 |
P < 0.05 vs. normoxia.
P < 0.05 vs. isocapnic hyperpnoea. Values are means ±s.e.m.
Table 2 shows heart rate and arterial pressure during all three conditions. While isocapnic hyperpnoea did not alter heart rate or pressure, isocapnic hypoxia was associated with an increase in both heart rate and arterial pressure. While there was a tendency for muscle sympathetic nerve activity to decrease during isocapnic hyperpnoea (1312 ± 642 vs. 1700 ± 536 total integrated units min−1, P = 0.13), isocapnic hypoxia was associated with an increase in sympathetic outflow (2032 ± 749 total integrated units min−1, P < 0.05) which represented an increase of 22 ± 13 % versus normoxia and 72 ± 21 % versus isocapnic hyperpnoea.
Table 2.
Heart rate and arterial pressure
| Normoxia | Isocapnic hyperpnoea | isocapnic hypoxia | |
|---|---|---|---|
| Heart rate (beats min−1) | 61.2 ± 1.8 | 61.9 ± 1.8 | 74.7 ± 2.7*† |
| Systolic arterial pressure (mmHg) | 143.4 ± 2.7 | 140.4 ± 3.4 | 150.8 ± 4.9† |
| Diastolic arterial pressure (mmHg) | 76.5 ± 1.6 | 75.9 ± 2.2 | 80.4 ± 3.1 |
| Mean arterial pressure (mmHg) | 98.8 ± 1.5 | 97.4 ± 2.0 | 103.9 ± 3.3† |
P < 0.05 vs. normoxia.
P < 0.05 vs. isocapnic hyperpnoea. Values are means ± s.e.m.
Cardiovascular regulation during isocapnic hyperpnoea and isocapnic hypoxia
An example from one subject of the arterial baroreflex response relationships for heart rate and muscle sympathetic nerve activity is shown in Fig. 1. Figure 2 shows the group average regressions between heart rate and systolic pressure and between sympathetic nerve activity and diastolic pressure. For both relationships, slopes tended to be less steep during isocapnic hyperpnoea compared to normoxia (P = 0.13-0.20). This tendency was reversed during isocapnic hypoxia so that there were no differences in slope between isocapnic hypoxia and normoxia (Fig. 3). If we analyse our data in terms of R-R interval, we also find there were no differences in slope across conditions (slope for R-R interval, normoxia: 18.5 ± 2.1; hyperpnoea: 17.1 ± 2.0; hypoxia: 15.0 ± 1.9 ms (mmHg)−1, P = 0.13).
Figure 1. Data from a representative subject showing baroreflex relationships.
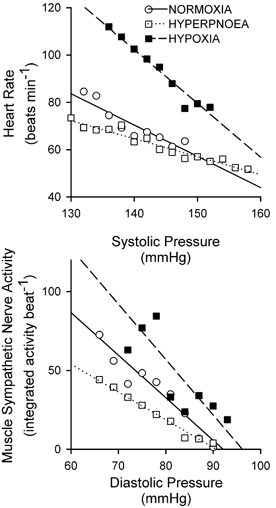
Upper panel, baroreflex relationship between heart rate and systolic pressure. Lower panel, baroreflex relationship between sympathetic nerve activity and diastolic pressure. ○, normoxia; □, isocapnic hyperpnoea; ▪, isocapnic hypoxia. The continuous line denotes the regression between pressure and effector response for normoxia (heart rate = 255 –(1.32 × pressure), r2 = 0.83; nerve activity = 249 –(2.7 × pressure), r2 = 0.94). The dotted line denotes the regression between pressure and effector response for isocapnic hyperpnoea (heart rate = 171 –(0.76 × pressure), r2 = 0.92; nerve activity = 160 –(1.8 × pressure), r2 = 0.97). The dashed line denotes the regression between pressure and effector response for isocapnic hypoxia (heart rate = 421 –(2.28 × pressure), r2 = 0.94; nerve activity = 338 –(3.5 × pressure), r2 = 0.69).
Figure 2. Group average regressions between heart rate and systolic pressure (upper panel) and between sympathetic nerve activity and diastolic pressure (lower panel).
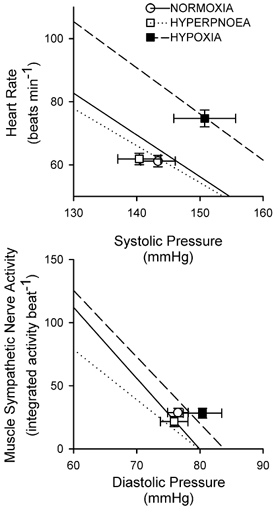
The operating points are indicated by symbol and error bars (mean ± s.e.m.) for each condition (○, normoxia; □, isocapnic hyperpnoea; ▪, isocapnic hypoxia). The continuous line denotes the regression between pressure and effector response for normoxia (heart rate = (255 ± 21) –(1.32 ± 0.14) × pressure, r2 = 0.91 ± 0.02; nerve activity = (449 ± 97) –(5.6 ± 1.2) × pressure, r2 = 0.82 ± 0.06). The dotted line denotes the regression between pressure and effector response for isocapnic hyperpnoea (heart rate = (230 ± 20) - (1.17 ± 0.13) × pressure, r2 = 0.93 ± 0.02; nerve activity = (317 ± 61) –(4.0 ± 0.8) × pressure, r2 = 0.74 ± 0.08). The dashed line denotes the regression between pressure and effector response for isocapnic hypoxia (heart rate = (295 ± 33) –(1.46 ± 0.21) × pressure, r2 = 0.88 ± 0.03; nerve activity = (442 ± 88) –(5.3 ± 1.2) × pressure, r2 = 0.85 ± 0.04). Values are means ± s.e.m. (n = 12 for heart rate; n = 10 for muscle sympathetic nerve activity).
Figure 3.
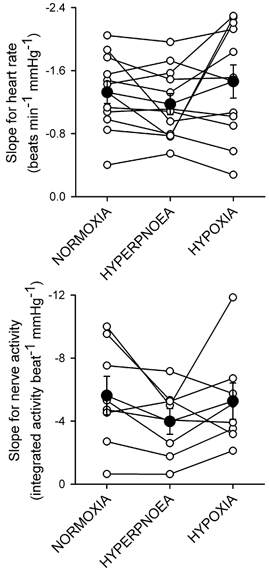
Individual (○) and group average (•) slopes for the relationship between heart rate and systolic pressure (upper panel) and between sympathetic nerve activity and diastolic pressure (lower panel) under normoxic, isocapnic hyperpnoea, and isocapnic hypoxia conditions.
Isocapnic hypoxia was associated with a shift in the baroreflex relationship upward and rightward as reflected by higher values for operating point pressure, heart rate, and sympathetic nerve activity relative to normoxia and hyperpnoea. To quantify the magnitude of this resetting, we determined the upward parallel shift in the baroreflex pressure-effector relation at an arbitrary pressure across all conditions for each individual. Based on this analysis, isocapnic hypoxia was associated with increases in heart rate of 20.9 ± 6.2 and 24.4 ± 7.0 beats min−1 at a given pressure versus normoxia and hyperpnoea (both P < 0.05). Isocapnic hypoxia was associated with increases in muscle sympathetic nerve activity of 2451 ± 1287 and 2982 ± 1607 total integrated units min−1 at a given pressure versus normoxia and hyperpnoea (both P < 0.05).
DISCUSSION
The purpose of this protocol was to study the effect of peripheral chemoreceptor activation with isocapnic hypoxia on baroreflex control of heart rate and muscle sympathetic nerve activity, and to differentiate between direct effects of chemoreflex activation and effects secondary to hyperventilation. We found that neither hyperpnoea nor hypoxia alter the sensitivity of the arterial baroreflex. However, hypoxia, but not hyperpnoea alone, resulted in a resetting of baroreflex control of both heart rate and sympathetic activity to higher pressures, higher heart rates, and higher levels of sympathetic vasoconstrictor outflow. Thus, the hypoxic resetting of the baroreflex that was previously observed (Halliwill & Minson, 2002) appears largely independent of breathing rate and tidal volume. This suggests that the increases in heart rate and sympathetic outflow observed at altitude or during exposure to hypoxia are indeed the result of peripheral chemoreflex activation per se and not secondary to the concomitant hyperpnoea and hypocapnia.
We were not the first to perform well controlled studies on the reflex control of heart rate during exposure to hypoxia. A preliminary report by Bhattacharya et al. (1973) similarly found resetting of baroreflex control of heart rate and an absence of changes in sensitivity during isocapnic hypoxia with controlled ventilation. Furthermore, Eckberg et al. (1982) demonstrated that brief (≈30 s) exposure to hypoxia leads to tachycardia via withdrawal of cardiac vagal tone, as reflected by reductions of respiratory sinus arrhythmia. Along these same lines, Gupta & Singh (1987) reported that hypoxia causes tachycardia in dogs when cardiac vagal tone is intact but not when cardiac vagal tone is reduced by anaesthetic agents. Our current findings are consistent with these previous studies.
Our new findings are also consistent with prior reports of elevated muscle sympathetic nerve activity during hypoxia in humans (Saito et al. 1988; Rowell et al. 1989; Somers et al. 1989; Halliwill & Minson, 2002). However, these results contrast with the effects of hypoxia on control of sympathetic nerve activity in animal models. In several animal models, hypoxia has been shown to cause baroreflex resetting with an increase in sensitivity of sympathetic outflow to the kidney (Pelletier & Shepherd, 1975; Iriki et al. 1977; Malpas et al. 1996) and skeletal muscle vascular beds (Pelletier & Shepherd, 1975). It is unclear whether these differences are species-related, or due to differences in study preparation. Some (Pelletier & Shepherd, 1975; Iriki et al. 1977) but not all (Malpas et al. 1996) of these animal studies have relied on anaesthetized, mechanically ventilated preparations. As mentioned above, it appears that some anaesthetic agents alter the response to hypoxia (Gupta & Singh, 1987). Some differences may be related to the degree of hypoxia. In the present study we used modest hypoxia, which was associated with small autonomic responses. However, more severe hypoxia that approximates what has been used in these animal studies (80 % vs. 85 % arterial O2 saturation) also failed to produce changes in baroreflex sensitivity in humans (Halliwill & Minson, 2002). We did notice a tendency in the present study for blunting of baroreflex sensitivity during hyperpnoea which was reversed during hypoxia, but differences were small and variable. In fact, sample size analysis of baroreflex sensitivity indicated that 38 subjects would need to be studied to demonstrate a difference in sensitivity. As such, it appears that the pathways involved in the human response to hypoxia do not alter sensitivity of the arterial baroreflex.
Role of increased respiratory effort
It has been suggested that increases in respiratory effort can produce sympathoexcitation via several pathways. For example, a central nervous system feed-forward pathway, analogous to the ‘central command’ implicated in cardiovascular responses to exercise, could be activated by additional recruitment of respiratory muscles during increases in respiratory effort (Victor et al. 1995). Also, feedback pathways involving mechanoreflexes or metaboreflexes arising from respiratory muscles could be activated during hyperventilation (St Croix et al. 2000). However, based on our data during hyperpnoea, it does not appear that either of these mechanisms is activated sufficiently to augment sympathetic outflow or cause baroreflex resetting under conditions of moderately increased respiratory effort. This would suggest that the sympathoexcitation and resetting of the baroreflex we observed during hypoxia is not related to a greater respiratory effort.
Role of peripheral chemoreceptors
We believe that within the context of our experimental controls, the current data support the notion that the resetting of the arterial baroreflex and associated increases in heart rate and sympathetic outflow to skeletal muscle vascular beds are mediated via stimulation of the peripheral chemoreceptors. This is based on the presumption that central chemoreceptors do not respond to modest hypoxia (Bruce & Cherniack, 1987). Further support for this notion comes from the observation that baroreceptor and chemoreceptor projections within the medulla often coincide, with the overlap of these medullary projections providing multiple locations in which interactions between these reflexes could occur (Loewy, 1990). Along these lines, classic studies have shown that activating the baroreflexes by increasing arterial pressure attenuates peripheral chemoreflex-mediated ventilatory (Heistad et al. 1974) and vascular responses to hypoxia (Heistad et al. 1975; Mancia, 1975). Likewise, the current results are consistent with the observations of Somers et al. (1991) who found that phenylephrine infusion to raise arterial pressure blunted the rise in muscle sympathetic nerve activity during hypoxia. Miura & Reis (1972) have localized similar interactions to the paramedian reticular nuclei. We can only speculate as to whether this location is involved in the baroreflex resetting that we have observed.
Based on descriptions of ‘classic’ resetting described by Korner (1979) and others (Eckberg & Sleight, 1992; Rowell, 1993), it would appear that the peripheral chemoreflex is causing changes in autonomic outflow via pathways that are both baroreflex-dependent and −independent, meaning that some of the affected autonomic outflow tracts which are being activated are not under baroreflex control but that others are being activated via the baroreflex. This may be analogous to the classical baroreflex resetting observed during exercise (Raven et al. 1997).
Limitations
In the present study, we have attempted to overcome certain limitations related to changes in ventilation, yet other limitations remain. A consistent confounding factor in studies of baroreflex control of heart rate is the inverse relationship between R-R interval and heart rate. When baseline heart rate is increased, there is a disproportionate reduction in R-R interval responses due to the non-linear relationship between R-R interval and heart rate. This issue has clouded many prior investigations on baroreflex control of heart rate, and obstructed the understanding of the baroreflex resetting that occurs during exercise for many years. Since heart rate (and not R-R interval) is linearly related to cardiac output, heart rate relates to correction of a change in pressure by the baroreflex. Thus, when baseline heart rate is changed, it is reasonable to consider the change in heart rate (and not R-R interval) in response to changes in arterial pressure in order to provide insight into whether or not baroreflex function has changed. In the context of moderate hypocapnic hypoxia (arterial saturations of 75-80 %), resetting of baroreflex control of heart rate occurs without alteration of the amplitude of the heart rate response to changes in pressure (i.e. no change in sensitivity) (Sagawa et al. 1997; Halliwill & Minson, 2002) but sensitivity in terms of the R-R interval response is reduced by moderate hypoxia. Unlike baroreflex control of sympathetic outflow, it appears that the sensitivity of arterial baroreflex control, when assessed in terms of R-R interval, is linked to changes in resting R-R interval such that a shortening of the resting R-R interval causes a reduction in R-R interval responses. However, heart rate responses appear analogous to the muscle sympathetic nerve activity responses. It is unclear if this is a reflection of the dual innervation of the heart or simply a mathematical artifact. Fortunately, in the context of the present study, the sensitivity of the baroreflex in terms of both heart rate and R-R interval shows analogous trends and thus the choice of variable becomes moot.
In animal models, it is often possible to assess baroreflex responses over a wide range of pressures so that response relations encompass the reflex from threshold to saturation. Such data can often be analysed by applying a sigmoidal model to the data. The modified Oxford method is not able to divulge the entire response relationship consistently in humans, as the non-linear threshold and saturation regions are variably present within the pressure ranges achieved. As such, we have restricted our analysis to the linear region of the reflex response that was evident in the collected data. Thus, the sensitivity as we define it may only be applicable to the pressure range that we have assessed. Further, cardiac response are probably dominated by changes in vagal outflow to the heart with negligible contribution of sympathetic outflow.
Conclusions
The current study was designed to address whether peripheral chemoreflex activation with isocapnic hypoxia resets baroreflex control of heart rate and muscle sympathetic nerve activity to higher pressures and higher levels of heart rate and sympathetic nerve activity. By tightly controlling for changes in ventilation, this study provides strong evidence that chemoreflex activation exerts an influence on arterial pressure, heart rate and sympathetic outflow without changes in sensitivity of the arterial baroreflex. This suggests that the increases in heart rate and sympathetic outflow observed at altitude or during exposure to hypoxia are indeed the result of peripheral chemoreflex activation per se and not secondary to the concomitant hyperpnoea and hypocapnia.
Acknowledgments
We thank Christopher P. Johnson, Karen P. Krucker, and Shelly K. Roberts for their technical assistance. We especially thank the subjects who volunteered for this study. This research was supported in part by a grant from the Wilderness Medical Society, National Institutes of Health (NIH) Grant HL-65305, and American Heart Association Grant 30403Z.
REFERENCES
- Banzett RB, Garcia RT, Moosavi SH. Simple contrivance ‘clamps’ end-tidal PCO2 and PO2 despite rapid changes in ventilation. J Appl Physiol. 2000;88:1597–1600. doi: 10.1152/jappl.2000.88.5.1597. [DOI] [PubMed] [Google Scholar]
- Bhattacharya J, Cunningham DJC, Howson MG, Lee MJR, Sleight P. The effects of mild hypoxia with constant PA,CO2 and ventilation on the setting and sensitivity of the baroreceptor-cardiac-depressor reflex in man. J Physiol. 1973;234:112–114P. [PubMed] [Google Scholar]
- Bowes G, Townsend ER, Kozar LF, Bromley SM, Phillipson EA. Effect of carotid body denervation on arousal response to hypoxia in sleeping dogs. J Appl Physiol. 1981;51:40–45. doi: 10.1152/jappl.1981.51.1.40. [DOI] [PubMed] [Google Scholar]
- Bruce EN, Cherniack NS. Central chemoreceptors. J Appl Physiol. 1987;62:389–402. doi: 10.1152/jappl.1987.62.2.389. [DOI] [PubMed] [Google Scholar]
- Dinenno FA, Joyner MJ, Halliwill JR. Failure of systemic hypoxia to blunt α-adrenergic vasoconstriction in the human forearm. J Physiol. 2003;549:985–994. doi: 10.1113/jphysiol.2003.042507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert TJ, Cowley AW., Jr Baroreflex modulation of sympathetic outflow during physiological increases of vasopressin in humans. Am J Physiol. 1992;262:H1372–1378. doi: 10.1152/ajpheart.1992.262.5.H1372. [DOI] [PubMed] [Google Scholar]
- Eckberg DL, Bastow H, III, Scruby AE. Modulation of human sinus node function by systemic hypoxia. J Appl Physiol. 1982;52:570–577. doi: 10.1152/jappl.1982.52.3.570. [DOI] [PubMed] [Google Scholar]
- Eckberg DL, Sleight P. Human Baroreflexes in Health and Disease. New York: Oxford University Press; 1992. [Google Scholar]
- Fitzgerald RS, Lahiri S. Reflex responses to chemoreceptor stimulation. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, section 3, The Respiratory System, Control of Breathing. II. Bethesda, MD, USA: American Physiological Society; 1986. pp. 313–362. [Google Scholar]
- Gupta PD, Singh M. Tachycardia of carotid chemoreceptors originates in apneic asphyxia in dogs. Am J Physiol. 1987;253:H591–597. doi: 10.1152/ajpheart.1987.253.3.H591. [DOI] [PubMed] [Google Scholar]
- Halliwill JR. Segregated signal averaging of sympathetic baroreflex responses in humans. J Appl Physiol. 2000;88:767–773. doi: 10.1152/jappl.2000.88.2.767. [DOI] [PubMed] [Google Scholar]
- Halliwill JR, Minson CT. Effect of hypoxia on arterial baroreflex control of heart rate and muscle sympathetic nerve activity in humans. J Appl Physiol. 2002;93:857–864. doi: 10.1152/japplphysiol.01103.2001. [DOI] [PubMed] [Google Scholar]
- Heistad DD, Abboud FM, Mark AL, Schmid PG. Interaction of baroreceptor and chemoreceptor reflexes. Modulation of the chemoreceptor reflex by changes in baroreceptor activity. J Clin Invest. 1974;53:1226–1236. doi: 10.1172/JCI107669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heistad DD, Abboud FM, Mark AL, Schmid PG. Effect of baroreceptor activity on ventilatory response to chemoreceptor stimulation. J Appl Physiol. 1975;39:411–416. doi: 10.1152/jappl.1975.39.3.411. [DOI] [PubMed] [Google Scholar]
- Iriki M, Dorward P, Korner PI. Baroreflex ‘resetting’ by arterial hypoxia in the renal and cardiac sympathetic nerves of the rabbit. Pflugers Arch. 1977;370:1–7. doi: 10.1007/BF00707938. [DOI] [PubMed] [Google Scholar]
- Korner PI. Central nervous control of autonomic cardiovascular function. In: Berne RM, Sperelakis N, Geiger SR, editors. Handbook of Physiology, section 2, The Cardiovascular System, The Heart. I. Bethesda, MD, USA: American Physiological Society; 1979. pp. 691–739. [Google Scholar]
- Loewy AD. Central autonomic pathways. In: Loewy AD, Spyer KM, editors. Central Regulation of Autonomic Function. New York: Oxford University Press; 1990. pp. 88–103. [Google Scholar]
- Malpas SC, Bendle RD, Head GA, Ricketts JH. Frequency and amplitude of sympathetic discharges by baroreflexes during hypoxia in conscious rabbits. Am J Physiol. 1996;271:H2563–2574. doi: 10.1152/ajpheart.1996.271.6.H2563. [DOI] [PubMed] [Google Scholar]
- Mancia G. Influence of carotid baroreceptors on vascular responses to carotid chemoreceptor stimulation in the dog. Circ Res. 1975;36:270–276. doi: 10.1161/01.res.36.2.270. [DOI] [PubMed] [Google Scholar]
- Minson CT, Halliwill JR, Young TM, Joyner MJ. Influence of the menstrual cycle on sympathetic activity, baroreflex sensitivity, and vascular transduction in young women. Circulation. 2000a;101:862–868. doi: 10.1161/01.cir.101.8.862. [DOI] [PubMed] [Google Scholar]
- Minson CT, Halliwill JR, Young TM, Joyner MJ. Sympathetic activity and baroreflex sensitivity in young women taking oral contraceptives. Circulation. 2000b;102:1473–1476. doi: 10.1161/01.cir.102.13.1473. [DOI] [PubMed] [Google Scholar]
- Miura M, Reis DJ. The role of the solitary and paramedian reticular nuclei in mediating cardiovascular reflex responses from carotid baro- and chemoreceptors. J Physiol. 1972;223:525–548. doi: 10.1113/jphysiol.1972.sp009861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier CL, Shepherd JT. Effect of hypoxia on vascular responses to the carotid baroreflex. Am J Physiol. 1975;228:331–336. doi: 10.1152/ajplegacy.1975.228.1.331. [DOI] [PubMed] [Google Scholar]
- Raven PB, Potts JT, Shi X. Baroreflex regulation of blood pressure during dynamic exercise. Exerc Sports Sci Rev. 1997;25:365–389. [PubMed] [Google Scholar]
- Rowell LB. Human Cardiovascular Control. New York: Oxford University Press; 1993. [Google Scholar]
- Rowell LB, Johnson DG, Chase PB, Comess KA, Seals DR. Hypoxemia raises muscle sympathetic activity but not norepinephrine in resting humans. J Appl Physiol. 1989;66:1736–1743. doi: 10.1152/jappl.1989.66.4.1736. [DOI] [PubMed] [Google Scholar]
- Rudas L, Crossman AA, Morillo CA, Halliwill JR, Tahvanainen KUO, Kuusela TA, Eckberg DL. Human sympathetic and vagal baroreflex responses to sequential nitroprusside and phenylephrine. Am J Physiol. 1999;276:H1691–1698. doi: 10.1152/ajpheart.1999.276.5.h1691. [DOI] [PubMed] [Google Scholar]
- Rutherford JD, Vatner SF. Integrated carotid chemoreceptor and pulmonary inflation reflex control of peripheral vasoreactivity in conscious dogs. Circ Res. 1978;43:200–208. doi: 10.1161/01.res.43.2.200. [DOI] [PubMed] [Google Scholar]
- Sagawa S, Torii R, Nagaya K, Wada F, Endo Y, Shiraki K. Carotid baroreflex control of heart rate during acute exposure to simulated altitudes of 3800 m and 4300 m. Am J Physiol. 1997;273:R1219–1223. doi: 10.1152/ajpregu.1997.273.4.R1219. [DOI] [PubMed] [Google Scholar]
- Saito M, Mano T, Iwase S, Koga K, Abe H, Yamazaki Y. Responses in muscle sympathetic activity to acute hypoxia in humans. J Appl Physiol. 1988;65:1548–1552. doi: 10.1152/jappl.1988.65.4.1548. [DOI] [PubMed] [Google Scholar]
- Seals DR, Suwarno NO, Dempsey JA. Influence of lung volume on sympathetic nerve discharge in normal humans. Circ Res. 1990;67:130–141. doi: 10.1161/01.res.67.1.130. [DOI] [PubMed] [Google Scholar]
- Seals DR, Suwarno NO, Joyner MJ, Iber C, Copeland JG, Dempsey JA. Respiratory modulation of muscle sympathetic nerve activity in intact and lung denervated humans. Circ Res. 1993;72:440–454. doi: 10.1161/01.res.72.2.440. [DOI] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Abboud FM. Interaction of baroreceptor and chemoreceptor reflex control of sympathetic nerve activity in normal humans. J Clin Invest. 1991;87:1953–1957. doi: 10.1172/JCI115221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Zavala DC, Abboud FM. Influence of ventilation and hypocapnia on sympathetic nerve responses to hypoxia in normal humans. J Appl Physiol. 1989;67:2095–2100. doi: 10.1152/jappl.1989.67.5.2095. [DOI] [PubMed] [Google Scholar]
- St Croix CM, Morgan BJ, Wetter TJ, Dempsey JA. Fatiguing inspiratory muscle work causes reflex sympathetic activation in humans. J Physiol. 2000;529:493–504. doi: 10.1111/j.1469-7793.2000.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundlöf G, Wallin BG. The variability of muscle nerve sympathetic activity in resting recumbent man. J Physiol. 1977;272:383–397. doi: 10.1113/jphysiol.1977.sp012050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor RG, Secher NH, Lyson T, Mitchell JH. Central command increases sympathetic nerve activity during intense intermittent isometric exercise in humans. Circ Res. 1995;76:127–131. doi: 10.1161/01.res.76.1.127. [DOI] [PubMed] [Google Scholar]
- Weisbrod CJ, Minson CT, Joyner MJ, Halliwill JR. Effects of regional phentolamine on hypoxic vasodilatation in healthy humans. J Physiol. 2001;537:613–621. doi: 10.1111/j.1469-7793.2001.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]