

ScpB from M. tuberculosis was crystallized using the sitting-drop vapour-diffusion method in the presence of 2 M NaCl and 10% PEG 6000 at 295 K. X-ray diffraction data were collected to a maximum resolution of 2.3 Å at a synchrotron beamline.
Keywords: ScpB, segregation and condensation proteins
Abstract
Structural maintenance of chromosome (SMC) proteins play diverse roles in cellular DNA reassembly by directly interacting with DNA. They require non-SMC proteins for their proper function; these include the conserved segregation and condensation proteins (Scps) in prokaryotes. ScpB from Mycobacterium tuberculosis was crystallized using the sitting-drop vapour-diffusion method in the presence of 2 M NaCl and 10% PEG 6000 at 295 K. X-ray diffraction data were collected to a maximum resolution of 2.3 Å at a synchrotron beamline. The crystal belongs to the hexagonal space group R32, with unit-cell parameters a = b = 136.69, c = 78.55 Å, γ = 120°. With one molecule per asymmetric unit, the crystal volume per unit protein weight (V M) is 2.95 Å3 Da−1. The structure was solved by the single anomalous dispersion method and structure refinement is in progress.
1. Introduction
Structural maintenance of chromosome (SMC) proteins are found in prokaryotes and eukaryotes and participate in various DNA processes such as sister chromatid cohesion, chromosome condensation and segregation, recombination and DNA repair (Hirano, 1999 ▶; Strunnikov & Jessberger, 1999 ▶). The N- and C-terminal domains of SMC proteins assemble to form a globular head domain, the structure of which is similar to the ATP cassettes of ABC transporters and which shows a weak ATPase activity (Hopfner et al., 2000 ▶; Lowe et al., 2001 ▶). The dynamic circular structure can be opened and closed through the intermolecular interaction of the head domain upon ATP binding (Lowe et al., 2001 ▶) or the hinge domain itself upon DNA binding (Gruber et al., 2006 ▶; Hirano & Hirano, 2006 ▶). SMC proteins form intermolecular multi-subunit protein complexes with non-SMC proteins for their diverse functions. In eukaryotes, the non-SMC protein Scc1 binds to the SMC head domain using its C-terminal winged-helix domain (cWHD) and forms a tertiary complex with an SMC protein (Hirano, 2006 ▶; Haering et al., 2004 ▶). Escherichia coli also has a functional analogue of an SMC, MukB, which requires two non-SMC proteins, MukE and MukF (Niki et al., 1991 ▶; Van Den Ent et al., 1999 ▶). The SMC protein in Bacillus subtilis interacts with two non-SMC proteins, ScpA and ScpB. The deletion of non-SMC proteins induces the same phenotypes as caused by deletion of SMC protein-coding genes. ScpA, which interacts directly with SMC proteins, mediates the ternary complex formation between SMC proteins and ScpB (Soppa et al., 2002 ▶; Lindow et al., 2002 ▶; Volkov et al., 2003 ▶). Recent structural studies of ScpB from Chlorobium tepidum (ScpB-Ct) revealed that the monomer is composed of two WHDs and a dimeric structure was formed through the interaction between the two sequence-conserved cWHDs (Kim et al., 2006 ▶). However, the biochemical function and biological implications of ScpB proteins could not be deduced from the reported structure alone.
A conserved protein in Mycobacterium tuberculosis, Rv1710, encodes a protein of 231 amino acids which appeared to be an ScpB protein upon amino-acid sequence analysis (36% amino-acid sequence identity with ScpB from C. tepidum) and is hereafter referred to as ScpB-Mt. A protein-family search using PFAM (http://www.sanger.ac.uk/Software/Pfam) also indicates that it is a member of the ScpB proteins, with the common structural motif of a DUF387 domain at the N-terminus (residues 30–188). As a step towards elucidating the detailed biochemical mechanisms, including DNA-binding properties and complex formation with ScpA, of ScpB from a pathogenic bacterium, we cloned its gene from the chromosome of M. tuberculosis and purified the protein. Crystals of ScpB-Mt protein were obtained by the sitting-drop method. The crystals diffracted well and data were collected to a resolution of 2.3 Å. Here, we describe the cloning, expression, purification, crystallization and X-ray crystallographic analysis of the ScpB-Mt protein.
2. Expression and purification of the recombinant ScpB-Mt protein
After detailed analysis of an amino-acid sequence comparison and secondary-structure prediction, the N-terminal 15 residues of ScpB-Mt were suspected to be a flexible region and the Rv1710 gene coding for the ScpB (16–231) was amplified from the chromosomal DNA of M. tuberculosis H37Rv strain by polymerase chain reaction (PCR). The PCR product was then subcloned into pPROEX HTa (Invitrogen) with His6 at the N-terminus and a recombinant TEV protease (rTEV) cleavage site. The resulting expression vector pPROEX HTa:scpB was transformed into E. coli B834 strain and the cells were grown in LB medium containing ampicillin at 310 K. After induction with 1.0 mM IPTG for a further 20 h at 295 K, the culture was harvested by centrifugation at 5000g at 277 K. The cell pellet was resuspended in ice-cold buffer A (50 mM Tris–HCl pH 8.0, 5 mM β-mercaptoethanol) and disrupted by ultrasonication. The cell debris was removed by centrifugation at 11 000g for 1 h and the lysate was bound to Ni–NTA agarose (Qiagen). After washing with buffer A containing 10 mM imidazole, the bound proteins were eluted with 300 mM imidazole in buffer A. The His6 tag was released from the ScpB by incubation with rTEV protease (Gibco). A trace amount of contamination was removed by applying HiTrap Q ion-exchange and Superdex75 size-exclusion chromatography. The purified protein with three-residue cloning artifact (Gly-His-Met) at the N-terminus showed ∼95% purity on SDS–PAGE (Fig. 1 ▶ a) and was concentrated to 20 mg ml−1 in 50 mM Tris–HCl pH 8.0 and stored at 193 K for crystallization trials. The SeMet-substituted protein was expressed in a minimal medium supplemented with SeMet and purified under the same conditions as the native protein.
Figure 1.

Purified protein and crystals of ScpB-Mt. (a) SDS–PAGE of purified ScpB (Rv1710) from M. tuberculosis. Molecular-weight markers are labelled in kDa on the left of the gel and purified ScpB-Mt (21–231) with molecular weight 27 kDa is indicated on the right of the gel. (b) Rhombohedral crystal of ScpB (Rv1710) from M. tuberculosis. Crystals of the best quality appeared in 5 d and reached maximal dimensions of approximately 0.2 × 0.2 × 0.2 mm.
3. Crystallization
Crystallization of the purified protein was initially performed with commercially available sparse-matrix screens from Hampton Research and Emerald BioSystems using the hanging-drop vapour-diffusion technique at 295 K. Each experiment consisted of mixing 2 µl protein solution (15 mg ml−1 in 20 mM Tris–HCl pH 7.0 and 5 mM β-mercaptoethanol) with 2 µl reservoir solution and then equilibrating it against 0.5 ml reservoir solution. ScpB-Mt crystals were observed in several crystallization screening conditions. After several steps that improved the crystallization process using the sitting-drop vapour-diffusion method, crystals of the best quality appeared in 5 d and reached maximal dimensions of approximately 0.2 × 0.2 × 0.2 mm using a reservoir solution containing 2 M NaCl and 10% PEG 6000 (Fig. 1 ▶ b). The volumes of the sitting drop and the reservoir solution were same as those in the hanging-drop vapour-diffusion technique used for the initial crystallization screening. SeMet crystals of ScpB-Mt were obtained from the same crystallization condition as used for the native protein crystals.
4. X-ray analysis
The crystals were transferred to cryoprotectant solution containing 2 M NaCl, 10% PEG 6000 and 30% glycerol, fished out with a loop larger than the crystals and flash-frozen by immersion in liquid nitrogen at 100 K. Data were collected to a resolution of 2.3 Å at the 6C1 beamline (MXII) at Pohang Accelerator Laboratory (PAL, Pohang, Korea) using a Quantum 210 CCD detector (ADSC, USA; Fig. 2 ▶). The data were indexed, integrated and scaled using the HKL-2000 suite (Otwinowski & Minor, 1997 ▶). The crystals belonged to space group R32, with unit-cell parameters a = b = 136.69, c = 78.55 Å, γ = 120°. Assuming the presence of one molecule of ScpB-Mt per asymmetric unit, the crystal volume per unit of protein weight was 2.95 Å3 Da−1 (Matthews, 1968 ▶), which corresponds to a solvent content of approximately 58.33%.
Figure 2.

Diffraction pattern of ScpB crystal from M. tuberculosis. The crystal diffracts to a maximum resolution of 2.3 Å.
SAD data were collected from an SeMet crystal at the 6C1 beamline (MXII) at PAL at a wavelength of 0.97950 Å. The data statistics are summarized in Table 1 ▶. Three of the four Se atoms in the asymmetric unit were identified using the program SOLVE (Terwilliger & Berendzen, 1999 ▶) at 2.9 Å resolution. The electron density was improved by density modification using RESOLVE (Terwilliger, 2000 ▶), resulting in 85% of the residues automatically being built. Crystallographic model building and refinement of the structure to 2.3 Å resolution are in progress.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| Native | Se peak | |
|---|---|---|
| Beamline | 6C1 (MXII), PAL | 6C1 (MXII), PAL |
| Wavelength (Å) | 0.97950 | 1.23985 |
| Temperature (K) | 100 | 100 |
| Oscillation (°) | 1.0 | 1.0 |
| Total rotation range (°) | 150 | 360 |
| Space group | R32 | R32 |
| Unit-cell parameters (Å, °) | a = b = 136.69, c = 78.55, γ = 120 | a = b = 136.93, c = 78.39, γ = 120 |
| Resolution limits (Å) | 50.0–2.3 (2.38–2.3) | 50.0–2.5 (2.59–2.5) |
| Total reflections | 263491 | 171875 |
| Unique reflections | 12625 | 9902 |
| Completeness (%) | 98.4 (97.0) | 99.7 (98.8) |
| Rmerge† (%) | 5.5 (21.4) | 4.6 (23.1) |
| I/σ(I) | 33.9 (4.7) | 52.9 (4.5) |
R
merge = 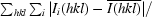
 , where I
i(hkl) and
, where I
i(hkl) and 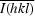 are the observed individual and mean intensities of a reflection, respectively.
are the observed individual and mean intensities of a reflection, respectively. 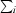 is the sum over the individual measurements of a reflection and
is the sum over the individual measurements of a reflection and  is the sum over all reflections.
is the sum over all reflections.
Acknowledgments
This work was supported by the 21C Frontier Microbial Genomics and Application Center Program, Ministry of Science and Technology, Republic of Korea.
References
- Gruber, S., Arumugam, P., Katou, Y., Kuglitsch, D., Helmhart, W., Shirahige, K. & Nasmyth, K. (2006). Cell, 127, 523–537. [DOI] [PubMed]
- Haering, C., Schoffnegger, D., Nishino, T., Helmhart, W., Nasmyth, K. & Lowe, J. (2004). Mol. Cell, 15, 951–964. [DOI] [PubMed]
- Hirano, M. & Hirano, T. (2006). Mol. Cell, 21, 175–186. [DOI] [PubMed]
- Hirano, T. (1999). Genes Dev.13, 11–19. [DOI] [PubMed]
- Hirano, T. (2006). Nature Rev. Mol. Cell Biol.7, 311–322. [DOI] [PubMed]
- Hopfner, K. P., Karcher, A., Shin, D. S., Craig, L., Arthur, L. M., Carney, J. P. & Tainer, J. A. (2000). Cell, 101, 789–800. [DOI] [PubMed]
- Kim, J. S., Shin, D. H., Pufan, R., Huang, C., Yokota, H., Kim, R. & Kim, S.-H. (2006). Proteins, 62, 322–328. [DOI] [PubMed]
- Lindow, J. C., Kuwano, M., Moriya, S. & Grossman, A. D. (2002). Mol. Microbiol.46, 997–1009. [DOI] [PubMed]
- Lowe, J., Cordell, S. C. & Van Den Ent, F. (2001). J. Mol. Biol.306, 25–35. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Niki, H., Jaffe, A., Imamura, R., Ogura, T. & Hiraga, S. (1991). EMBO J.10, 183–193. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Soppa, J., Kobayashi, K., Noirot-Gros, M. F., Oesterhelt, D., Ehrlich, S. D., Dervyn, E., Ogasawara, N. & Moriya, S. (2002). Mol. Microbiol.45, 59–71. [DOI] [PubMed]
- Strunnikov, A. V. & Jessberger, R. (1999). Eur. J. Biochem.263, 6–13. [DOI] [PubMed]
- Terwilliger, T. C. (2000). Acta Cryst. D56, 965–972. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed]
- Van Den Ent, F., Lockhart, A., Kendrick-Jones, J. & Lowe, J. (1999). Structure Fold. Des.7, 1181–1187. [DOI] [PubMed]
- Volkov, A., Mascarenhas, J., Andrei-Selmer, C., Ulrich, H. D. & Graumann, P. L. (2003). Mol. Cell. Biol.23, 5638–5650. [DOI] [PMC free article] [PubMed]