

Abstract
Synthesis and antiviral activity of the title fluoromethylenecyclopropane analogues 15a, 15b, 16a and 16b is described. Methylenecyclopropane carboxylate was first transformed to 2,2-bis-hydroxymethylmethylenecyclopropane. Selective monoacetylation followed by introduction of fluorine gave 2-acetoxymethyl-2-fluoromethylmethylenecyclopropane as the key intermediate. The synthesis of analogues 15a, 15b, 16a and 16b then followed alkylation-elimination procedure as described previously for other methylenecyclopropane analogues. The adenine Z-isomer 15a was found to be a potent inhibitor of Epstein-Barr virus (EBV) in vitro with EC50/CC50 (µM) 0.5/55.7. Compounds 15b, 16a and 16b were also active but at higher concentrations, EC50/CC50 (µM) 3.2–7.5/53.6–64.1. Analogue 15a inhibited hepatitis C virus by virtue of its cytotoxicity and it moderately inhibited replication of the Towne strain of human cytomegalovirus (HCMV). The E-isomer 16a was a substrate for adenosine deaminase whereas the Z-isomer 15a was not deaminated.
Keywords: Methylenecyclopropanes, Nucleoside analogues, Alkylation-elimination, Methylenecyclopropane-methylenecyclobutane rearrangement, Antiviral agents, Adenosine deaminase
1. Introduction
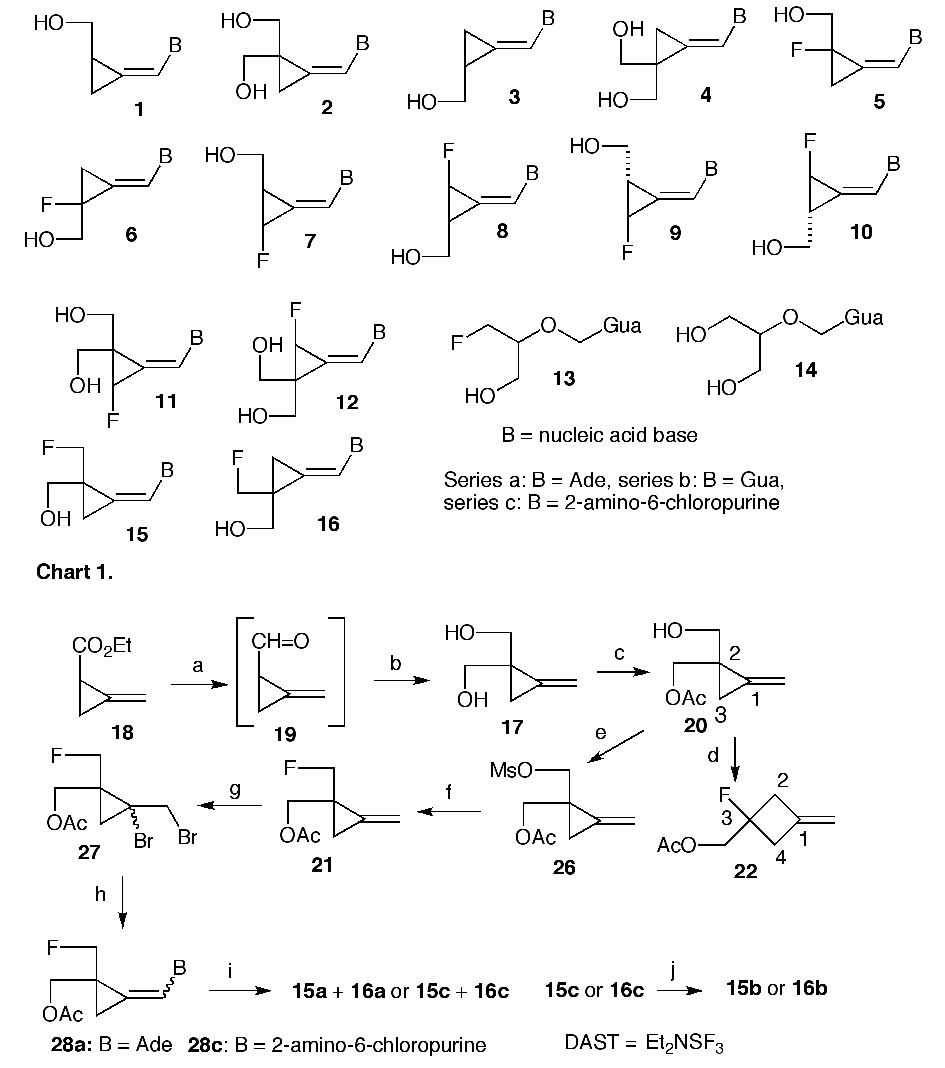
The Z-methylenecyclopropane analogues of purine nucleosides 1 and 2 are effective antiviral agents whereas the E-isomers 3 and 4 (Chart 1) are either inactive or of limited potency.1–3 The guanine analogue 2b (cyclopropavir) is currently under preclinical investigation as a possible drug against infections caused by human cytomegalovirus (HCMV).4,5 It is also effective in vitro6 against Epstein-Barr virus (EBV) and human herpes viruses (HHV) 6 and 8. Structure-activity relationships (SAR) studies have indicated that introduction of fluorine into the cyclopropane moiety of 1 and 3 can also provide new antiviral agents. Thus, purine and/or pyrimidine Z- and E-2-fluoro analogues 5 and 6 were effective against HCMV, EBV or varicella zoster virus (VZV).7 Purine 3-fluoroanalogues 7, 8, 9 and 10 had more narrow antiviral effects or they were less potent.8 This trend was also reflected in the bis(2,2-hydroxymethyl)-3-fluoro derivatives9 11 and 12.
Fluorine can mimic both a hydrogen atom and a hydroxy group because of its small van der Waals radius and polarity of the carbon-fluorine bond.10 Although all possible monofluoromethylenecyclopropane analogues (5 through 12) derived by replacement of hydrogens of the cyclopropane moiety were investigated,7–9 compounds having the hydroxy group(s) replaced with fluorine have not been described. Similar fluoro analogue of ganciclovir 13 exhibited activity11 against herpes simplex virus 1 (HSV-1). Because cyclopropavir 2b can be regarded as a rigid bioisostere of anti-HCMV drug ganciclovir4 14 it was of interest to synthesize and investigate biological activity of purine fluoromethylenecyclopropane analogues 15a, 15b, 16a and 16b.
2. Results and discussion
2.1. Synthesis
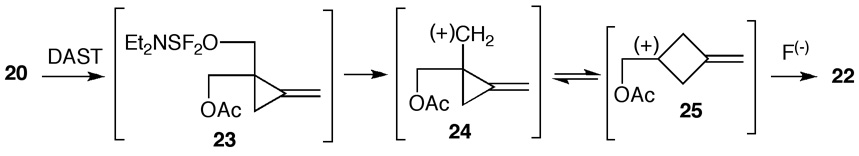
Methylenecyclopropane diol 17 was chosen as a convenient starting material for synthesis of analogues 15 and 16. For the present purpose, compound 17 was obtained by an alternate approach. Methylenecyclopropane carboxylate12 18 was reduced using a less than a stoichiometric amount of diisobutylaluminum hydride (DIBALH). The intermediary aldehyde 19 was not isolated but it was subjected in situ to aldol and crossed Cannizzaro reaction with formaldehyde to give diol 17 in 64% yield. It should be noted that this is a new synthesis of an important intermediate for cyclopropavir (2b).4,13,14 Acetylation of 17 via the corresponding cyclic orthoester13 gave monoacetate 20 in 78% yield. Reaction of 20 with diethylaminosulfur trifluoride (DAST)15,16 using pyridine in CH2Cl2 at −78 °C did not give the expected fluoro derivative 21 but it led instead to a ring-expanded fluorocyclobutane 22 as the only product in 87% yield. It is important to note that this is a new synthesis of methylenefluorocyclobutane skeleton. The parent compound17 is accessible only by reaction of [1.1.1]propellane with XeF2. Recently, ring expansion of prolinols to fluoropiperidines effected by DAST was described.18 Nevertheless, the reaction course was not uniform and ratio of five-membered to six-membered products was about 2 : 3.
The reaction is initiated by transformation of 20 by DAST to intermediate 23 (Scheme 2). In the next step, a non-classical19 cyclopropylmethyl carbocation 24 existing in equilibrium with cyclobutonium ion 25 reacts with fluoride ion to give methylenefluorocyclobutane 22. The reported solvolysis20 of methylenecyclopropylmethyl chloride and deamination21 of methylenecyclopropylmethylamine led also to methylenecyclobutanes in addition to methylenecyclopropyl methyl derivatives.
Scheme 2.
It was then clear that avoiding formation of intermediary carbocation might lead to a successful synthesis of methylenefluorocyclopropane 21. Therefore, monoacetate 20 was converted to methylsulfonate 26 (81%) using methylsulfonyl chloride (MsCl) which, in turn, was smoothly transformed to fluorocyclopropane 21 (72%) using tetrabutylammonium fluoride (NBu4F) in THF. Addition of bromine via pyridinium tribromide gave dibromo derivative 27 which was used for alkylation elimination1–3 of nucleic acid heterocycles. The reaction of 27 with adenine gave the Z,E-isomeric mixture methylenecyclopropanes 28a in 65% yield. The yield of 28c obtained with 2-amino-6-chloropurine was lower (46%). Deacetylation of 28a using K2CO3 in 90% aqueous methanol at room temperature furnished the target analogues 15a and 16a after chromatographic separation in 49 and 43% yield, respectively. In a similar fashion, deacetylation of intermediate 28c at 0 °C afforded the Z- and E-isomers 15c and 16c (46 and 54%). Hydrolytic dechlorination of 15c and 16c using 80% formic acid at 80 °C provided guanine analogues 15b and 16b (84 and 91%).
2.2. The Z,E-Isomeric Assignment
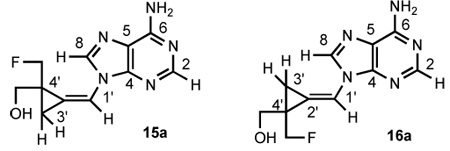
As in previous cases of methylenecyclopropane analogues,2,4 the NMR spectroscopy was indispensable to confirm the Z,E-isomeric structure of analogues 15a, 15b and 16a, 16b. The chemical shift patterns of relevant protons parallell those of analogues 2a, 2b and 4a, 4b (Table 1). Thus, the 1H NMR signals of OH and H8 of the Z-isomers 15a, 15b are more deshielded than those of the E-isomers 16a, 16b whereas an opposite pattern was found in the alkene H1’ signals. In the 13C NMR spectra, the cyclopropane C4’ of the Z-isomers 15a, 15b is located at a lower field than in the E-isomers 16a, 16b in contrast to the corresponding C3’ shifts. The final confirmation of the Z.E-isomeric assignment came from the NOE experiments performed with adenine analogues 15a and 16a (Table 2). In the Z-isomer 15a, the NOE enhancements were found between the cis-arranged H1’ and H3’ protons as well as between the H8 and protons of OH, CH2F and CH2O groups. By contrast, in the E-isomer 16a a strong NOE interaction occurs between the cis-located H3’ and H8. Also, the NOE enhancements were found between the H3’ and OH, CH2F and CH2O groups.
Table 1.
Comparison of chemical shifts (δ) of the relevant 1H and 13C NMR signals of the (Z)-and (E)-2,2-bis(hydroxymethyl)- and 2-fluoromethyl-2-hydroxymethylmethylenecyclopropanes 2a, 4a, 2b, 4b, 15a, 16a, 15b and 16b
| Compounda | Isomer | OH | H1’ | H8 | C3’ | C4’ |
|---|---|---|---|---|---|---|
| 2a | Z | 5.07 | 7.37 | 8.82 | 11.7 | 31.4 |
| 4a | E | 4.76 | 7.48 | 8.49 | 14.4 | 29.7 |
| 2b | Z | 4.99 | 7.07 | 8.41 | 11.5 | 31.3 |
| 4b | E | 4.76 | 7.21 | 8.03 | 14.3 | 29.5 |
| 15a | Z | 5.37 | 7.45 | 8.57 | 12.0 | 29.4 |
| 16a | E | 5.02 | 7.52 | 8.49 | 15.0 | 27.6 |
| 15b | Z | 5.31 | 7.16 | 8.15 | 11.9 | 29.2 |
| 16b | E | 5.01 | 7.26 | 8.04 | 14.8 | 27.5 |
Table 2.
The NOE enhancements of relevant 1H NMR signals of (Z)- and (E)-2-fluoromethyl-2-hydroxymethyltmethylenecyclopropanes 15a and 16a
 | |||||
|---|---|---|---|---|---|
| Compound | Hirr | δ | Hobs | δ | NOE (%) |
| 15a | H1’ | 7.45 | H3’ | 1.54 | 1.83 |
| H3’ | 1.54 | H1’ | 7.45 | 2.17 | |
| OH | 5.37 | H8 | 8.57 | 4.0 | |
| H8 | 8.57 | OH | 5.37 | 1.71 | |
| CH2F | 4.44–4.70 | H8 | 8.57 | 3.16 | |
| CH2O | 3.48–3.80 | H8 | 8.57 | 3.84 | |
| 16a | H3’ | 1.76 | H8 | 8.49 | 2.75 |
| OH | 5.02 | H1’ | 7.52 | 1.42 | |
| CH2F | 4.47 | H1’ | 7.52 | 1.34 | |
| CH2O | 3.46 | H1’ | 7.52 | 1.87 | |
2.3. Antiviral Activity
Compounds 15a, 15b, 16a and 16b were tested against the following viruses: herpes simplex virus 1 and 2 (HSV-1 and HSV-2), human cytomegalovirus (HCMV, Towne and AD 169 strains), varicella zoster virus (VZV), Epstein-Barr virus (EBV), human immunodeficiency virus (HIV-1), hepatitis B and C virus (HBV and HCV). They were all effective against EBV in Akata cells using a DNA hybridization assay.22 The adenine analogue 15a was the most potent (Table 3) and least cytotoxic. It was more effective than cyclopropavir (2b). The E-isomer 16a and guanine derivatives 15b, 16b were less effective than 15a. Interestingly, a somewhat similar anti-EBV activity pattern was found with Z- and E-isomers of fluoroanalogues7 5a, 5b, 6a and 6b which can be regarded as lower homologues of 15a, 15b, 16a and 16b. However, an exact comparison is not possible because of the differences in assays. In the series of fluoroanalogues 7 through 12 only adenine Z-isomer8 9a was effective against EBV. It is likely that the mechanism of anti-EBV action of analogues 15a, 15b, 16a and 16b includes their phosphorylation to triphosphates which then inhibit the viral DNA polymerase as suggested for other fluorinated methylenecyclopropane analogues.7,8
Table 3.
Inhibition of Replication of EBV with Fluoromethyl Methylenecyclopropane Nucleoside Analoguesa
| Compound | EC50/CC50 (µM)b | Selectivity Index |
|---|---|---|
| 2b | 2.5/>100c | >40 |
| 5a | 6.8/>213 | >31.3 |
| 5b | 8.0/>199 | >24.9 |
| 6a | 167/>209d | >1.25 |
| 6b | 29.1/>199e | >6.8 |
| 15a | 0.5/55.7 | 111 |
| 15b | 7.5/59.7 | 8 |
| 16a | 3.4/53.6 | 15.8 |
| 16b | 3.2/64.1 | 20 |
Akata cells, DNA hybridization assay. For details see Experimental. Acyclovir as a control had EC50 1.7 µM.
Results for analogues 5a - 6b were taken from from ref.7 (DNA hybridization assay in Daudi cells).
Data from ref.19
EC50 2.3 µM in viral capsid immunofluorescence (VCA) ELISA and 3.6 µM in H-1 cells (DNA hybridization).
EC50 <0.32 µM in VCA ELISA.
Compound 15a also inhibited HCV in Huh7 AVA5 cells23 (replicon assay) with EC50/CC50 (µM) 6.5/11 using 2’-methylcytidine as a control (EC50/CC50 1.8/>300) but the antiviral activity was poorly separated from cytotoxicity. Compound 15a moderately inhibited the replication of HCMV Towne strain but not AD169 strain (plaque reduction assay) in human foreskin fibroblast (HFF) cells /EC50/CC50 (µM) 46/>100, ganciclovir (14) as a control exhibited EC50/CC50 2.5/>100. No significant activity against the rest of tested viruses was detected.
2.4. Adenosine Deaminase (ADA)
Adenine analogues 15a and 16a were investigated as substrates for adenosine deaminase. In agreement with the general trend in the series of methylenecyclopropane analogues,1,2 the E-isomer 16a was a moderate substrate and it was deaminated after 28 h, whereas the Z-isomer 15a was resistant to deamination.
3. Conclusion
Fluoromethylenecyclopropane analogues 15a, 15b, 16a and 16b were synthesized and evaluated for antiviral activity. All analogues were inhibitors of replication of EBV in Akata cells with adenine derivative 15a being the most potent with EC50/CC50 (µM) 0.5/55.7. Against HCMV, only compound 15a had a moderate effect whereas its potency against HCV was offset by cytotoxicity. No activity was observed against other tested viruses. The E-isomer 15b was a moderate substrate for adenosine deaminase whereas Z-isomer 15a was not deaminated.
4. Experimental
4.1. General Methods
The UV spectra were measured in ethanol and NMR spectra were determined at 300 or 400 MHz (1H), 75 or 100 MHz (13C) and 376 MHz (19F) in CD3SOCD3 unless stated otherwise. For 19F NMR, CFCl3 was used as a reference. Mass spectra were determined in electron-impact (EI-MS) or electrospray ionization (ESI, methanol - NaCl) mode. Thin-layer chromatography (TLC) was performed on Analtech aluminum foils coated with silica gel F254.
4.2. 2,2-Bis(hydroxymethyl)methylenecyclopropane (17)
A solution of DIBALH in hexane (1 M, 26 mL, 26 mmol) was added dropwise to ethyl 8 methylenecyclopropane carboxylate12 18 (4.12 g, 32.7 mmol) in dichloromethane at −78 °C with stirring. The stirring was continued for 1 h. TLC (hexane - AcOEt 4 : 1) indicated the presence of aldehyde 19 as the major product accompanied by minor amounts of the faster moving starting ester 18 and slower moving methylenecyclopropylmethanol. The reaction was quenched with saturated aqueous NH4Cl (100 mL). The mixture was stirred for 6 h, the aqueous layer was extracted with ether (2 × 100 mL), the combined organic phase was dried (MgSO4) and it was concentrated to about 10 mL by distillation at <45 °C at an atmospheric pressure. A mixture of this product, aqueous formaldehyde (37%, 65 mL, 0.8 mol) and KOH (18.3 g, 0.33 mmol) in methanol (60 mL) was stirred for 5 days at room temperature. Methanol was removed in vacuo and the aqueous portion was extracted with ethyl acetate (10 × 100 mL). The organic phase was dried (MgSO4) and concentrated. The precipitated paraformaldehyde was filtered off using a short silica gel column which was then washed with AcOEt - hexanes (4 : 1). The solvents were evaporated and the residue was refluxed in 1 M HCl (5 mL) for 2 h. The volatile components were evaporated and the crude product was chromatographed on a silica gel column using AcOEt - hexanes (1 : 1) to give diol 17 (1.88 g, 64% based on DIBALH) as a yellow oil. TLC (AcOEt - hexanes, 2 : 1) and 1H NMR spectrum were identical with those of authentic samples.4,13
4.3. 2-Acetoxymethyl-2-hydroxymethyl-1-methylenecyclopropane (20)
A mixture of of diol 17 (1.80 g, 15.8 mmol), trimethyl orthoacetate (2.9 g, 23.7 mmol) and p-toluenesulfonic acid (2 mg) in CH2Cl2 (20 mL) was stirred for 1 h at room temperature. The reaction was quenched with Et3N (0.1 mL) and solvent was evaporated. The residue was dissolved in 80% acetic acid (5 mL) and the solution was allowed to stand at room temperature for 30 min whereupon it was diluted with dichloromethane (200 mL). The organic phase was 9 washed with saturated NaHCO3 (2 × 200 mL, caution!) and water (2 × 200 mL). It was dried (MgSO4) and the solvent was removed to give product 20 (1.87 g, 78%) as a colorless oil. 1H NMR (CDCl3) δ 5.52 (t, 1H, J = 3.1 Hz), 5.43 (t, 1H, J = 1.8 Hz, CH2=), 4.15, 4.10 (AB, 2H, J = 11.6 Hz, CH2OAc), 3.56, 3.51 (AB, 2H, J = 11.6 Hz, CH2OH), 2.09 (s, 3H, CH3), 1.25 (t, 2H, J = 2.4 Hz, H3). 13C NMR 171.9 (C=O), 134.9 (C=), 105.2 (CH2=), 66.7, 65.2 (CH2O), 26.2 (C2), 21.2 (CH3), 13.8 (C3). ESI-MS 179 (84.8, M + Na), 157 (26.6, M + H), 97 (100.0). Anal. Calcd for C8H12O3 × 0.25 H2O: C,59.80; H, 7.84 H. Found: C, 59.74; H, 7.73.
4.4. 3-Acetoxymethyl-3-fluoro-1-methylenecyclobutane (22)
DAST (0.16 mL, 0.81 mmol) was added dropwise to a stirred solution of acetate 20 (75 mg, 0.48 mmol) and pyridine (0.16 mL, 2 mmol) in CH2Cl2 (20 mL) at −78 °C. The temperature was allowed to raise, the solvent was evaporated and the crude product was chromatographed on a silica gel column using hexanes - ether (4 : 1) to give compound 22 (70 mg, 87%) as a colorless oil. 1H NMR (CDCl3) δ 4.97 (m, 2H, CH2=), 4.26 (d, 2H, J = 22.8 Hz, CH2O), 3.05 (dt, J = 19.0, 2.9 Hz, 2H), 2.84 (m, 2H, H2, H4), 2.11 (s, 3H, CH3). 13C NMR 171.1 (C=O), 136.9 (d, J = 15.7 Hz, C=), 109.7 (d, J = 8.2 Hz, CH2=), 91.0 (d, J = 216.4 Hz, C3), 66.1 (d, J = 23.1 Hz, CH2O), 41.7 (d, J = 23.1 Hz, C2, C4), 21.0 (CH3). 19F NMR -149.32 (m). EI-MS 138 (34.5, M - HF), 116 (22.6, M - CH2CO), 97 (100.0). HRMS calcd for C8H10O2 (M - HF) 138.0681. Found: 138.0682. Anal. Calcd for C8H11FO2: C, 60.75; H. 7.01. Found: C, 61.02; H, 7.07.
4.5. 2-Acetoxymethyl-2-methylsulfonyloxymethylmethylenecyclopropane (26)
Methylsulfonyl chloride (0.90 mL 11.5 mmol) was added dropwise with stirring and external ice cooling to a solution of acetate 20 (1.80 g, 11.5 mmol) and triethylamine (3.3 mL, 23 mmol) in CH2Cl2 (20 mL). The stirring was continued for 1 h, the mixture was diluted with ether (150 mL), the organic phase was washed with water (100 mL), saturated NaHCO3 (2 × 100mL), water 10 (2 × 100 mL) and it was dried with MgSO4. The solvent was evaporated to give compound 26 (2.2 g, 81%) as a colorless oil. 1H NMR (CDCl3) δ 5.59 (t, 1H, J = 3.1 Hz), 5.51 (t, 1H, J = 1.8 Hz, CH2=), 4.20, 4.16 (AB, 2H, J = 10.5 Hz), 4.14, 4.05 (AB, 2H, J = 11.6 Hz, CH2O), 3.01 (s, 3H, CH3SO2), 2.07 (s, 3H, CH3CO), 1.42 (t, 2H, J = 1.8 Hz, H3). 13C NMR 171.1 (C=O), 132.9 (C=), 107.0 (CH2=), 72.1 (CH2OMs), 65.5 (CH2OAc), 37.8 (CH3SO2), 23.1 (C2), 21.1 (CH3 of AcO), 14.7 (C3). EI-MS (MeOH + LiCl) 241 (M + Li, 100.0), 475 (2M + Li, 48.8). Anal. Calcd for C9H14O5S × H2O: C, 42.85; H. 6.39. Found: C 42.97; H, 6.40.
4.6. 2-Acetoxymethyl-2-(fluoromethyl)methylenecyclopropane (21)
A solution of Bu4NF (1M, 35 mL, 35 mmol) in THF was added with stirring to compound 26 (1.65 g, 7 mmol) in THF (100 mL) under N2 at room temperature. The stirring was continued for 6 h, the mixture was diluted with ether (200 mL), the organic phase was washed with saturated NaHCO3 (2 × 200 mL), water (2 × 200 mL) and it was dried (MgSO4) The solvent was removed by distillation at an atmospheric pressure. The crude product was chromatographed on a silica gel column using 1-pentane - ether (15 : 1) to give compound 21 (0.80 g, 72%) as a colorless oil. 1H NMR (CDCl3) δ 5.57 (t, 1H, J = 2.4 Hz), 5.49 (poorly resolved dd, 1H, CH2=), 4.43, 4.40 and 4.30, 4.28 (2AB, 2H, JH,F = 48.8 Hz, JAB = 9.8 Hz, CH2F), 4.17, 4.10 (AB, 2H, JAB = 12.0 Hz, CH2OAc), 2.09 (s, 3H, CH3), 1.38 (m, 2H, H3). 13C NMR 171.2 (C=O), 133.2 (C=), 106.3 (CH2=), 85.5 (d, J = 172.3 Hz, CH2F), 65.7 (CH2OAc), 24.3 (d, J = 23.1 Hz, C2), 21.2 (CH3), 14.0 (d, J = 6.7 Hz, C3). 19F NMR −216.52 (poorly resolved tt, J = 48.8, 2,6 Hz). EI-MS 138 (16.7, M - HF), 116 (23.8, M - CH2CO), 97 (100.0). HRMS calcd for C8H10O2 (M - HF) 138.0681. Found 138.0687.
4.7. (Z,E)-1-Acetoxymethyl-1-fluoromethyl-2-bromo-2-bromomethylcyclopropane (27)
A mixture of of pyridinium tribromide (2.12 g, 6.6 mmol) and compound 21 (0.7 g (4.4 mmol) in CH2Cl2 (20 mL) was stirred at 0 °C for 1 h. The solid portion was filtered off and it was washed with CH2Cl2 (5 mL). The filtrate was diluted with ether (100 mL), the organic phase was washed with saturated Na2S2O3 (2 × 100 mL) and water (2 × 100 mL) and it was dried with Na2SO4. The solvents were evaporated and the residue was chromatographed on a silica gel column using AcOEt - hexanes (1 : 10) to give product 27 (1.12 g, 80%) as a colorless oil. 1H NMR (CDCl3) δ 4.80 - 4.20 (cluster of m, 6H, CH2F, CH2Br, CH2OAc), 2.08, 2.07 (2s, 3H, CH3), 1.46, 1.32 (2m, 2H, H3). 13C NMR 170.9 (C=O), 86.8, 81.6 (2d, J = 173.8 Hz, CH2F), 67.9 (d, J = 1.5 Hz), 61.7 (d, J = 1.5 Hz, CH2OAc), 42.1 (d, J = 4.5 Hz), 42.0 (d, J = 4.3 Hz, CH2Br), 41.3, 41.2 (C1), 33.1 (d, J = 23.1 Hz), 32.8 (d, J = 20.9 Hz, C2), 26.5, 26.3 (2d, J = 6.7 Hz, C3), 21.09, 21.06 (CH3). 19F NMR −219.24 (dt, J = 48.9, 6.2 Hz), −219.92 (tt, 47.4, 9.0, 4.1 Hz). ESIMS 339, 341, 343 (53.3, 100.0, 51.8, M + Na). Anal. Calcd for C8H11Br2FO2: C, 30.22; H. 3.49. Found: C, 30.61; H, 3.50.
4.8. (Z,E)-9-[(2-Acetoxymethyl-2-fluoromethylcyclopropylidene)methyl]adenine (28a)
A mixture of of dibromide 27 (400 mg, 1.26 mmol), adenine (170 mg, 1.26 mmol) and K2CO3 (1.8 g, 12.6 mmol) in DMF (25 mL) was stirred for 5 h at 110–115 °C. After cooling, solids were filtered off and they were washed with DMF (5 mL). The filtrate was concentrated in vacuo and the residue was chromatographed on a silica gel column using CH2Cl2 - methanol (200 : 5) to give compound 28a (240 mg, 65%) as a white solid. The Z/E ratio was 1 : 1 as determined by 1H NMR, mp 189–196 °C. UVλmax 277 nm (ε 8,400), 263 (ε 11,800), 227 (ε 24,900). 1H NMR δ 8.50, 8.34 (1H, 2s, 1H, H8), 8.19, 8.18 (2s, 1H, H2), 7.58 (t, J = 2.5 Hz), 7.51 (m, 1H, H1’), 7.39 (bs, 2H, NH2), 4.79, 4.65 and 4.62, 4.49 (2AB, JH,F = 49.0 Hz, JAB = 10.1 Hz), 4.46 (d, 2H, J = 49.2 Hz, CH2F), 4.40, 4.13 and 4.20, 4.14 (2AB, 2H, J = 11.7 Hz, CH2OAc), 2.06, 1.91 (2s, 3H, CH3), 1.95, 1.70 (2m, 2H, H3). 13C NMR 171.1, 170.6 (C=O), 156.8 (C6), 153.9 (C2), 149.0, 12 148.9 (C4), 138.0 (C8), 119.1 (C5), 115.6 (d, J = 8.0 Hz), 115.3 (d, J = 7.0 Hz, C2’), 113.1, 112.9 (C1’), 86.4 (d, J = 169.4 Hz), 85.5 (d, J = 169.2 Hz, CH2F), 65.8, 65.2 (CH2OAc), 26.6 (d, J = 23.2 Hz), 24.6 (d, J = 23.2 Hz, C4’), 21.4, 21.1 (CH3), 15.9 (d, J = 6.8 Hz), 12.8 (d, J = 7.1 Hz, C3’). 19F NMR −215.10, −214.94 (2 overlapped t, J = 48.2 Hz). ESI-MS 292 (100.0, M + H), 314 (44.4, M + Na). Anal. Calcd for C13H14FN5O2: C, 53.60; H, 4.84; N, 24.04. Found: C, 53.51; H, 4.89; N, 23.87.
4.9. (Z)-9-[(2-Fluoromethyl-2-hydroxymethylcyclopropylidene)methyl]adenine (15a) and (E)-9-[(2-Fluoromethyl-2-hydroxymethylcyclopropylidene)methyl]adenine (16a)
mixture of compound 28a (220 mg, 0.76 mmol) and K2CO3 (200 mg, 1.45 mmol) in methanol - water (9 : 1, 20 mL) was stirred for 1 h at room temperature. The solvent was evaporated and the residue was chromatographed on a silica gel column using CH2Cl2 - methanol (20 : 1) to give the Z-isomer 15a (93 mg, 49%), followed by E-isomer 16a (80 mg, 43%).
Z-isomer 15a
Mp 234–236 °C. UV λmax 278 nm (ε 7,700), 261 (ε 10,700), 227 (ε 22,800). 1H NMR ε 8.57 (s, 1H, H8), 8.17 (s, 1H, H2), 7.45 (s, 1H, H1’), 7.37 (bs, 2H, NH2), 5.37 (t, 1H, J = 5.4 Hz, OH), 4.70, 4.56 and 4.58, 4.44 (2 partly overlapped AB, 1H, JH,F = 47.8 Hz, JAB = 8.8 Hz, CH2F), 3.80 (dd, 1H, J = 10.4, 4.8 Hz), 3.48 (dd, 1H, J = 11.6, 5.6 Hz, CH2OH), 1.54 (m, 2H, H3’). 13C NMR 156.7 (C6), 153.8 (C2), 148.7 (C4), 138.0 (C8), 119.1 (C5), 116.2 (d, J = 9.0 Hz, C2’), 112.2 (C1’), 85.5 (d, J = 168.6 Hz, CH2F), 62.6 (CH2OH), 29.4 (d, J = 23.1 Hz, C4’), 12.0 (d, J = 7.5 Hz, C3’). 19F NMR −216.38 (t, J = 48.2 Hz). ESI-MS 250 (100.0, M + H), 272 (13.7, M + Na). Anal. Calcd for C11H12FN5O: C, 53.01; H, 4.85; N, 28.10. Found: C, 52.99; H, 4.82; N, 27.81.
E-isomer 16a
Mp 251–253 °C. UV λmax 277 nm (ε 7,800), 262 (ε 11,000), 226 (ε 24,200). 1H NMR δ 8.49 (s, 1H, H8), 8.16 (s, 1H, H2), 7.52 (, 1H, H1’), 7.37 (bs, 2H, NH2), 5.02 (t, 1H, J = 5.6 Hz, OH), 4.47 (d, 2H, J = 47.8 Hz, CH2F), 3.54 (dd, 1H, J = 11.0, 6.2 Hz), 3.46 (dd, 1H, J = 11.2, 5.8 Hz, CH2O), 1.76 (m, 2H, H3’). 13C NMR 156.7 (C6), 153.8 (C2), 149.0 (C4), 137.9 (C8), 119.1 (C5), 117.1 (d, J = 9.0, C2’), 112.0 (C1’), 85.1 (d, J = 168.6 Hz, CH2F), 62.5 (CH2O), 27.6 (d, J = 23.1 Hz, C4’), 15.0 (d, J = 7.5 Hz, C3’). 19F NMR −215.42 (poorly resolved dt, J = 48.8, 3.0 Hz). ESI-MS 250 (100.0, M + H), 272 (29.8, M + Na). Anal. Calcd for C11H12FN5O: C, 53.01; H, 4.85; N, 28.10. Found: C, 53.25; H, 4.89; N, 28.18.
4.10. (Z,E)-2-Amino-6-chloro-9-[(2-acetoxymethyl-2-fluoromethylcyclopropylidene)- methyl]purine (28c)
A mixture of dibromide 27 (470 mg, 1.48 mmol), 2-amino-6-chloropurine (256 mg, 1.48 mmol) and K2CO3 (2.08 g, 15 mmol) in DMF (25 mL) was stirred for 5 h at 110–115 °C. After cooling, the solid portion was filtered off and it was washed with DMF (5 mL). Filtrate was concentrated in vacuo and the residue was chromatographed on a silica gel column using CH2Cl2 - methanol (200 : 1) to give product 28c (220 mg, 46%) as a white solid. The Z/E ratio was 1 : 1 as determined by 1H NMR, mp 153–170 °C. UV λmax 311 nm (ε 7,900), 231 (ε 29,900). 1H NMR δ 8.45, 8.25 (2s, 1H, H8), 7.42 (d, J = 2.4 Hz), 7.34 (bs, 1H, H1’), 7.06, 7.05 (2bs, 2H, NH2), 4.76 – 4.33 (cluster of m, 4H, CH2F, CH2OAc), 2.05, 1.89 (2s, 3H, CH3), 1.94 (poorly resolved t), 1.70 (bs, 2H, H3’). 13C NMR 171.0, 170.7 (C=O), 160.8 (C6), 153.3, 153.2 (C2), 150.4 (C4), 140.5, 140.3 (C8), 123.78, 123.75 (C5), 116.7 (d, J = 8.2 Hz), 116.6 (d, J = 9.7 Hz, C2’), 112.8, 112.6 (C1’), 86.2 (d, J = 168.6 Hz), 85.5 (d, J = 170.1 Hz, CH2F), 65.7, 65.2 (CH2OAc), 26.6, 24.7 (2d, J = 23.1 Hz, C4’), 21.4, 21.1 (CH3), 16.1, 12.9 (2d, J = 6.7 Hz, C3’). 19F NMR −214.99 (2 overlapped dt). ESI-MS 191 (100.0), 326, 328 (6.5, 2.0, M + H), 348, 350 (5.9, 2.0, M + Na). Anal. Calcd for C13H13 ClFN5O2: C, 47.94; H, 4.02: N. 21.50. Found: C, 47.93; H, 4.08; N, 21.23.
4.11. (Z)-2-Amino-6-chloro-9-[(2-hydroxymethyl-2-fluoromethylcyclopropylidene)methyl]- purine (15c) and (E)-2-Amino-6-chloro-9-[(2-hydroxymethyl-2-fluoromethylcyclopropylidene) methyl]purine (16c)
A mixture of compound 28c (210 mg, 0.65 mmol) and K2CO3 (178 mg, 1.30 mmol) in methanol - water (9 : 1, 30 mL) was stirred for 1 h at 0 °C. The solvent was evaporated and the residue was chromatographed on a silica gel column using CH2Cl2 - methanol (100 : 3) to give the Z-isomer 15c (85 mg, 46%), followed by E-isomer 16c (100 mg, 54%).
Z-isomer 15c
Mp 206–208 °C. UV λmax 310 nm (ε 7,000), 232 (ε 28,600). 1H NMR δ 8.53 (s, 1H, H8), 7.26 (s, 1H, H1’), 7.04 (2H, bs, NH2), 5.33 (t, 1H, J = 5.2 Hz, OH), 4.69, 4.53 and 4.57, 4.41 (2AB, 2H, JH,F = 48.1 Hz, JAB = 9.8 Hz, CH2F), 3.80 (dd, 1H, J = 11.2, 4.8 Hz), 3.44 (dd, 1H, J = 11.2, 5.6 Hz, CH2O), 1.54 (m, 2H, H3’). 13C NMR 160.8 (C6), 153.1 (C2), 150.4 (C4), 140.1 (C8), 123.7 (C5), 117.1 (d, J = 8.2 Hz, C2’), 111.7 (C1’), 85.4 (d, J = 167.9 Hz, CH2F), 62.6 (CH2OH), 29.4 (d, J = 23.1 Hz, C4’), 12.1 (d, J = 6.7 Hz, C3’). 19F NMR −216.29 (t, J = 48.0 Hz). ESI-MS (MeOH + KOAc) 123 (100.0), 284, 286 (M + H, 11.0, 2.7), 322, 324 (18.2, 6.9, M + K) 605, 607 (14.9, 11.0, 2M + K). Anal. Calcd for C11H11ClFN5O: C, 46.57; H, 3.91; N, 24.69. Found: C, 46.54; H, 3.91; N, 24.45.
E-isomer 16c
Mp 216 °C (decomp). UV λmax 311 nm (ε6,700), 232 (ε 27,000). 1H NMR δ 8.45 (s, 1H, H8), 7.36 (poorly resolved d, 1H, H1’), 7.04 (2H, bs, NH2), 5.04 (poorly resolved t, 1H, OH), 4.52, 4.39 (2 poorly resolved ddd, 2H, JH,F = 48.6 Hz, CH2F), 3.54 – 3.52, 3.46 – 3.44, (2m, 2H, CH2O), 1.76 (m, 2H, H3’). 13C NMR 160.8 (C6), 153.3 (C2), 150.4 (C4), 140.2 (C8), 123.7 (C5), 118.1 (d, J = 9.7 Hz, C2’), 111.6 (C1’), 85.0 (d, J = 168.6 Hz, CH2F), 62.4 (CH2OH), 27.8 (d, J = 23.9 Hz, C4’), 15.1 (d, J = 6.7 Hz, C3’). 19F NMR −215.47 (t, J = 48.2 Hz). ESI-MS (MeOH + KOAc) 123 (100.0), 322, 324 (22.9, 8.0, M + K), 605, 607 (6.0, 4.8, 2M + K). Anal. 15 Calcd for C11H11ClFN5O × 0.5H2O: C, 45.13; H, 4.13; N, 23.93. Found: C, 45.28; H, 4.14; N, 23.57.
4.12. (Z)-9-[(2-hydroxymethyl-2-fluoromethylcyclopropylidene)methyl]guanine (15b)
A solution of the Z-isomer 15c (85 mg, 0.3 mmol) in formic acid (80%, 15 mL) was heated at 80 °C for 4 h. The solvent was removed and the crude product was dissolved in methanolic NH3 (20%, 30 mL) at 0 °C with stirring which was continued for 5 h. The volatile components were evaporated and methanol was evaporated from the residue (3 times). The resultant solid was washed with methanol (5 mL) to give product 15b (67 mg, 84%) as a white solid, mp >280 °C. UV λmax 273 nm (ε 10,200), 230 (ε 25,200). 1H NMR δ 10.68 (s, 1H, CONH), 8.15 (s, 1H, H8), 7.16 (s, 1H, H1’), 6.54 (bs, 2H, NH2), 5.31 (t, 1H, J = 5.0 Hz, OH), 4.64 – 4.42 (2 overlapped AB, CH2F), 3.74 (poorly resolved dd, 1H), 3.46 (dd, 2H, J = 11.2, 4.8 Hz, CH2O), 1.49 (m, 2H, H3’). 13C NMR 157.3 (C6), 154.7 (C2), 150.4 (C4), 134.5 (C8), 116.9 (C5), 115.7 (d, J = 7.1 Hz, C2’), 112.0 (C1’), 85.3 (d, J = 168.6 Hz, CH2F), 62.4 (CH2O), 29.2 (d, J = 23.1 Hz, C4’), 11.9 (d, J = 6.7 Hz, C3’). 19F NMR −216.48 (t, J = 47.9 Hz). ESI-MS 266 (100.0, M + H), 288 (48.2, M + Na). Anal. Calcd for C11H12FN5O2 × 0.2 H2O: C, 49.14; H, 4.65; N, 26.04. Found: C, 49.13; H, 4.54; N, 25.83.
4.13. (E)-9-[(2-hydroxymethyl-2-fluoromethylcyclopropylidene)methyl]guanine (16b)
The procedure described for the Z-isomer 15b was followed with E-isomer 16c (128 mg, 0.45 mmol). The product was recrystallized from methanol (20 mL) to give compound 16b (109 mg, 91%) as a white solid, mp >300 °C. UV λmax 272 nm (ε 10,200), 229 (ε 26,700). 1H NMR δ 10.70 (s, 1H, CONH), 8.04 (s, 1H, H8), 7.26 (s, 1H, H1’), 6.53 (bs, 2H, NH2), 5.01 (t, 1H, J = 5.8 Hz, OH), 4.52, 4.47 and 4.40, 4.35 (2AB, 2H, JH,F = 48.1 Hz, JAB = 9.8, 8.8 Hz, CH2F), 3.51, 3.43 (2dd, 2H, J = 11.0, 5.6 Hz, CH2O), 1.71 (m, 2H, H3’). 13C NMR 157.4 (C6), 154.6 (C2), 16 150.6 C4), 134.4 (C8), 117.0 (C5), 116.7 (d, J = 8.9 Hz, C2’), 111.9 (C1’), 85.1 (d, J = 167.6 Hz, CH2F), 62.4 (CH2OH), 27.5 (d, J = 23.0 Hz, C4’), 14.8 (d, J = 6.7 Hz, C3’). 19F NMR −215.38 (t, J = 48.9 Hz). ESI-MS 266 (100.0, M + H), 288 (73.2, M + Na). Anal. Calcd for C11H12N5O2F × 0.2 H2O: C, 49.14; H, 4,65; N, 26.04. Found: C, 49.35; H, 4.57; N, 25.96.
4.14. Adenosine deaminase (ADA) Assay7
The Z- and E-isomers 15a and 16a (4.2 – 4.4 µmol) were incubated with ADA from calf intestine (Worthington, Lakewood, New Jersey, USA, 1.5 unit/mL) in 0.05 M Na2HPO4 (pH 7.5, 1.2 mL) with magnetic stirring at room temperature. Aliquots were withdrawn, they were diluted with the buffer (0.2 mL/10 mL) and the UV spectra were recorded. The UV maximum of 16a at 260 nm completely disappeared after 28 h, whereas the spectrum of 15a was unchanged (UVmax 260 nm).
4.15. Antiviral Assays
The antiviral assays, with the exception of EBV and HCV, were described previously.7
4.15.1. EBV DNA Hybridization Assay.22
Akata cells were maintained in RPMI 1640, (Mediatech, Inc, Herndon, Va.) supplemented with 10% fetal bovine serum (Hyclone, Logan, Utah), L-glutamine, penicillin and gentamicin at 37 °C in a humidified 5% CO2 atmosphere. Latently infected cells were induced to undergo a lytic infection by adding a F(ab′)2 fragment of goat anti-human IgG antibody (MP Biomedicals, Aurora, OH). Total DNA from the cells was purified and genome copy number was quantified by Real-Time PCR. The primers 5’-CGG AAG CCC TCT GGA CTT C-3’ and 5’-CCC TGT TTA TCC GAT GGA ATG-3’ were used with the fluorescent probe, 6FAM-TGT ACA CGC ACG AGA AAT GCG CC-TAMRA corresponding to coordinates 155959-155981 in the EBV genome (Applied Biosystems). The PCR was performed in an optical 96 well plate using an ABI 7300 Real-Time PCR system. The PCR reaction contained 900 nM primers, 200 nM probe, 12.5 µL Taqman Universal Master Mix (Applied Biosystems, Foster City, CA), and 5 µL target DNA in a final volume of 25 µL. Each sample was run in duplicate and EC50 values were calculated by standard methods.
4.15.2. HCV Studies
Antiviral activity of test compounds was assessed in the stably-expressing HCV replicon cell line, AVA5 (subgenomic CON1, genotype 1b)23 maintained at sub-confluent cultures on 96-well plates as previously described.24 Antiviral activity was determined by blot hybridization analysis of intracellular HCV RNA and cytotoxicity was assessed by neutral red red dye uptake after 3 days of treatment. Compounds were added each day in fresh medium. Intracellular RNA levels and cytotoxicity were assessed 24 h after the last dose of compound.
Scheme 1.
Reagents: a. DIBALH, CH2Cl2, −78 °C, b. 1. 37% CH2O, MeOH, 2. 1M HCI, Δ, c. 1. MeC(OMe)3, cat. TsOH, CH2CI2. 2. NEt3. 3. 80% AcOH, d. DAST, pyridine, CH2CI2. −78 °C → rt. E. MsCI, NEt3, CH2CI2. f. Bu4NF, THF. g. Pyridine, HBr3, CH2CI2, 0 °C. h. B-H, K2CO3, DMF, Δ. i. K2CO3, MeOH - H2O (9 : 1), rt or 0 °C. j. 1. 80% HCO2, Δ. 2. NH3, MeOH, 0 °C.
Acknowledgments
We thank L. M. Hryhorczuk from the Central Instrumentation Facility of the Department of Chemistry, Wayne State University for mass spectra. The work described herein was supported by grant RO1-CA32779 from the National Cancer Institute and contract NO1-AI-30049 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland, USA. We also thank a reviewer for reference 18.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zemlicka J. In: Recent Advances in Nucleosides: Chemistry and Chemotherapy. Chu CK, editor. Amsterdam: Elsevier; 2002. pp. 327–357. [Google Scholar]
- 2.Zemlicka J, Chen X. In: Frontiers in Nucleosides and Nucleic Acids. Schinazi RF, Liotta DC, editors. Tucker, Georgia: IHL Press; 2004. pp. 267–307. [Google Scholar]
- 3.Zemlicka J. In: Advances in Antiviral Drug Design. De Clercq E, editor. Vol. 5. Amsterdam, The Netherlands: Elsevier; 2007. pp. 113–165. [Google Scholar]
- 4.Zhou S, Breitenbach JM, Borysko KZ, Drach JC, Kern ER, Gullen E, Cheng Y-C. J. Med. Chem. 2004;47:566. doi: 10.1021/jm030316s. [DOI] [PubMed] [Google Scholar]
- 5.Kern ER, Bidanset DJ, Hartline CB, Yan Z, Zemlicka J, Quenelle DC. Antimicrob. Agents Chemother. 2004;48:4745. doi: 10.1128/AAC.48.12.4745-4753.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kern ER, Kushner NL, Hartline CB, Williams-Azziz SL, Harden EA, Zhou S, Zemlicka J, Prichard MN. Antimicrob. Agents Chemother. 2005;49:1039. doi: 10.1128/AAC.49.3.1039-1045.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou S, Kern ER, Gullen E, Cheng Y-C, Drach JC, Matsumi S, Mitsuya H, Zemlicka J. J. Med. Chem. 2004;47:6964. doi: 10.1021/jm040093l. [DOI] [PubMed] [Google Scholar]
- 8.Zhou S, Kern ER, Gullen E, Cheng Y-C, Drach JC, Tamiya S, Mitsuya H, Zemlicka J. J. Med. Chem. 2006;49:6120. doi: 10.1021/jm0607404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou S, Zemlicka J, Kern ER, Drach JC. Nucleosides, Nucleotides & Nucleic Acids. 2007;26:231. doi: 10.1080/15257770701257210. [DOI] [PubMed] [Google Scholar]
- 10.Welch JT, Eswarakrishnan S. Fluorine in Bioorganic Chemistry. New York: John Wiley & Sons; 1991. p. 2. [Google Scholar]
- 11.Martin JC, McGee DPC, Jeffrey GA, Hobbs DW, Smee DF, Matthews TR, Verheyden JPH. J. Med. Chem. 1986;29:1384. doi: 10.1021/jm00158a011. [DOI] [PubMed] [Google Scholar]
- 12.Guan H-P, Ksebati MB, Cheng Y-C, Drach JC, Kern ER, Zemlicka J. J. Org. Chem. 2000;65:1280. doi: 10.1021/jo991030r. [DOI] [PubMed] [Google Scholar]
- 13.Yan Z, Kern ER, Gullen E, Cheng Y-C, Drach JC, Zemlicka J. J. Med. Chem. 2005;48:91. doi: 10.1021/jm040149b. [DOI] [PubMed] [Google Scholar]
- 14.Tiruchinapally G, Zemlicka J. Synth. Commun. in press. [Google Scholar]
- 15.Middleton WJ. J. Org. Chem. 1975;40:574. [Google Scholar]
- 16.Hudlicky M. Org. React. 1988;35:513. [Google Scholar]
- 17.Adcock JL, Gakh AA. J. Org. Chem. 1992;57:6206. [Google Scholar]
- 18.Dechamps I, Pardo DG, Cossy J. Eur. J. Org. Chem. 2007:4224. [Google Scholar]
- 19.Smith MB, March J. March’s Advanced Organic Chemistry. New Jersey: Wiley, Hoboken; 2007. pp. 450–468. [Google Scholar]
- 20.Bottini AT, Christensen JE. Tetrahedron. 1974;30:393. [Google Scholar]
- 21.Nishimura A, Kato H, Ohta M. J. Am. Chem. Soc. 1967;89:5083. [Google Scholar]
- 22.Prichard MN, Daily SL, Jefferson GL, Perry AL, Kern ER. J. Virol. Methods. 2007;144:86. doi: 10.1016/j.jviromet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blight KJ, Kolykhalov AA, Rice CM. Science. 2000;290:1972. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 24.Okuse C, Rinaudo JA, Farrar K, Wells F, Korba BE. Antiviral Res. 2005;65:23. doi: 10.1016/j.antiviral.2004.09.002. [DOI] [PubMed] [Google Scholar]