

Abstract
Hypothalamic neuropeptide Y (NPY) is thought to be important in the regulation of feeding and also in the release of Adrenocorticotrophic hormone (ACTH). Intracerebroventricular administration of NPY to male rats significantly increased plasma ACTH 10 min after injection and stimulated 2-h food intake. A series of analogues of NPY that have a greatly reduced affinity for the Y1 [human pancreatic polypeptide (human PP), NPY(3–36)], the Y2 ([Pro34]NPY, human PP), the Y3 (peptide YY), and the Y6 (human PP) receptor, all markedly stimulated ACTH release. Rat PP, which binds with high affinity to the Y4 receptor, was unable to stimulate ACTH release. A novel analogue fragment [Pro34]NPY(13–36) was synthesized as a ligand with low Y1 and Y2 receptor affinity. Interestingly, neither [Pro34]NPY(13–36) nor the selective Y5 receptor agonist [d-Trp32]NPY stimulated food intake, whereas both significantly increased plasma ACTH. Thus the hypothalamic NPY receptor mediating increases in plasma ACTH has a fragment activation profile unlike the Y1–Y4 or Y6 receptors and appears distinct from the NPY receptor controlling food intake.
Keywords: intracerebroventricular, hypothalamus, hypathalamo-pituitary-adrenal axis
Neuropeptide Y (NPY) is a 36-amino acid peptide of the pancreatic polypeptide family, with homology to pancreatic polypeptide (PP, ≈50%) and peptide YY (PYY, ≈70%) (1). NPY is one of the most abundant neuropeptides in the central and peripheral nervous system of many mammals, including humans (2). NPY immunoreactivity is present throughout the brain, particularly in the hypothalamus (2). Within the hypothalamus NPY is implicated in the activation of the hypothalamic pituitary adrenal (HPA) axis (3), the regulation of growth, and sexual function (4), and is the most potent stimulant of feeding yet reported (5, 6).
Several NPY receptor subtypes have been identified and characterized by their ability to bind NPY, PYY, and PP fragments and analogues. The NPY Y1 receptor binds with high affinity full-length NPY or analogues, such as [Pro34]NPY or [Leu31Pro34]NPY (7). The Y1 receptor has a reduced affinity for C-terminal fragments such as NPY(2–36), NPY(3–36), and NPY(13–36) (8). This receptor has been cloned from the human (9) and rat (10) central nervous system and is expressed in the human neuroblastoma cell line, SK-N-MC (9). The Y2 receptor has a high affinity for C-terminal fragments such as NPY(13–36) and NPY(3–36) (8) but a low affinity for [Pro34]NPY, and has been cloned from human hippocampus (11) and from the human neuroblastoma cell line, SMS-KAN (12). Human neuroblastoma cell lines are considered the de facto standards for the characterization of ligands for Y1 and Y2 receptors. PYY binds with high affinity to both the Y1 and Y2 receptor subtypes, whereas human and rat PP bind with very low affinities (9, 11, 13). A group of receptors with disparate pharmacological profiles have been labeled Y3 and are characterized by a low affinity for PYY in comparison to NPY (14–16). Hypotension, bradycardia, and inhibition of glutamate effects in response to unilateral injection of NPY into the nucleus tractus solitarius of the rat are associated with Y3 receptor activation in the central nervous system (17, 18). The Y4 receptor is characterized by a very high affinity for PP compared with NPY and NPY(13–36) (13, 19). The Y4 receptor has been cloned and its mRNA is expressed in the human brain, coronary artery, and ileum (19). An NPY receptor designated Y6 has been cloned from mouse (20), human, chimpanzee, gorilla, and rabbit (21) but is inactivated in primates by a frame-shift mutation. The Y6 receptor has a high affinity for NPY and PYY and a low affinity for human PP (20, 21).
NPY has been proposed to activate a hypothalamic “feeding” receptor (YFEEDING) distinct from previously cloned NPY receptors (22, 23). This receptor was initially suggested to be Y1-like because [Pro34]NPY mediates a potent feeding response (23, 24). However, NPY(2–36), which has been reported to be less potent than intact NPY for most functions of Y1 receptors (25, 26), was at least as potent a stimulant of feeding as NPY (22, 23). A further receptor, termed Y5, has recently been cloned from rat hypothalamus (13, 27). Y5 receptor mRNA is found primarily in the central nervous system, including the paraventricular nucleus of the hypothalamus (PVN) and the lateral hypothalamus, areas of the brain that have been implicated in the control of appetite (28). The profile of peptide analogues and fragments which inhibited adenylyl cyclase through the Y5 receptor expressed in HEK-293 cells corresponded well to the NPY peptide profile that stimulated food intake in rats. This result led the authors to conclude that this Y5 receptor was the postulated hypothalamic YFEEDING receptor (13).
Immunohistochemical studies in the rat have indicated that NPY immunoreactive axon terminals have synaptic connections with parvocellular corticotrophin-releasing hormone (CRH) neurons of the PVN (29, 30). NPY evokes the release of CRH from hypothalamic explants incubated in vitro (31). Intracerebroventricular (ICV) administration of NPY produces a marked release of pituitary adrenocorticotrophic hormone (ACTH), and this effect is significantly attenuated by prior administration of the CRH antagonist, α-helical CRH(9–41) (32). Thus CRH is the likely mediator of the action of NPY on ACTH release. ICV immunoneutralization techniques using specific NPY antibodies inhibited the ACTH and cortisol release in response to hypoglycaemia, suggesting the involvement of endogenous NPY in this effect (33).
The NPY receptor subtype(s) mediating increases in plasma ACTH (YACTH) have yet to be determined but it has been suggested previously that the entire NPY molecule is required for HPA axis activation (34). We administered by ICV injection, NPY, PYY, human PP, rat PP, NPY(3–36), NPY(13–36), NPY(1–24), [Pro34]NPY, [d-Trp32]NPY, desamido NPY, [Pro34]NPY(3–36) and the novel NPY fragment analogue, [Pro34]NPY(13–36) and measured plasma ACTH levels 10 min later. [Pro34]NPY(13–36) was synthesized as a ligand with potential low Y1 receptor affinity due to the N-terminal deletion and low Y2 affinity due to the Pro34 substitution. The effect of [Pro34]NPY(13–36) on 2-h food intake and [d-Trp32]NPY on 1-h food intake were also investigated. One-hour food intake was investigated for the [d-Trp32]NPY experiment to be compatible with the timing used in previous studies (35).
MATERIALS AND METHODS
Materials.
All tissue culture materials were supplied by GIBCO/BRL (Life Technologies, Paisley, U.K.). Unless specified other reagents were supplied by Merck or Sigma. Peptides were synthesized using fmoc chemistry on an Advanced ChemTech 396 MPS peptide synthesizer (Advanced ChemTech). The products each comprised one major peak which was purified to homogeneity by reversed phase HPLC on a C8 column (Phenomenex, Macclesfield, U.K.) using a gradient of acetonitrile (20–50%) in 0.1% aqueous trifluoroacetic acid. The identity of the peptides was confirmed by Electrospray mass spectrometry. All peptides used in these studies were based on the porcine sequence unless otherwise stated.
Cell Culture.
SK-N-MC and SMS-KAN cells were kindly donated by S. Legon (Royal Postgraduate Medical School, London) and T. Schwartz (Rigshospitalet, Copenhagen), respectively. Both cell lines were routinely maintained at 37°C in 50/50 DMEM/Ham’s F-12, supplemented with 10% heat inactivated fetal bovine serum, 2 mM glutamate, 1× nonessential amino acids, 100 μg/ml streptomycin, and 100 units/ml penicillin at 37°C in a 5% CO2 atmosphere. HEK-293 cells were cultured in DMEM containing 4.5 mg/ml glucose and 1 mM sodium pyruvate supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 mg/ml streptomycin in a 5% CO2 atmosphere. Medium was changed every 48 h, and cells were passaged when they reached 70% confluence (≈7 days).
Y5 Stable Expression Studies.
No cell lines have been identified which naturally express Y5 receptors. HEK-293 cells were therefore transfected with rat Y5 cDNA (13) as a model for the Y5 receptor. Transfections were carried out using calcium phosphate coprecipitation (36) by the method of Sambrook et al. (37) with the cells plated at a density of 1 × 104/cm2 in 60-mm dishes. The cells were incubated at 37°C in 5% CO2 with the precipitate for 18 h and allowed to recover for a further 24 h. Cells stably transfected were selected by the addition of 600 mg/ml G418S (Sigma) to the medium. For further studies, groups of 100 clones were subcultured and assayed for NPY binding. One of these groups was used for all subsequent work.
Peptide Iodination.
Porcine PYY was iodinated using the iodogen method (38) as described (39). Iodination products separated by reversed phase C18 HPLC (Waters Novapak column, Millipore), developed with a 15–45% acetonitrile/water/0.05% trifluoroacetic acid gradient. Fractions (1.5 ml) were collected, and radioactive peaks were assayed for receptor binding activity. Active fractions were aliquoted, freeze-dried, and stored at −20°C. The specific activity of the radioligand was 27 Bq/fmol.
Membrane Preparation.
Neuroblastoma and cell membranes were prepared by osmotic lysis and differential centrifugation as described (39). For rat hippocampal and cortical membranes, male Wistar rats (250–300 g) (Interfauna, Huntingdon, U.K.) were killed by CO2 asphyxiation, and brain regions were rapidly dissected, frozen in liquid nitrogen, and stored at −80°C until used. Tissue membranes were prepared by homogenization and differential centrifugation as described (39).
Receptor Binding.
Membrane binding. Membranes (100 μg protein) were incubated with 40 pM (1,000 Bq) 125I-labeled PYY in the presence or absence of unlabeled peptides as indicated as described (22, 39). Total specific binding was defined as the difference in counts between assays in the presence (nonspecific) and absence (total) of 200 nM NPY. Analysis of equilibrium competition data was performed using receptor fit programs (Lundon Software, Cleveland, OH) to give Ki values for each ligand.
Intact cell binding.
HEK-293 cells stably expressing the Y5 receptor were grown to confluence in 24-well plates. Medium was removed, and the cells were washed with 1 ml of the assay buffer [20 mM Hepes, pH 7.4/5 mM CaCl2/1 mM MgCl2/1% (wt/vol) BSA]. This was replaced with 1 ml assay buffer containing 40 pM (1,000 Bq) 125I-labeled PYY and unlabeled peptide as required. After a 90-min incubation at 30°C, the assay buffer was removed and the cells washed with a further 1 ml of ice cold assay buffer. Cells were then solubilised in 1 ml 0.1 M NaOH, and counted in a γ-counter.
ACTH Studies.
Adult male Wistar rats (250–300 g) were maintained in individual cages under controlled temperature (21–23°C) and light (10 h light, 14 h dark), with ad libitum access to food and water. Rats were cannulated ICV as described (22, 40). All animals were allowed 7 days recovery after surgery, and then an ICV injection (10 μl) of angiotensin II (150 ng) was administered. Animals not demonstrating a prompt and sustained drinking response were excluded (<10%). Animals were acclimatized to the procedure of injection and guillotine apparatus, with sham decapitations were performed daily for 3 days before the study, to minimize stress and its metabolic consequences on the day of the study.
Circadian rhythms for ACTH and corticosterone have been previously demonstrated in the male rat with nadir values during the light phase and peaks around the light/dark interface (22.00) (41). All studies were carried out at the beginning of the light phase (08.00–11.00). Peptides were administered via a stainless steel injector, projecting 0.5 mm below the guide cannulae. All peptides were dissolved in 0.9% saline and injected in a 10 μl volume. In pilot studies (data not shown), plasma ACTH was significantly increased 10 min following ICV injection of NPY (7.2 nmol). This time point was chosen for all subsequent ACTH studies. Ten minutes after injection, rats were decapitated and trunk blood collected in plastic lithium heparin tubes containing 0.6 mg aprotinin. Plasma was separated by centrifugation, frozen on dry ice, and stored at −70°C until assay. Plasma ACTH was determined by immunoradiometric assay (Biogenesis, Poole, Dorset, U.K.) (42).
Groups of rats (n = 7–8) were injected with NPY, [Pro34]NPY (six doses, 0.024–7.2 nmol), NPY(3–36), NPY(13–36) (five doses, 0.024–7.2 nmol), PYY, human PP, rat PP, desamido-NPY, [Pro34]NPY(13–36), [Pro34]NPY(3–36), [d-Trp32]NPY (three dose, 0.072–7.2 nmol), or NPY(1–24) (7.2 nmol only), or a control and then decapitated 10 min later. Only three doses of PYY, human PP, rat PP, desamido NPY, [Pro34]NPY(13–36), [Pro34]NPY(3–36), and [d-Trp32]NPY were used due to the constraint of total animal numbers. The doses administered for these fragments and analogues were determined from the initial NPY dose-response experiment (0.024–7.2 nmol). The doses chosen (0.072, 0.72 and 7.2 nmol) spanned the full dose response of the NPY effect (Fig. 1). None of the doses of peptides administered caused any abnormal behavior in the animals. Two control groups were used, saline and a random sequence of amino acids (peptide control) synthesized using the same methodology as the NPY fragments. Each NPY fragment or analogue tested had an individual saline control group. Other nonrelated peptides, glucagon-like peptide-1 (GLP-1 7–36 NH2) (7.2 nmol) and adrenomedullin (0.3 nmol), again synthesized “in house” were administered ICV and plasma ACTH measured at 10 min after injection.
Figure 1.
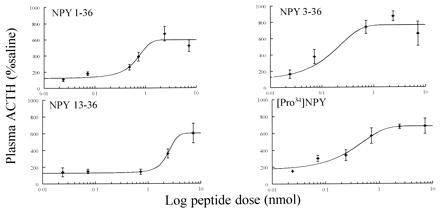
Dose-response (0.024–7.2 nmol) effects of NPY, [Pro34]NPY, NPY(3–36), and NPY(13–36) on plasma ACTH following ICV administration. Rats were injected ICV with NPY (or selected compounds) at the start of the light phase. Rats (n = 7–8 per group) were decapitated 10 min postinjection and plasma ACTH was measured. All results are expressed as a mean ± SEM percentage of saline control. The saline response (100%) was 50 ± 5 pg/ml.
[Pro34]NPY(13–36) Feeding Study.
[Pro34]NPY(13–36) was administered by ICV injection at four doses (0.24, 2.4, 24, and 50 nmol), and 2.4 nmol NPY was given as a positive control (22, 43). Rats (n = 7–8 per group) were injected at the beginning of the light phase. Following injection, rats were returned to cages containing preweighed chow and 2-h food intake for each rat was measured.
[d-Trp32]NPY Feeding Study.
To investigate the effects of [d-Trp32]NPY on food intake we administered two ICV injections 15 min apart. Rats were injected at the beginning of the light phase. Four groups of animals (n = 17 per group) received the following injections: saline plus saline, saline plus NPY, [d-Trp32]NPY plus saline, and [d-Trp32]NPY plus NPY. NPY was administered at a dose of 2.4 nmol and [d-Trp32]NPY at a dose of 12 nmol. Following the second ICV injection rats were returned to cages containing preweighed chow and observed for behavioral changes and 1-h food intake for each rat was measured.
Statistical Analysis.
All feeding data are presented as mean food intake ± SEM. In ACTH studies each NPY fragment or analogue tested had its own saline control group. To normalize the plasma ACTH data the results are presented as a percentage of this saline (saline = 100%) control. ANOVA (post-hoc comparisons were made using Tukey’s test) was performed for each fragment or analogue (three doses) using the individual saline control. Statistical significance was taken as P < 0.05. The results from this statistical analysis are displayed in Table 2.
RESULTS
Receptor Binding.
Binding assays with 125I-labeled PYY routinely gave >80% specific binding to all membranes (SK-N-MC, 94 ± 1%, n = 10; cortex, 86 ± 1%, n = 10; SMS-KAN, 86 ± 2%, n = 10; and hippocampus, 90 ± 2%, n = 10). Binding was saturable, temperature, and time dependent (results not shown). Competition curves constructed using SK-N-MC and SMS-KAN human cell membranes were mirrored closely by those using rat cortex (predominately Y1) and hippocampus (predominately Y2). The human and rodent Y1 and Y2 binding data were highly reproducible; for example, for NPY Y1, the Ki for SK-N-MC cells was 0.58 ± 0.11 nM and for cortex membranes the Ki was 0.15 ± 0.04 nM, in close agreement to previously published values (22). Results obtained (Table 1) confirmed the high affinity of NPY and PYY for both Y1 and Y2 receptors, and the specificity of [Pro34]NPY for the Y1 receptor and of NPY(3–36) and NPY(13–36) for the Y2 receptor (Table 1). The Y1 agonist [Pro34]NPY showed the greatest degree of specificity, with a >1,000-fold difference between its affinity for the Y1 and Y2 receptors. NPY(3–36) and NPY(13–36) showed a lower degree of specificity, with, respectively, 12-fold and 4-fold higher affinities for the Y2 receptor. [Pro34]NPY(13–36) (Table 1) and [Pro34]NPY(3–36) [as previously described (22)] showed similar binding to the corresponding unsubstituted C-terminal fragments in SK-N-MC cells, but were unable to compete for binding to SMS-KAN cell membranes. Human and rat PP, NPY(1–24), [d-Trp32]NPY and desamido NPY were all unable to compete for 125I-labeled PYY binding to either rat or human Y1 or Y2 receptors.
In Y5-expressing HEK-293, cells ligands bound with affinities in the rank order NPY > [Pro34]NPY > NPY(3–36) = hPP = [Pro34]NPY(13–36) > [d-Trp32]NPY = NPY(13–36) > rPP = desamido NPY (Table 1). Binding to Y5-expressing HEK-293 cells gave 55 ± 2.5% specific binding. Competition curves showed a similar ligand profile to that reported elsewhere for the Y5 receptor (27).
ACTH Studies.
Six-point dose-response curves were constructed for [Pro34]NPY and NPY and five-point dose-response curves were constructed for NPY(13–36) and NPY(3–36) (Fig. 1).
ANOVA and post hoc Tukey’s comparison showed that [Pro34]NPY and NPY(3–36) (P < 0.01) caused a significant increase in plasma ACTH at the 0.072 nmol dose compared with the individual saline control (Table 2). NPY, PYY, human PP, [Pro34]NPY, [d-Trp32]NPY, the novel NPY fragments [Pro34]NPY(13–36) and [Pro34]NPY(3–36) (P < 0.01) and NPY(3–36)(P < 0.001), caused a significant rise in plasma ACTH at the 0.72 nmol dose compared with the individual saline control (Table 2). NPY, PYY, NPY(3–36), human PP (P < 0.001), NPY(13–36), [Pro34]NPY, [Pro34]NPY(13–36) (P < 0.01), and [Pro34]NPY(3–36) (P < 0.05) caused a significant rise in plasma ACTH at the 7.2 nmol dose compared with the saline control (Table 2). Rat PP and desamido-NPY, although increasing plasma ACTH, did not cause a significant increase in plasma ACTH at any of the doses tested (Table 2). NPY(1–24) and the peptide control, a random sequence of amino acids synthesized using the same methodology as the NPY fragments, administered at the 7.2 nmol dose also did not cause significant increases in plasma ACTH [NPY(1–24), 95 ± 30 and peptide control, 125 ± 22].
The structurally unrelated NPY peptides, GLP-1 7–36 NH2 and adrenomedullin, synthesized using the same methodology as the NPY fragments, had no effect on plasma ACTH compared with the saline control 10 min after central injection [adrenomedullin (0.3 nmol), 42 ± 9 pg/ml vs. saline, 35 ± 16 pg/ml and GLP-1 7–36 NH2 (7.2 nmol), 51 ± 17 pg/ml vs. saline, 46 ± 7 pg/ml 10 min after ICV injection].
[Pro34]NPY(13–36) Feeding Study.
NPY (2.4 nmol) caused a significant increase in 2-h food intake compared with saline control (NPY, 5.6 ± 0.6 g; saline, 0.8 ± 0.2 g; P < 0.0001). Doses of the novel fragment [Pro34]NPY(13–36) up to 50 nmol had no effect on feeding {[Pro34]NPY(13–36), 0.7 ± 0.3 vs. saline, 0.8 ± 0.2 g, P = NS} whereas significantly stimulating plasma ACTH at the 0.72 and 7.2 nmol dose (Table 2).
[d-Trp32]NPY Feeding Study.
NPY (2.4 nmol) increased 1-h food intake in satiated animals [2.8 ± 0.4 g vs. control, 0.5 ± 0.1 g; (F(64,3) = 10.4, P < 0.0001] (Fig. 2). [d-Trp32]NPY (12 nmol) did not increase 1-h food intake compared with control. [d-Trp32]NPY caused a significant attenuation in 1-h food intake induced by NPY ([d-Trp32]NPY and NPY, 1.4 ± 0.4 g vs. NPY, 2.8 ± 0.4 g, P < 0.01).
Figure 2.
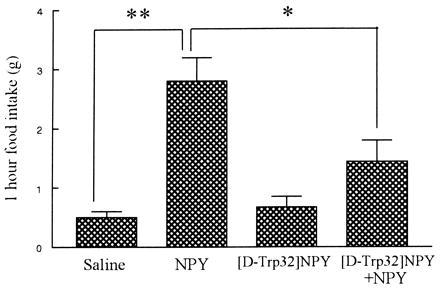
The effects of [d-Trp32]NPY on 1-h food intake following ICV administration. Rats (n = 17 per group) were injected at the beginning of the light phase (08.00–11.00). Two ICV injections were administered 15 min apart. Four groups of animals received the following injections: saline plus saline, saline plus NPY, [d-Trp32]NPY plus saline, and [d-Trp32]NPY plus NPY. NPY was administered at a dose of 2.4 nmol and [d-Trp32]NPY at a dose of 12 nmol. Following the second ICV injection rats were returned to cages containing preweighed chow, observed for behavioral changes and 1-h food intake in grams for each rat was measured. The results are expressed as mean 1-h food intake ± SEM of groups of 17 animals. ∗∗, P < 0.0001; ∗, P < 0.01.
DISCUSSION
The pharmacological profile of the NPY receptor controlling activation of the HPA axis (YACTH) is unlike that previously described for the Y1–Y4 or Y6 NPY receptors. NPY, PYY, human PP, NPY(3–36), NPY(13–36), [Pro34]NPY, [d-Trp32]NPY, and the novel NPY fragments [Pro34]NPY(13–36) and [Pro34]NPY(3–36) all stimulate ACTH release (Table 2). To assess if this might be a nonspecific effect of ICV peptide injection, NPY(1–24), GLP-1 7–36 NH2, adrenomedullin, and a peptide control (a random sequence of amino acids) were administered and found to have no significant effect on plasma ACTH. Central Y3 receptors, located in the nucleus tractus solitarius, are characterized by having a low affinity for PYY in comparison to NPY (15, 17). It is unlikely that the Y3 receptor mediates the activation of the HPA axis because PYY significantly stimulates plasma ACTH (Table 2) and the effects of NPY on plasma ACTH are thought to be localized to the PVN (29, 30). Rat PP, a more potent Y4 agonist than NPY (19, 44), was tested at all three doses and also failed to significantly stimulate plasma ACTH. The Y1 receptor is characterized by having low affinity for NPY(3–36) and PP (Table 1), the Y2 receptor has a low affinity for [Pro34]NPY and PP (Table 1), and the Y6 receptor has a low affinity for human PP (21). Thus, because NPY, PYY, NPY(3–36), [Pro34]NPY, and human PP, but not rat PP, all stimulate plasma ACTH following ICV administration, the NPY receptor mediating release of ACTH shows a different profile of activation to the Y1–Y4 or Y6 receptors. The compounds [Pro34]NPY(13–36), NPY(13–36), and [d-Trp32]NPY increase plasma ACTH (Table 2) but fail to stimulate feeding (22) (Fig. 2) following central administration. This suggests that YACTH and the postulated hypothalamic NPY feeding receptor (YFEEDING) may well be different NPY receptor subtypes.
The binding profile of the recently cloned Y5 receptor (13, 27), appears closer to the YACTH receptor than the YFEEDING receptor for the following reasons.
(i) Human PP has been reported to be a potent Y5 agonist in both functional studies and in receptor binding studies. Rat PP, however, was active at the Y5 receptor only at high concentrations, binding with an affinity 600 times lower than NPY (27) and activating the receptor at concentrations 260 times higher than NPY (13). Similarly, we found human PP bound to the Y5 receptor with a higher affinity than did rat PP (Table 1). Here we have found that human PP significantly increased plasma ACTH, whereas rat PP did not.
(ii) NPY(13–36) was shown to activate the Y5 receptor (13). However, several groups have reported that NPY(13–36) does not stimulate feeding (4, 22, 45) even when administered directly into the PVN (24). For example, NPY (13–36) when administered ICV at a dose of up to 50 nmol (70 times the minimal effective dose of NPY) was not found to stimulate feeding (22). We found that NPY(13–36) significantly increased plasma ACTH.
(iii) [d-Trp32]NPY is found to be a selective Y5 agonist in vitro (13, 27). Gerald et al. (13) also reported that, when administered ICV (2 nmol), [d-Trp32]NPY weakly stimulated feeding. Balasubramaniam et. al. (35), however, have previously demonstrated that [d-Trp32]NPY (2.4 nmol), administered directly into the PVN, did not significantly affect feeding and reduced the 1-h food intake induced by NPY (0.24 nmol, 1:10 ratio). In agreement with the findings of Balasubramaniam et. al. (35), we found that [d-Trp32]NPY administered ICV did not significantly affect feeding but did reduce the stimulation of 1-h food intake induced by NPY (1:5 ratio). Thus the efficacy of [d-Trp32]NPY at the YFEEDING receptor appears questionable. We find here that [d-Trp32]NPY does bind to the Y5 receptor (13) and significantly increases plasma ACTH. Although both our study and that of Balasubramaniam et al. (35) used porcine [d-Trp32]NPY, whereas Gerald et al. (13) used the human/rat peptide, we feel it unlikely that this is responsible for the different findings because the species analogues differ only by a single amino acid at position 17, and other orthologous fragments and analogues behave in an essentially identical manner. For example, Gerald et al. (13) demonstrated in an in vitro NPY Y5 receptor assay (inhibition of forskolin stimulated cAMP production) that human NPY and porcine NPY had an EC50 of 0.96 ± 0.19 and 0.66 ± 0.15, respectively. The stimulation of plasma ACTH by [d-Trp32]NPY is peculiar in that it appears to have a bell-shaped dose-response curve. This is a consistent finding, but difficult to explain. It would be interesting to investigate whether a similar effect is seen in a pure Y5 in vitro system, or whether it is specific to the complex in vivo model investigated here.
Rat PP was active at the Y5 receptor only at high concentrations (13, 27). We have shown that this peptide is unable to significantly increase plasma ACTH at any of the doses tested (Table 2) and did not compete at the Y5 receptor at a dose of 1 μM (Table 1). In neither our Y5 receptor binding assay or our functional studies would an agonist of this low affinity be expected to be active. Desamido-NPY, although increasing plasma ACTH, failed to achieve statistical significance even at the maximum dose tested and did not compete at the Y5 receptor at a dose of 1 μM (Table 1). It is possible that, similarly to rat PP, our Y5 receptor assay is not sensitive enough to pick up this low affinity ligand.
The YACTH receptor has an interesting fragment activation profile. Unlike the Y1 and Y2 NPY receptors, YACTH is activated by both [Pro34]NPY and N terminally truncated NPY fragments such as NPY(13–36) and NPY(3–36). The YACTH receptor does not seem to require an intact N terminus of the NPY molecule for activation, and truncation beyond position 3 reduces the ability of NPY to stimulate plasma ACTH. Deamidation of the C terminal of the NPY molecule (desamido NPY) causes loss of much of the biological activity of NPY at the YACTH receptor. In addition, NPY(1–24) ICV fails to stimulate plasma ACTH at the 7.2 nmol dose. The smallest NPY fragment to retain biological activity at the YACTH receptor has yet to be established.
The use of agonists does make firm identification of receptors theoretically difficult. At present very few specific NPY-receptor antagonists are available. Among these is BIBP 3226, a specific NPY Y1 antagonist (22, 46). We have carried out preliminary investigations with this compound. A total of 30 nmol of BIBP 3226 (dissolved in 70% ethanol vehicle) was administered ICV to rats (n = 7–8) followed 30 min later by 4.8 nmol of NPY, the animals were decapitated 10 min after the second injection, and plasma ACTH was measured. Prior ICV administration of 30 nmol BIBP 3226 failed to block the NPY induced increases in plasma ACTH (BIBP 3226/NPY 235 ± 47 pg/ml vs. Ethanol/NPY 223 ± 61 pg/ml ns). A total of 30 nmol BIBP 3226 alone cause no significant rise in plasma ACTH. The ratio of BIBP 3226/NPY used in this experiment was a ratio of 6.25:1. These preliminary data suggest that the NPY Y1 antagonist BIBP 3226 has no effect on NPY-induced rises in plasma ACTH, further suggesting that YACTH is not the Y1 receptor. Studies are currently underway investigating a higher ratio of BIBP/NPY. To date no specific NPY Y5-receptor antagonists are available to us, but the effect of these, when developed, will be illuminating.
In this discussion we have, for simplicity, assumed that one receptor subtype mediates the increase in plasma ACTH, but it is possible that several NPY receptors are involved in the same functional response. For example, it has recently been demonstrated that activation of the Y1, and at least one other NPY receptor subtype, participate in the suppression of synaptic transmission within the arcuate nucleus (49). In the absence of selective antagonists to five of the six known NPY receptor subtypes, and the observation that a broad range of NPY related peptides increase plasma ACTH when administered ICV, the possibility that multiple NPY receptor subtypes are involved must be considered.
ICV infusion of NPY, but not NPY(13–36), has been shown to profoundly affect two other pituitary hormones, namely growth hormone (GH) and luteinizing hormone (LH), in both male (4) and female (47) rats. Acute ICV injections of NPY stimulate the gonadotrophic axis, but chronic injection inhibits both the somatotrophic and the gonadotrophic axes. The analogue [Leu31Pro34]NPY acutely stimulated the gonadotrophic axis, and it has been concluded that the effects of NPY on LH secretion are mediated by the Y1 receptor (48). Although further studies with other fragments and analogues are now required to confirm these observations, the finding that NPY(13–36) was completely inactive on either the gonadotrophic or somatotrophic axes (4, 48) suggests that the receptor which mediates these effects is distinct from the receptor that activates the HPA axis.
NPY was first identified as a neuropeptide with widespread distribution in the mammalian brain (2). It has now been established as a potent regulator of a number of physiological systems including feeding, reproductive function, growth, and activation of the HPA axis. We have provided evidence to suggest the possibility that the YACTH receptor is distinct from the Y FEEDING receptor. In addition, of the identified NPY receptor subtypes, the profile of the YACTH receptor appears most similar to that of the recently described Y5 receptor (13).
Table 2.
Stimulation of plasma ACTH by ICV NPY and analogues
| Peptide | 0.072 nmol | 0.72 nmol | 7.2 nmol |
|---|---|---|---|
| NPY(1–36) | 180 ± 24 | 389 ± 55** | 520 ± 72*** |
| NPY(3–36) | 378 ± 87** | 738 ± 80*** | 654 ± 147*** |
| NPY(13–36) | 151 ± 20 | 143 ± 32 | 600 ± 116** |
| [Pro34]NPY | 302 ± 36** | 568 ± 91** | 681 ± 90** |
| [Pro34]NPY(13–36) | 74 ± 17 | 385 ± 76** | 383 ± 52** |
| [Pro34]NPY(3–36) | 183 ± 21 | 295 ± 50** | 263 ± 49* |
| [d-Trp32]NPY | 233 ± 92 | 531 ± 78** | 310 ± 61 |
| Human PP | 436 ± 131 | 608 ± 106** | 729 ± 131*** |
| Rat PP | 135 ± 38 | 301 ± 70 | 288 ± 102 |
| PYY | 329 ± 108 | 453 ± 82** | 612 ± 68*** |
| Desamido-NPY | 136 ± 66 | 206 ± 97 | 318 ± 67 |
Effects of NPY, PYY, human PP, rat PP, and fragments NPY(3–36), NPY(13–36), [Pro34]NPY, [d-Trp32]NPY, desamido-NPY, [Pro34]NPY(3–36) and [Pro34]NPY(13–36) on plasma ACTH following ICV administration. Rats were injected ICV with NPY (or selected compounds) at the start of the light phase. Three doses of peptide were used 0.072, 0.72, and 7.2 nmol. Rats (n = 7–8 per group) were decapitated 10 min postinjection and plasma ACTH was measured. All results are expressed as a mean ± SEM percentage of saline control. The saline response ( 100%) was 50.3 ± 5 pg/ml. ∗∗∗, P > 0.001, ∗∗, P < 0.01, and ∗, P < 0.05.
Table 1.
Binding of NPY fragments and analogues to Y1, Y2, and Y5 receptors (Ki, nM)
| Y5, HEK-293 cells | Y1
|
Y2
|
|||
|---|---|---|---|---|---|
| SK-N-MC | Cortex | SMS-KAN | Hippocampus | ||
| NPY | 8.0 ± 0.7 | 0.58 ± 0.11 | 0.15 ± 0.04 | 0.67 ± 0.15 | 0.5 ± 0.2 |
| PYY | ND | 0.19 ± 0.05 | 0.05 ± 0.01 | 0.29 ± 0.02 | 0.2 ± 0.2 |
| Human PP | 95 ± 7 | >1,000 | >1,000 | >1,000 | >1,000 |
| Rat PP | >1,000 | >1,000 | >1,000 | >1,000 | >1,000 |
| NPY(3–36) | 60 ± 5 | 20 ± 2 | 12 ± 1 | 1.7 ± 0.7 | 3.2 ± 0.4 |
| NPY(13–36) | 540 ± 30 | 12 ± 3 | 9.8 ± 1.4 | 2.8 ± 0.4 | 2.9 ± 0.2 |
| NPY(1–24) | ND | >1,000 | >1,000 | >1,000 | >1,000 |
| [d-Trp32]NPY | 226 ± 50 | >1,000 | >1,000 | >1,000 | >1,000 |
| Desamido-NPY | >1,000 | >1,000 | >1,000 | >1,000 | >1,000 |
| [Pro34]NPY | 22 ± 3 | 0.49 ± 0.14 | 0.48 ± 0.16 | >1,000 | >1,000 |
| [Pro34]NPY(13–36) | 86 ± 11 | 4.3 ± 0.4 | 7.4 ± 2.8 | >1,000 | >1,000 |
Competition for I25I-labeled PYY binding by NPY, PYY (Y1 and Y2 only), human PP, rat PP, NPY(3–36), NPY(13–36), NPY(1–24)(Y1 and Y2 only), [d-Trp32]NPY, desamido-NPY, [Pro34]NPY, and [Pro34]NP(13–36)in membranes prepared from SK-N-MC cells and rat cortex that express NPY Y1 receptors, in membranes prepared from SMS-KAN cells and rat hippocampus that express NPY Y2 receptors and intact HEK-293 cells transfected with rat Y5 cDNA. Ki (inhibition constant) is shown as mean ± SEM (nM). Values are from three experimental curves, with each point performed in triplicate. ND, not done.
Acknowledgments
We thank Miss W. Callinan and Dr. P. Byfield for their help with the synthesis of peptides, Miss M. Turton for her help with the [d-Trp32]NPY feeding study, and Dr. J. Howard for her help with the animal experiments. We also thank the Medical Research Council for program support. C.J.S. is a Wellcome Trust Prize student. K.M. and A.P.G. are U.K. Medical Research Council Research Fellows, D.O.S. is a Wellcome Trust Research Fellow, and D.G.A.M., M.M.H. and G.M.T. are U.K. Medical Research Council students.
ABBREVIATIONS
- NPY
neuropeptide Y
- [Pro34]NPY
Pro-34 neuropeptide Y, where Pro has been substituted for Gln at position 34 of the molecule [fragments of NPY are denoted by NPY(3–36), NPY(13–36) indicating the remaining amino acids]
- PP
pancreatic polypeptide
- PYY
peptide YY
- CRH
corticotrophin-releasing hormone
- PVN
paraventricular nucleus of the hypothalamus
- ICV
intracerebroventricular
- ACTH
adrenocorticotrophic hormone
- HPA
hypothalamic pituitary adrenal
- Y5
the receptor cloned in ref. 13
- Y6
- YFEEDING
the hypothalamic NPY receptor mediating feeding
- YACTH
the hypothalamic NPY receptor mediating increases in plasma ACTH
References
- 1.Tatemoto K, Carlquist M, Mutt V. Nature (London) 1982;296:659–660. doi: 10.1038/296659a0. [DOI] [PubMed] [Google Scholar]
- 2.Allen Y S, Adrian T E, Allen J M, Tatemoto K, Crow T J, Bloom S R, Polak J M. Science. 1983;221:877–879. doi: 10.1126/science.6136091. [DOI] [PubMed] [Google Scholar]
- 3.Suda T, Tozawa F, Iwai I, Sato Y, Sumitomo T, Nakano Y, Yamada M, Demura H. Brain Res Mol Brain Res. 1993;18:311–315. doi: 10.1016/0169-328x(93)90094-6. [DOI] [PubMed] [Google Scholar]
- 4.Pierroz D D, Catzeflis C, Aebi A C, Rivier J E, Aubert M L. Endocrinology. 1996;137:3–12. doi: 10.1210/endo.137.1.8536627. [DOI] [PubMed] [Google Scholar]
- 5.Clark J T, Kalra P S, Crowley W R, Kalra S P. Endocrinology. 1984;115:427–429. doi: 10.1210/endo-115-1-427. [DOI] [PubMed] [Google Scholar]
- 6.Stanley B G, Leibowitz S F. Proc Natl Acad Sci USA. 1985;82:3940–3943. doi: 10.1073/pnas.82.11.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuhlendorff J, Gether U, Aakerlund L, Langeland Johansen N, Thogersen H, Melberg S G, Olsen U B, Thastrup O, Schwartz T W. Proc Natl Acad Sci USA. 1990;87:182–186. doi: 10.1073/pnas.87.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grundemar L, Krstenansky J L, Hakanson R. Eur J Pharmacol. 1993;232:271–278. doi: 10.1016/0014-2999(93)90784-f. [DOI] [PubMed] [Google Scholar]
- 9.Larhammar D, Blomqvist A G, Yee F, Jazin E, Yoo H, Wahlestedt C. J Biol Chem. 1992;267:10935–10938. [PubMed] [Google Scholar]
- 10.Eva C, Keinanen K, Monyer H, Seeburg P, Sprengel R. FEBS Lett. 1990;271:81–84. doi: 10.1016/0014-5793(90)80377-u. [DOI] [PubMed] [Google Scholar]
- 11.Gerald C, Walker M W, Vaysse P J, He C, Branchek T A, Weinshank R L. J Biol Chem. 1995;270:26758–26761. doi: 10.1074/jbc.270.45.26758. [DOI] [PubMed] [Google Scholar]
- 12.Rose P M, Fernandes P, Lynch J S, Frazier S T, Fisher S M, Kodukula K, Kienzle B, Seethala R. J Biol Chem. 1995;270:22661–22664. doi: 10.1074/jbc.270.39.22661. [DOI] [PubMed] [Google Scholar]
- 13.Gerald C, Walker M W, Criscione L, Gustafson E L, Batzl-Hartmann C, Smith K E, Vaysse P, Durkin M M, Laz T M, Linemeyer D L, Schaffhauser A O, Whitebread S, Hofbauer K G, Taber R I, Branchek T A, Weinshank R L. Nature (London) 1996;382:168–171. doi: 10.1038/382168a0. [DOI] [PubMed] [Google Scholar]
- 14.Wahlestedt C, Regunathan S, Reis D J. Life Sci. 1992;50:PL7–PL12. doi: 10.1016/0024-3205(92)90342-m. [DOI] [PubMed] [Google Scholar]
- 15.Dumont Y, Cadieux A, Pheng L H, Fournier A, St Pierre S, Quirion R. Brain Res Mol Brain Res. 1994;26:320–324. doi: 10.1016/0169-328x(94)90105-8. [DOI] [PubMed] [Google Scholar]
- 16.Grundemar L, Hakanson R. Trends Pharmacol Sci. 1994;15:153–159. doi: 10.1016/0165-6147(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 17.Grundemar L, Wahlestedt C, Reis D J. Neurosci Lett. 1991;122:135–139. doi: 10.1016/0304-3940(91)90211-b. [DOI] [PubMed] [Google Scholar]
- 18.Grundemar L, Wahlestedt C, Reis D J. J Pharmacol Exp Ther. 1991;258:633–638. [PubMed] [Google Scholar]
- 19.Bard J A, Walker M W, Branchek T A, Weinshank R L. J Biol Chem. 1995;270:26762–26765. doi: 10.1074/jbc.270.45.26762. [DOI] [PubMed] [Google Scholar]
- 20.Weinberg D H, Sirinathsinghji D J S, Tan C P, Shiao L-L, Morin N, Rigby M R, Heavens R H, Rapoport D R, Bayne M L, Cascieri M A, Strader C D, Linemeyer D L, MacNeil D J. J Biol Chem. 1996;271:16435–16438. doi: 10.1074/jbc.271.28.16435. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto M, Nomura T, Momose K, ikeda Y, Kondou Y, Akiho H, Togami J, Kimura Y, Okada M, Yamaguchi T. J Biol Chem. 1996;271:27217–27220. doi: 10.1074/jbc.271.44.27217. [DOI] [PubMed] [Google Scholar]
- 22.O’Shea D, Morgan D G, Meeran K, Edwards C M B, Turton M D, Choi S J, Heath M M, Gunn I, Taylor G M, Howard J K, Bloom C I, Small C J, Haddo O, Ma J J, Callinan W, Smith D M, Ghatei M, Bloom S R. Endocrinology. 1997;138:196–201. doi: 10.1210/endo.138.1.4899. [DOI] [PubMed] [Google Scholar]
- 23.Stanley B G, Magdalin W, Seirafi A, Nguyen M M, Leibowitz S F. Peptides. 1992;13:581–587. doi: 10.1016/0196-9781(92)90093-i. [DOI] [PubMed] [Google Scholar]
- 24.Leibowitz S F, Alexander J T. Peptides. 1991;12:1251–1260. doi: 10.1016/0196-9781(91)90203-2. [DOI] [PubMed] [Google Scholar]
- 25.Grundemar L, Jonas S E, Morner N, Hogestatt E D, Wahlestedt C, Hakanson R. Br J Pharmacol. 1992;105:45–50. doi: 10.1111/j.1476-5381.1992.tb14208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cadieux A, Pheng L H, St Pierre S, Fournier A, Benchekroun M T, Dumont Y, Satoh H, Taoudi Benchekroun M, Quirion R. Regul Pept. 1993;109:902–904. [Google Scholar]
- 27.Hu Y, Bloomquist B T, Cornfield J L, DeCarr L B, Flores-Riveros J R, Friedman L, Jiang P, Lewis-Higgins L, Sadlowski Y, Schaefer J, Velazquez N, McCaleb M J. J Biol Chem. 1996;271:26315–26319. [PubMed] [Google Scholar]
- 28.Stanley B G, Kyrkouli S E, Lampert S, Leibowitz S F. Peptides. 1986;7:1189–1192. doi: 10.1016/0196-9781(86)90149-x. [DOI] [PubMed] [Google Scholar]
- 29.Liposits Z, Sievers L, Paull W K. Histochemistry. 1988;88:227–234. doi: 10.1007/BF00570278. [DOI] [PubMed] [Google Scholar]
- 30.Wahlestedt C, Skagerberg G, Ekman R, Heilig M, Sundler F, Hakanson R. Brain Res. 1987;417:33–38. doi: 10.1016/0006-8993(87)90176-4. [DOI] [PubMed] [Google Scholar]
- 31.Tsagarakis S, Rees L H, Besser G M, Grossman A. Brain Res. 1989;502:167–170. doi: 10.1016/0006-8993(89)90472-1. [DOI] [PubMed] [Google Scholar]
- 32.Inoue T, Inui A, Okita M, Sakatani N, Oya M, Morioka H, Mizuno N, Oimomi M, Baba S. Life Sci. 1989;44:1043–1051. doi: 10.1016/0024-3205(89)90556-0. [DOI] [PubMed] [Google Scholar]
- 33.Inui A, Inoue T, Nakajima M, Okita M, Sakatani N, Okimura Y, Chihara K, Baba S. Brain Res. 1990;510:211–215. doi: 10.1016/0006-8993(90)91369-r. [DOI] [PubMed] [Google Scholar]
- 34.Miura M, Inui A, Teranishi A, Hirosue Y, Nakajima M, Okita M, Inoue T, Baba S, Kasuga M. Neuropeptides. 1992;23:15–18. doi: 10.1016/0143-4179(92)90004-g. [DOI] [PubMed] [Google Scholar]
- 35.Balasubramaniam A, Sheriff S, Johnson M E, Prabhakaran M, Huang Y, Fischer J E, Chance W T. J Med Chem. 1994;37:811–815. doi: 10.1021/jm00032a015. [DOI] [PubMed] [Google Scholar]
- 36.Graham F L. Virology. 1973;52:456. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 38.Fraker P J, Speck J C. Biochem Biophys Res Commun. 1978;80:849–857. doi: 10.1016/0006-291x(78)91322-0. [DOI] [PubMed] [Google Scholar]
- 39.Morgan D G, Lambert P D, Smith D M, Wilding J P H, Bloom S R. J Neuroendocrinol. 1996;8:283–290. doi: 10.1046/j.1365-2826.1996.04565.x. [DOI] [PubMed] [Google Scholar]
- 40.Taylor G M, Meeran K, O’Shea D, Smith D M, Ghatei M A, Bloom S R. Endocrinology. 1996;137:3260–3264. doi: 10.1210/endo.137.8.8754748. [DOI] [PubMed] [Google Scholar]
- 41.Siaud P, Mekaouche M, Maurel D, Givalois L, Ixart G. Brain Res. 1994;652:273–278. doi: 10.1016/0006-8993(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 42.Hodgkinson S C, Allolio B, Landon J, Lowry P J. Biochem J. 1984;218:703–711. doi: 10.1042/bj2180703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi S J, Meeran K, O’Shea D, Lambert P D, Bloom S R. Brain Res. 1996;729:223–227. doi: 10.1016/s0006-8993(96)00423-4. [DOI] [PubMed] [Google Scholar]
- 44.Lundell I, Blomqvist A G, Berglund M M, Schober D A, Johnson D, Statnick M A, Gadski R A, Gehlert D R, Larhammar D. J Biol Chem. 1995;270:29123–29128. doi: 10.1074/jbc.270.49.29123. [DOI] [PubMed] [Google Scholar]
- 45.Kalra S P, Dube M G, Fournier A, Kalra P S. Physiol Behav. 1991;50:5–9. doi: 10.1016/0031-9384(91)90490-f. [DOI] [PubMed] [Google Scholar]
- 46.Jacques D, Cadieux A, Dumont Y, Quirion R. Eur J Pharmacol. 1995;278:R3–5. doi: 10.1016/0014-2999(95)00179-o. [DOI] [PubMed] [Google Scholar]
- 47.Catzeflis C, Pierroz D D, Rohner Jeanrenaud F, Rivier J E, Sizonenko P C, Aubert M L. Endocrinology. 1993;132:224–234. doi: 10.1210/endo.132.1.8380374. [DOI] [PubMed] [Google Scholar]
- 48.Kalra S P, Fuentes M, Fournier A, Parker S L, Crowley W R. Endocrinology. 1992;130:3323–3330. doi: 10.1210/endo.130.6.1317781. [DOI] [PubMed] [Google Scholar]
- 49.Rhim H, Kinney G A, Emmerson P J, Miller R J. J Neurosci. 1997;17:2980–2989. doi: 10.1523/JNEUROSCI.17-09-02980.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]