

Abstract
The spread of bacteria resistant to antimicrobial agents calls for population-wide treatment strategies to delay or reverse the trend toward antibiotic resistance. Here we propose new criteria for the evaluation of the population-wide effects of treatment protocols for directly transmitted bacterial infections and discuss different usage patterns for single and multiple antibiotic therapy. A mathematical model suggests that the long-term benefit of single drug treatment from introduction of the antibiotic until a high frequency of resistance precludes its use is almost independent of the pattern of antibiotic use. When more than one antibiotic is employed, sequential use of different antibiotics in the population (“cycling”) is always inferior to treatment strategies where, at any given time, equal fractions of the population receive different antibiotics. However, treatment of all patients with a combination of antibiotics is in most cases the optimal treatment strategy.
The appearance and spread of antibiotic resistance is becoming an increasingly serious public health problem. Antibiotic resistance has become clinically important in such community-acquired organisms as Streptococcus pneumoniae (1), Neisseria gonorrhoeae (2), and Mycobacterium tuberculosis (3) and in nosocomial pathogens including Staphylococcus aureus (4), Enterococcus spp. (4, 5), and Klebsiella spp. (6). These resistant organisms not only compromise the success and increase the cost of treating individual patients, but can also be transmitted to other hosts, resulting in the epidemic spread of antibiotic-resistant infections.
In response to the spread of resistant bacteria from patient to patient, a number of measures have been proposed and tried, with varying success. These include improvement of hospital hygiene (7), the use of vaccines (8), controls on or reductions of antibiotic use (9), and cycling of different antibiotics (4, 10). The general goal of these interventions is to reduce the incidence of resistant infections, and thereby to prolong or restore the effectiveness of existing antibiotics. Mathematical models have been used to evaluate the competition between sensitive and resistant bacteria (11) and the community-wide effects of treating tuberculosis under various assumptions about treatment success rates (12). Thus far, however, there are (to our knowledge) no quantitative models that evaluate the population wide effects of different patterns of antibiotic use on the number of infections that occur during the useful “lifetime” of one or more antibiotics.
Here we present and analyze a series of mathematical models to generate predictions concerning the effects of various patterns of drug treatment at the population level. Two models are considered. First, we consider treatment with a single drug and resistance to that drug and analyze the model to predict the consequences of different usage patterns. The second model analyzes the population-level consequences of different usage patterns of the two drugs. The goal of analyzing such models is to understand how antibiotic usage patterns may be optimized to preserve or restore antibiotic effectiveness.
Evaluation of Antibiotic Policies
Before proceeding, it is necessary to define precisely what would constitute an optimal antibiotic policy. One criterion might be to choose a policy that maximizes the time before resistant bacteria constitute some fixed fraction of all bacteria of a given species. Clearly this criterion by itself is useless, as it would be best achieved by never using the antibiotic. Therefore, although the rate of ascent of resistance to a drug is of interest, we would like a criterion of optimality that balances the value of preserving a drug’s effectiveness with the value of treating patients successfully with the drug. Thus, another measure of the efficacy of an antibiotic use policy is the extent to which it increases the total number of uninfected hosts and/or reduces the total number of infected hosts over a defined period. Mathematically, this means that the optimality criterion for a treatment policy is to maximize the number of uninfected hosts, integrated over time, or to minimize the corresponding integral for infected hosts. This criterion gives equal weight to infections prevented or cured in the short term due to use of the drug, as well as to infections prevented or cured in the long term due to the preservation of the drug’s effectiveness. In our analysis of these models, we consider all three of these criteria: time until resistance reaches a particular fraction of the bacterial population, number of hosts infected with the bacterium, and number of uninfected hosts.
Single Antibiotic Therapy
We first consider a simple compartment model in which patients with bacterial infections may be treated with a single antibiotic. The model is depicted in Fig. 1A. Uninfected hosts, of density x, enter the population at a rate λ and are removed from the population (die) at a per capita rate d. They can be infected by bacteria that are either sensitive (wt) or resistant (res) to the treating antibiotic (or drug for short). The densities of wt- and res-infected patients are yw and yr, respectively. Uninfected hosts become infected at a rate proportional to their density, x, the density of infecteds, yw + yr, and a transmission rate parameter b. This reflects direct, contact-dependent transmission of infectious agents from diseased hosts to uninfected hosts. Infected hosts die at rate c, which includes natural and disease-associated mortality. We assume that in the absence of treatment patients infected with wt and res bacteria recover from infection at rates rw and rr, respectively. Once an infection is cleared, the surviving patient immediately returns to the susceptible subpopulation. Patients infected with wt-bacteria are removed from the wt-infected compartment at a rate fh, where f is a scaling parameter (between 0 and 1) reflecting the fraction of patients treated and h is the maximum rate when all patients are treated. A fraction s of treated wt-infecteds develop resistance during treatment. Such resistance is referred to as “acquired” or “de novo” resistance, in contrast to “primary” resistance (infection by a resistant organism). The remainder recover and become susceptible again. In terms of ordinary differential equations the model is
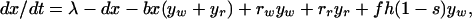 |
1 |
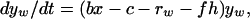 |
2 |
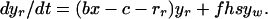 |
3 |
Figure 1.

(A) Graphical illustration of the single antibiotic treatment model. The variables and parameters are explained in the main text. (B) Multiple antibiotic treatment model: The variables are x for the susceptibles, and yw, ya, ya, and yab for patients infected with wild-type (wt), A-res, B-res, and AB-res bacteria. The model is dx/dt = λ − dx − b(yw + ya + yb + yab)x + rwyw + raya + rbyb + rabyab + h(1 − q)fabyw + h(1 − s)((fa + fb)yw + fayb + fbya + fab(ya + yb)); dyw/dt = (bx − c − rw − h(fa + fb + fab))yw; dya/dt = (bx − c − ra − h(fb + fab))ya + hsfayw; dyb/dt = (bx − c − rb − h(fa + fab))yb + hsfbyw; dyab/dt = (bx − c − rab)yab + hs(fab(ya + yb) + fayb + fbya) + qhfabyw. The parameters are rw, ra, rb, and rab, for the recovery rates of wt, A-res, B-res and AB-res infecteds, respectively; fa, fb, and fab, for the fraction of patients treated with antibiotic A, B, or AB; and s and q are the fractions of hosts that become resistant when treated with a single drug or both drugs simultaneously. The parameters fa, fb, and fab reflect the fraction of patients treated with antibiotics, A, B, and AB. (Note that the parameters fa, fb, fab, s, q represent fractions and are therefore restricted to be between 0 and 1. The parameters fa, fb, and fab additionally must fulfill fa + fb + fab ≤ 1. If their sum is smaller than 1, than this reflects that some fraction of the patients are not treated at all.) The equilibrium levels of susceptibles and infecteds are derived in Appendix B1.
Assumptions
This model makes several assumptions. (i) The fitness cost associated with resistance is manifest by a higher rate of clearance of the infection (recovery) of hosts infected with resistant bacteria relative to those infected with sensitive (rr > rw). In fact, this cost of resistance could also be manifest in a lower rate of transmission b, and the conclusions about optimal treatment protocols would not be altered. (ii) We assume that patients who are treated and cured become immediately susceptible again. We thus neglect temporary or life-long immunity. We have checked, however, that incorporating a class of immune hosts we can derive results that are completely analogous to those presented in this paper. (iii) We assume that in a fraction, s, of wt-infected patients, there is a pre-existing, small subpopulation of resistant bacteria. When these patients receive therapy, the resistant population will grow and will quickly dominate the infection. We define this process as “acquired resistance” and assume that acquisition of resistance happens only in treated hosts. (iv) The model does not include superinfection of wt-infecteds by resistant bacteria. The effect of superinfection can be safely neglected as long as the frequency of infecteds is low.
Long-Term Consequences of Treatment
There are two different long-term outcomes depending on the efficacy of the antibiotic policy (see Appendix A1). If the selection pressure imposed by the antibiotic on sensitive infections, fh, outweighs the cost of resistance, Δr = rr − rw, then resistant infections will prevail in the long term and sensitive infections will disappear. Otherwise sensitive infections will prevail, but resistant infections will coexist at low levels. In the following we will only consider cases where the selection pressure exerted by the antibiotic treatment protocol is strong enough that there is a net selection advantage for the resistant bacteria in the presence of treatment.
Dynamics of Resistance
Resistant organisms will account for a fraction σ of all infections at a time given by: Tσ = (1/(fh − Δr))ln[1 + (σ/(1 − σ))((fh − Δr)/fhs)] (see Appendix A2). Faster rates of antibiotic treatment and cure (fh) accelerate the emergence of resistance in the population. Tσ depends inversely on the net advantage of resistant bacteria in the presence of treatment, fh − Δr, and logarithmically on the fraction, s, of patients that acquire resistance when treated. Therefore small changes in f or h may have a strong effect on Tσ, whereas even large changes in s will only weakly affect Tσ.
The parameter s determines the dynamics of the initial appearance of resistant infections. Soon after the start of therapy, however, the majority of res-infecteds are due to epidemic transmission rather than acquired resistance, and the subsequent rise of resistance in the population is caused by an epidemic of resistant bacteria. Once most resistant cases are due to epidemic transmission, the time necessary for res-infecteds to increase from a fraction ρ0 to a fraction ρe is given by Ti = (1/(fh − Δr))ln[((1 − ρ0)/ρ0)(ρe/1( − ρe))] (see Appendices A3 and A4). When treatment is withdrawn (f = 0), the reversal time until the res-infecteds have decreased from a fraction ρe to a fraction ρ0 is given by Td = (1/Δr)ln[((1 − ρ0)/ρ0)(ρe/(1 − ρe))]. Hence, the ratio of the times necessary to decrease from ρe to ρ0 and to increase from ρ0 to ρe is Td/Ti = ((fh − Δr)/Δr). Because the fitness difference, Δr, between wt and res bacteria in absence of treatment is typically much smaller than the difference, fh − Δr, in the presence of treatment, this ratio will be much larger than one. Thus, the emergence of resistance in response to treatment is usually much faster than the reversion to wt when treatment is withdrawn. A similar result has been obtained for pesticide resistance (13).
Benefit of Treatment
Fig. 2 shows a simulation of the single antibiotic therapy model. The shaded area reflects the total gain of uninfecteds that is achieved by treatment before the antibiotic therapy fails due to resistance. The question is, what treatment policy maximizes the total gain of uninfecteds?
Figure 2.

Emergence of antibiotic resistance in the patient population. The shaded area reflects the total gain of uninfecteds during treatment. If there is no cost of resistance (Δr = rr − rw = 0), then the total gain of uninfecteds is independent of the rate fh at which patients are treated and cured. Hence all treatment protocols result in the same total gain of uninfecteds. If there is a cost of resistance (Δr > 0), then the total gain is maximized if a maximal fraction of patients receive therapy. The parameters of the simulation are given in Appendix C.
The total gain of uninfecteds is given by G = (1/b)[−ln(s) +ΔrT + ln(γ) + ln(1 − s − (Δr/fh))], where T is the time past the start of therapy and γ is the ratio between the density of infecteds at time T and their density at equilibrium before therapy (see Appendix A3).
If there is no significant cost to resistance (i.e., Δr ≈ 0), then the total gain of uninfecteds until resistant infections dominate in the patient population is G = −ln(s)/b. The gain is independent of the rate at which patients are treated and cured, fh, and therefore independent of the treatment policy. Hence, whether a small or large fraction of patients is treated, whether treatment of the population is continuous or intermittent, in the absence of a significant cost to resistance all treatment policies eventually result in the same total gain of uninfecteds.
If, on the other hand, there is a fitness cost to resistance (Δr > 0), then increasing the rate of treatment and cure (fh) increases the gain of uninfected hosts. However, this gain is negligible unless the drug induced selection pressure against wt only just compensates the cost of resistance (i.e., Δr ≈ fh).
Imagine that the rise of resistant infections has progressed to a point where the contribution of acquired resistance to the overall prevalence of resistance is negligible compared to the epidemic spread of resistance. From this time onwards, the total gain of uninfecteds is given by G = (1/b)[ln(γ/ρ0) + ΔrT], where ρ0 is the initial fraction of resistant infections before treatment (see Appendix A4). The total gain is independent of fh, and hence, even in the presence of a cost to resistance, the total benefit of treatment is independent of the treatment protocol, once most resistant infections arise from primary resistance (infection with a resistant organism). Put another way, the effect of treatment patterns on the total gain of resistance is limited to the period during which de novo acquisition of resistance is numerically important by comparison to the spread of resistant infections.
All these results were derived by assessing the benefit of a treatment schedule in terms of the total gain of uninfecteds. However, all results can be obtained in complete analogy by measuring the benefit of treatment in terms of the total reduction of infected hosts (see Appendix A3).
Multiple antibiotics
Let us consider treatment strategies using several antibiotics. In the following we assume that we have two antibiotics, A and B, but the results can easily be generalized for more than two antibiotics. Fig. 1B shows a natural extension of the single antibiotic therapy model to incorporate two antibiotics and resistance to either or both antibiotics. The question now is, what is the optimal treatment policy using more than one antibiotic.
We distinguish three scenarios: case I, the majority of resistant infections are caused by transmission of resistant organisms; case II, the majority of resistant infections initially result from acquired resistance, and only later does transmission of these organisms become important; and case III, multiple resistance is initially absent, and its appearance has a very low probability. For these three scenarios we compare three treatment protocols: (i) drugs are cycled periodically (cycling treatment); (ii) equal proportions of the infected host population receive each drug (50-50 treatment); and (iii) drugs are given simultaneously to each infected host (combination treatment).
For the sake of simplicity we generally assume in the following that the cost of single drug resistance is the same for drug A and B (i.e., ra = rb). If, however, the costs of resistance are different for drug A and B, then the drug for which resistance is more costly should be used more to equalize the selection pressure on both resistant types (see Appendix B3 for the relationship between cost and use of drug).
Case I: Infectious Transmission of Resistance.
Suppose that before population-wide treatment is started a sufficiently large fraction of patients have resistant bacteria, such that infectious transmission of resistance results in many more resistant cases than resistance acquired during treatment. (Mathematically this amounts to q, s ≈ 0 and ya, yb, yab > 0 at the start of therapy in the model of Fig. 1B.) Under these conditions, all treatment strategies will eventually result in the same benefit as measured by the total gain of uninfected hosts or the total reduction of infected hosts (see Figs. 3 A–C). This case is completely analogous to the case of single antibiotic therapy (Appendix A4). The total gain of uninfecteds is given by G = (1/b)[ln(γ/ρ0) + ΔrT), where ρ0 is the initial fraction of resistants before the start of therapy and Δr = rab − rw. Hence, as long as the initial incidence of primary resistance is considerably greater than the incidence of acquired resistance, the long-run benefit of treatment is independent of the treatment protocol.
Figure 3.

Multiple antibiotic treatment policies. The solid, dotted, and dashed lines show the densities of uninfecteds, wt-infecteds and AB-res infecteds, respectively. The dot-dashed lines show the densities of single resistants. The shaded area reflects the total gain of uninfecteds, G. G1/2 is the total gain of uninfecteds before 50% of the infecteds are AB-res. T1/2 is the time necessary until 50% of the infecteds are AB-res. Three treatment strategies (cycling where drugs are alternated every 5 time units, 50-50 treatment, and combination treatment) are compared. In A–C we assume that the prevalence or resistance has progressed to a point where the contribution of acquired resistance is negligible to primary resistance (infection by a resistant organism). In D–G we assume that resistant infections are initially rare, such that the contribution of acquired resistance is initially numerically important by comparison to the epidemic spread of resistance. In H–J we assume that multiple resistance is not initially present and is not generated during treatment. Measured in terms of the total gain of uninfecteds (or total reduction of infecteds) cycling is always worse that 50-50 treatment and combination therapy is superior to cycling and 50-50 treatment except when q > s2. Note, however, that T1/2 is shorter for combination therapy.
Case II: Acquired Resistance.
We now consider the case where resistant infections are rare before the start of population-wide therapy and initially most cases of treatment failure are due to acquired resistance. If there is no cost to resistance (rw = ra = rb = rab) then cycling and 50-50 treatment result in the same total gain of uninfecteds (see Appendix B2). However, if there is a cost to resistance (rab > ra = rb > rw), then 50-50 treatment is marginally superior to cycling drugs, both with respect to the gain of uninfecteds within a given time span and with respect to the gain of uninfecteds before a given fraction of patients is AB-res (see Fig. 3 D and E). Numerical simulations show that 50-50 treatment is superior to cycling regardless of how frequently the drugs are cycled.
The intuitive reason why cycling is worse than 50-50 treatment is that the optimal policy is to treat at any time point with that antibiotic for which there is least resistance in the patient population. Imagine we start with antibiotic A. Treating with A increases the proportion of the population resistant to A. Eventually, the frequency of resistance to A would exceed that to B and, hence, it would be better to use B. But doing so would increase resistance to B, and therefore force a switch back to A. At this point the optimal policy is to switch back and forth each time a new patient is treated. This is equivalent to the 50-50 policy.
Fig. 3 E–G compares 50-50 treatment with combination therapy. If there is no cost to resistance, then the total gain of uninfecteds is G = −(1/b)ln(s2) for 50-50 treatment and G = −(1/b)ln(q) for combination treatment, where s and q are the fraction of patients that acquire resistance in response to single and combination treatment (see Appendix B2). Thus, the relationship between q and s2 determines whether 50-50 treatment or combination treatment is superior.
If q < s2 then combination therapy is superior to 50-50 treatment with respect to both the total gain of uninfecteds and the time taken until given percentage of the infections are resistant to both antibiotics (see Fig. 3 E and F).
If q > s2, then combination treatment accelerates the emergence of resistance and reduces the total gain of uninfecteds, compared to 50-50 treatment (see Fig. 3 E and G).
For mutation-borne resistance, we would expect q ≪ s2 for the following reason. Assume there are N bacteria within a host and assume that on average a fraction p of these carry mutations to resist drug A and a fraction p carry resistance to B. Assuming the resistance mutations are independent, a fraction of about p2 should carry both mutations. The probability s that single-drug treatment will cause the emergence of resistance is approximately min{1, Np}. By the same reasoning, the probability q that double resistance will emerge is approximately min{1, Np2}. Because initially an infection is presumably clonal, sensitive and resistant bacteria are likely not to be in a selection-mutation equilibrium. Therefore p will be approximately given by the mutation rate p = 10−10–10−7. Biologically realistic values for N are ≤1011. Hence, Np2 will typically be much smaller than one and therefore q ≪ s2. If, however, sensitive and resistant bacteria are in mutation-selection equilibrium then the estimate for p might be considerably higher (14–16) and consequently the merit of combination therapy relative to 50-50 therapy smaller.
This argument may not hold for cases where resistance is acquired by horizontal transfer of accessory elements (e.g., plasmids). Under these conditions, the simultaneous acquisition of multiple resistance determinants is very common (17). This could increase q considerably; if the resistance determinants for both antibiotics are on the same plasmid, then the probabilities of acquiring single and multiple resistance would be approximately equal (i.e., s = q). On the other hand, if both resistance determinants do not occur on the same plasmid, then the chances of the same bacterium acquiring both resistance determinants may be vanishingly small: q ≪ s2. Thus it is difficult to predict the relative likelihoods of the acquisition of single and multiple resistance when resistance is conferred by plasmids.
Case III: Multiply Resistant Bacteria Are Not Present.
Imagine that resistance to both drugs requires such a rare genetic change that it is very unlikely to be generated, even over a time span of several years. In this case minimizing the number of singly resistant cases (or maximizing the number of uninfecteds) is a reasonable criterion for a policy to forestall the advent of multiple resistance, because each new singly resistant case provides an opportunity for the emergence of double resistance. To maximize the number of uninfected hosts in the absence of doubly resistant bacteria, combination therapy is obviously superior to cycling and 50-50 treatment, because it will successfully treat all infections without giving an advantage to either singly resistant strain (see Fig. 3J). In the absence of multiple resistance, cycling is generally worse than 50-50 treatment with respect to the total gain of uninfecteds for the same reason as discussed for acquired resistance in the previous section (see Fig. 3 H and I). If there is no cost to resistance, then both strategies are equally effective.
Discussion
Our main conclusions are as follows. For directly transmitted bacterial infections, the long-term benefit of using a single drug from the time it is introduced until resistance makes it ineffective is almost independent of the pattern of use (although there is a slight increase in benefit if the drug is used heavily in the early stages). When using two agents, the simultaneous use of two drugs at the population level (but only one for each patient), which we have called 50-50 treatment, always produces at least as much, or more, benefit than a policy of cycling between two drugs. In most cases, treatment of all hosts with a combination of both drugs is better than either of these policies; the only exception is the case in which resistance to the two drugs is carried on the same plasmid.
In light of the model’s predictions, two of the diseases for which the model is most appropriate provide an interesting contrast. For treatment of tuberculosis, combination therapy is the norm (12, 18), while gonorrhea is usually treated with a single drug, which may be a member of any one of several drug classes (19, 20). Although problems of nonadherence and the rise of tuberculosis among immunocompromised persons complicate the picture, the consistent use of multidrug therapy and its general success (until recently) in stemming the spread of tuberculosis in developed countries accords with the predictions of the model. With gonorrhea, there has been considerable spread of resistance to a number of antibiotic classes, which might have been preventable with the more widespread use of combination therapy.
Another finding of the model, which agrees with the predictions of most models of resistance (13, 21), is that the spread of resistance due to drug use will be considerably faster than its decline when selection (treatment) is removed. This is because the cost of resistance in the absence of the selecting agent (antibiotic) is generally much less than the benefit of resistance in the presence of the antibiotic. Furthermore, laboratory experiments have shown that the cost of resistance is quickly reduced by compensatory mutations (22, 23). Hence, the time necessary for reversion from resistant to sensitive infections after treatment is withdrawn might be very long.
Strictly speaking, the model considered here applies directly only to those bacterial infections, such as tuberculosis, gonorrhea, and some diarrheal diseases, in which the recovery from the infection coincides with the termination of carriage and transmission of the infectious organisms. Many of the organisms causing nosocomial infections are not obligate pathogens of this kind, but are organisms that colonize the nose, nasopharynx, or gut of healthy patients and cause disease when they enter and proliferate in normally sterile sites (24). As a result, infection, colonization, and shedding (transmission) are distinct states, and treatment of an infection may or may not terminate colonization or transmission. For such organisms, a different model may be more appropriate (25). It is not straightforward whether the conclusions of this model about the general inferiority of cycling will extend to these pathogens (or to the commensal bacterial flora or sexually recombining pathogens). However, in the absence of a specific reason cycling of antibiotics should be done with caution.
The goal of this paper was to discuss and evaluate different patterns of antibiotic use in a very general fashion and thereby lay the basis for future research that addresses the specific properties of particular pathogens in greater detail. Mathematical models may provide very useful tools to develop a rationale to extend the effective life of existing and newly introduced antimicrobial agents.
Acknowledgments
S.B. is supported by the Wellcome Trust. M.L. and B.R.L. acknowledge support from National Institutes of Health Grant GM-33782 to B.R.L.
Appendix
A: Single Drug Treatment Model
A1: Equilibrium Analysis.
In absence of therapy (fh = 0), the equilibrium is given by x̂ = (c + rw)/b; ŷw = (λ/c) − (d/b) − (drw/bc); ŷr = 0. In the presence of treatment there are two stable equilibria depending on the efficacy of antibiotic therapy. If the selection pressure against the sensitive bacteria, fh, is smaller than the cost of resistance, Δr = rr − rw, then the equilibrium density of uninfecteds is given by x̃ = (c + rw + fh)/b. The total density of infecteds ỹw + ỹr = (λ/c) − (d/b) − [d(fh + rw)/bc], of which a fraction (Δr − fh)/(Δr − fh(1 − s)) is sensitive to the antibiotic. If the selection pressure exceeds the cost of resistance (i.e., fh > Δr), then the equilibrium is given by x* = (c + rr)/b; y*w = 0; y*r = (λ/c) − (d/b) − (drr/bc).
A2: Rise of Resistance and Tσ.
Substitute y for yw + yr and ρ for yr/y in Eqs. 1–3. We obtain dx/dt = λ − dx − bxy + (rw + fh(1 − s))(1 − ρ)y + rrpy; dy/dt = (bx − c −rw − (rr − rw)ρ − fh(1 −s)(1 − ρ))y; dρ/dt = (fh − Δr)ρ(1 − ρ) + fhs(1 − ρ)2, where Δr = rr − rw. The solution for the frequency of resistant infections is ρ(t) = (e(fh−Δr)t − 1)/(e(fh−Δr)t − 1 + (fh − Δr)/(fhs)). The time necessary until a fraction σ of the patients are infected with resistant virus is given by ρ(Tσ) = σ. We obtain Tσ = (1/(fh − Δr))ln[1 + (σ/(1 − σ))((fh − Δr)/fhs)]. If fh ≫ Δr and s, σ ≪ 1 we get Tσ ≈ (1/fh)ln(σ/s).
A3: Total Benefit of Treatment.
We obtain the total gain of uninfecteds (in units of time) by integrating over (1/y)(dy/dt) (see Appendix A2). Provided that the duration of treatment is sufficiently long {i.e., T ≫ (1/(fh − Δr))ln[1 + 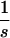 (1 − (Δr/fh))]}, we get after a series of algebraic transformations for the total gain of uninfecteds G = ∫0T (x − x̂)dt = 1/b{−ln(s) + ln[1 − s − (Δr/fh)] + ΔrT + ln(γ)}, where x̂ = (c + rw)/b is the equilibrium density of uninfecteds in absence of treatment (see equilibrium analysis) and γ is the factor of reduction in the total equilibrium density of infecteds at time T compared to baseline before therapy [i.e., γ = y(T)/y(0)]. Provided that T is sufficiently large, such that the resistants have reached equilibrium during therapy, the ratio of the densities of infecteds after a time T and at equilibrium at the start of therapy is given by y(T)/y(0) = [(λ/c) − (d/b) − (drr/bc)][(λ/c) − (d/b) − (drw/bc)]. If there is no cost to resistance (Δr = 0) then y(T) = y(0) and the total gain is of uninfecteds is given by G = (1/b)ln((1 − s)/s). We obtain the total reduction in the density of infecteds by integrating over dx/dt + dy/dt. This yields ∫0T (y − ŷ)dt = d/c ∫0T (x − x̂)dt. Therefore all results can be obtained in complete analogy for the total reduction of infecteds. There is an interesting parallel to Haldane’s (26) derivation of the cost of natural selection.
(1 − (Δr/fh))]}, we get after a series of algebraic transformations for the total gain of uninfecteds G = ∫0T (x − x̂)dt = 1/b{−ln(s) + ln[1 − s − (Δr/fh)] + ΔrT + ln(γ)}, where x̂ = (c + rw)/b is the equilibrium density of uninfecteds in absence of treatment (see equilibrium analysis) and γ is the factor of reduction in the total equilibrium density of infecteds at time T compared to baseline before therapy [i.e., γ = y(T)/y(0)]. Provided that T is sufficiently large, such that the resistants have reached equilibrium during therapy, the ratio of the densities of infecteds after a time T and at equilibrium at the start of therapy is given by y(T)/y(0) = [(λ/c) − (d/b) − (drr/bc)][(λ/c) − (d/b) − (drw/bc)]. If there is no cost to resistance (Δr = 0) then y(T) = y(0) and the total gain is of uninfecteds is given by G = (1/b)ln((1 − s)/s). We obtain the total reduction in the density of infecteds by integrating over dx/dt + dy/dt. This yields ∫0T (y − ŷ)dt = d/c ∫0T (x − x̂)dt. Therefore all results can be obtained in complete analogy for the total reduction of infecteds. There is an interesting parallel to Haldane’s (26) derivation of the cost of natural selection.
A4: Transmitted Resistance.
Suppose epidemic transmission of resistance outweighs acquisition of resistance during treatment. Hence s ≈ 0 and yr > 0 at the start of treatment. We get for the total gain of uninfecteds G = ∫0T (x − x̂)dt = 1/b[ln(γ/ρ0) + ΔrT], where γ is the factor of reduction of the equilibrium densities of infecteds before and during treatment [i.e., γ = y(T)/y(0)] and ρ0 is the fraction of resistants when therapy is started {i.e., ρ0 = [yr(0)/(yr(0) + yw(0))]}. Note that the total gain of uninfecteds is independent of the treatment strategy. For the fraction of resistants we obtain ρ(t) = ρ0/[ρ0 + (1 − ρ0)e−(fh−Δr)t], where ρ0 is the initial fraction of resistant infections before therapy is started. The time necessary until a fraction ρe of the patients are resistant is given by Ti = (1/(fh − Δr))ln[(1 − ρ0)/ρ0)(ρe/(1 − ρe))]. After treatment is withdrawn (f = 0) the time necessary to decrease resistance from a fraction ρe to a fraction ρ0 is Td = (1/Δr)ln[((1 − ρ0)/ρ0)(ρe/(1 − ρe))].
B: Multiple Antibiotic Treatment Model
B1: Equilibrium Analysis.
The multiple antibiotic therapy model is described in Fig. 1B. In absence of treatment (fa = fb = fab = 0) the system converges to a stable equilibrium given by x̂ = (c + rw)/b; ŷw = (λ/c) − (d/b) − (drw/bc); ŷa = ŷb − ŷab = 0. In presence of sufficiently strong treatment [i.e., b/(c +rab) larger than b/(c + rw + h(fa + fb + fab)), b/(c + ra + h(fb + fab)), or b/(c + rb + h(fa + fab))] the system converges to x̂ = (c + rab)/b; ŷw = ŷa = ŷb = 0; ŷab = (λ/c) − (d/b) − (drab/bc).
B2: Total Benefit and Tσ.
Assume that there is no cost to resistance (rw = ra = rb = rab). Substitute y for yw + ya + yb + yab, φ for (ya + yb)/y, and ρ for yab/y. For combination therapy (fab = f, fa = fb = 0) we get dρ/dt = fh(q + (1 − q)ρ)(1 − ρ), which is solved by ρ(t) = q(efht − 1)/(q(efht − 1) + 1). Hence, the time until a fraction σ of the infecteds are multiply resistant is Tσ = (1/fh)ln[(1/q)(σ/(1 − σ)) + 1]. If σ, q ≪ 1 then Tσ ≈ (1/fh)ln(σ/q). For 50-50 therapy (fa = fb = f/2, fab = 0) we get dφ/dt = fh[−φ/2 + s(1 − φ − ρ) + (1 − s)φ(1 − φ/2 − ρ)] and dρ/dt = fh[sφ/2 + (1 − s)ρ(1 − φ/2 − ρ)]. The solutions are φ(t) = 2s(efht/2 − 1)/[1 + s(efht/2 − 1)]2 and ρ(t) = [s(efht/2 − 1)/(1 + s(efht/2 − 1))]2. Thus Tσ = (2/fh)ln[(1/s)(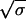 /(1 −
/(1 − 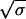 )) + 1]. By integrating over dy/dt we get for the gain of uninfecteds G = −(1/b)ln(q) for multiple antibiotic therapy and G = −(1/b)ln(s2) for 50-50 treatment (or cycling). Hence if q = s2 all treatment strategies result in the same gain or uninfecteds (although there is a difference in Tσ for combination therapy and cycling or 50-50 treatment. If q < s2 then the gain is larger for multiple treatment. If q > s2 then the gain is smaller for multiple treatment.
)) + 1]. By integrating over dy/dt we get for the gain of uninfecteds G = −(1/b)ln(q) for multiple antibiotic therapy and G = −(1/b)ln(s2) for 50-50 treatment (or cycling). Hence if q = s2 all treatment strategies result in the same gain or uninfecteds (although there is a difference in Tσ for combination therapy and cycling or 50-50 treatment. If q < s2 then the gain is larger for multiple treatment. If q > s2 then the gain is smaller for multiple treatment.
B3: Unequal Cost to Resistance.
Assume rb > ra without loss of generality. The ratio of the times necessary for A and B resistance to increase from a fraction ρ0 to a fraction ρe is Ta/Tb = (hfb − rb − rw/hfa − ra − rw) (see Appendix A4). Hence, for cycling the respective time intervals should relate to each other as Ta to Tb. The fractions treated for an adjusted 50-50 therapy are obtained by assuming Ta/Tb = 1. Hence, the fractions of patients treated relate to each other as fb = ((ra − rb)/h) + fa. Note, that ((ra − rb)/h) < 1, because we assume that there is a net benefit for resistance in presence of treatment (i.e., hfa > ra − rw and hfb > rb − rw).
C: Parameters
All parameters are given in arbitrary units. Fig. 2: λ = 100, d = 1, c = 1.5, h = 1., rw = 0, s = 10−3, and rr = 0 in A and rr = 0.1 in B. The simulation is started in the equilibrium in absence of treatment (see Appendix A1). Figure 3: In all simulations λ = 100, d =1, c = 1.5, h = 1, rw = 0, ra = rb = 0.1, rab = 0.2. Cycling: fa or fb = 1, fab = 0, switching every five time units. 50-50: fa = fb = 0.5, fab = 0. Combination: fa = fb = 0, fab = 1. De novo generation of resistance: s = q = 0 in A–C and H–J, s = 10−3 in D–G, q = 10−8 in D–F, q = 10−5 in G. Initial frequency of resistants at start of therapy: ρa = ρb = 10−3 in A–C and H–J, ρab = 10−6 in A–C, ρa = ρb = ρab = 0 in D–G.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: wt, wild type; res, resistant.
References
- 1.Doern G V, Brueggemann A, H P, Holley J, Rauch A M. Antimicrob Agents Chemother. 1996;40:1208–1213. doi: 10.1128/aac.40.5.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohen M L. Trends Microbiol. 1994;2:422–425. doi: 10.1016/0966-842x(94)90623-8. [DOI] [PubMed] [Google Scholar]
- 3.Bloom B R, Murray C J L. Science. 1992;257:1055–1064. doi: 10.1126/science.257.5073.1055. [DOI] [PubMed] [Google Scholar]
- 4.Swartz M N. Proc Natl Acad Sci (USA) 1994;91:2420–2427. doi: 10.1073/pnas.91.7.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arthur M, Courvalin P. Antimicrob Agents Chemother. 1993;37:1563–1571. doi: 10.1128/aac.37.8.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacoby G A. Ann Rev Med. 1996;47:169–179. doi: 10.1146/annurev.med.47.1.169. [DOI] [PubMed] [Google Scholar]
- 7.Murray B E. N Engl J Med. 1994;330:1229–1230. doi: 10.1056/NEJM199404283301710. [DOI] [PubMed] [Google Scholar]
- 8.Jernigan D B, Cetron M S, Breiman R F. J Am Med Assoc. 1996;275:206–209. [PubMed] [Google Scholar]
- 9.Anonymous. Am J Inf Contr. 1995;23:87–94. [Google Scholar]
- 10.J. E. McGowan J. Infect Control. 1986;7:573–576. doi: 10.1017/s0195941700065401. [DOI] [PubMed] [Google Scholar]
- 11.Massad E, Lundberg S, Yang H M. Int J Bio-Med Comput. 1993;33:65–81. doi: 10.1016/0020-7101(93)90060-j. [DOI] [PubMed] [Google Scholar]
- 12.Blower S M, Small P M, Hopewell P C. Science. 1996;273:497–500. doi: 10.1126/science.273.5274.497. [DOI] [PubMed] [Google Scholar]
- 13.May R M, Dobson A P. Pesticide Resistance: Strategies and Tactics for Management. Washington, DC: Natl. Acad. Sci.; 1986. pp. 170–193. [Google Scholar]
- 14.Lipsitch M, Levin B R. Antimicrob Agents Chemother. 1997;41:363–373. doi: 10.1128/aac.41.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonhoeffer S, Nowak M A. Proc R Soc London B. 1997;264:631–637. doi: 10.1098/rspb.1997.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonhoeffer S, May R M, Shaw G M, Nowak M A. Proc Natl Acad Sci USA. 1997;94:6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falkow S. Infectious Multiple Drug Resistance. London: Pion; 1975. [Google Scholar]
- 18.Centers for Disease Control (1989) Morbid. Mortal. Wkly. Rep. 38, (Suppl. 3), 38.
- 19.Kam K M, Luey K Y, Fung S M, Yiu P P, Harden T J, Cheung M M. Antimicrob Agents Chemother. 1995;39:2667–2670. doi: 10.1128/aac.39.12.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moran, J. S. & Levine, W. C. (1995) Clin. Infect. Dis. 20 (Suppl. 1), S47–S65. [DOI] [PubMed]
- 21.Anderson R M, May R M. Infectious Diseases of Humans. Oxford: Oxford Univ. Press; 1991. [Google Scholar]
- 22.Bouma J E, Lenski R E. Nature (London) 1988;335:351–352. doi: 10.1038/335351a0. [DOI] [PubMed] [Google Scholar]
- 23.Schrag S J, Perrot V. Nature (London) 1996;381:120–121. doi: 10.1038/381120b0. [DOI] [PubMed] [Google Scholar]
- 24.Fekety J. Medicine. 1964;45:593–613. [Google Scholar]
- 25.Levin, B. R., Lipsitch, M., Perrot, V., Schrag, S., Antia, R., Simonsen, L., Moore, N. & Stewart, F. M. (1997) Clin. Infect. Dis. 24, Suppl. 1, S9–S16. [DOI] [PubMed]
- 26.Haldane J. J Genet. 1957;55:511–524. [Google Scholar]