

Abstract
Previous research in our laboratory comparing the three-dimensional structural elements of two highly homologous alcohol dehydrogenases, one from the mesophile Clostridium beijerinckii (CbADH) and the other from the extreme thermophile Thermoanaerobacter brockii (TbADH), suggested that in the thermophilic enzyme, an extra intrasubunit ion pair (Glu224-Lys254) and a short ion-pair network (Lys257-Asp237-Arg304-Glu165) at the intersubunit interface might contribute to the extreme thermal stability of TbADH. In the present study, we used site-directed mutagenesis to replace these structurally strategic residues in CbADH with the corresponding amino acids from TbADH, and we determined the effect of such replacements on the thermal stability of CbADH. Mutations in the intrasubunit ion pair region increased thermostability in the single mutant S254K- and in the double mutant V224E/S254K-CbADH, but not in the single mutant V224E-CbADH. Both single amino acid replacements, M304R- and Q165E-CbADH, in the region of the intersubunit ion pair network augmented thermal stability, with an additive effect in the double mutant M304R/Q165E-CbADH. To investigate the precise mechanism by which such mutations alter the molecular structure of CbADH to achieve enhanced thermostability, we constructed a quadruple mutant V224E/S254K/Q165E/M304R-CbADH and solved its three-dimensional structure. The overall results indicate that the amino acid substitutions in CbADH mutants with enhanced thermal stability reinforce the quaternary structure of the enzyme by formation of an extended network of intersubunit ion pairs and salt bridges, mediated by water molecules, and by forming a new intrasubunit salt bridge.
Keywords: Alcohol dehydrogenase, Clostridium beijerinckii, Thermoanaerobacter brockii, site-directed mutagenesis, ion-pair network, salt bridge, thermostability
Discerning and manipulating the structurally different elements of mesophilic and thermophilic members of homologous protein families is the key to understanding the adaptation of hyperthermophilic proteins to extreme temperatures (for review, see Kumar et al. 2000b, 2001; Szilagyi and Zavodszky 2000; Sterner and Liebl 2001). Theoretical studies were based on amino acid sequences and three-dimensional structures of functionally related thermophilic and mesophilic proteins (e.g., Perutz and Raidt 1975; Argos et al. 1979; Menéndez-Arias and Argos 1989; Warren and Petsko 1995), whereas experimental studies involved the use of site-directed mutagenesis, pioneered by Matthews (1993) and Fersht (Fersht and Serrano 1993). Structural comparisons suggest that among the molecular interactions that could explain the unusual properties of hyperthermophiles, salt-bridge networks that are energetically more favorable than isolated ion pairs (Horovitz et al. 1990) are the most prominent (Yip et al. 1995; Hennig et al. 1997; Pappenberger et al. 1997).
Salt bridging is important in the structure and function of folded proteins (Perutz 1970; Barlow and Thornton 1983; Musafia et al. 1995; Xu et al. 1997a, b), yet the results of theoretical studies have been inconsistent. The contribution of salt bridges to protein stability has been variously reported as being small or insignificant (e.g., Singh 1988; Serrano et al. 1990; Barril et al. 1998), stabilizing (e.g., Perutz and Raidt 1975; Horovitz and Fersht 1992; Marqusee and Sauer 1994; Pervushin et al. 1996; Lounnas and Wade 1997; Kursula et al. 2002), or destabilizing (e.g., Dao-pin et al. 1991; Sun et al. 1991; Hendsch and Tidor 1994; Honig and Nicholls 1995; Waldburger et al. 1995; Phelan et al. 2002).
With the long-term goal of determining the molecular basis of protein thermostability, we are focusing on two functionally related, highly homologous, tetrameric alcohol dehydrogenases (ADHs): one from the extremely thermophilic Thermoanaerobacter brockii (TbADH; Peretz and Burstein 1989), first isolated from the Yellowstone Park hot springs (Lamed and Zeikus 1981), and the other from its mesophilic counterpart Clostridium beijerinckii (CbADH; Ismaiel et al. 1993). The enzymes in this reliable model system share 75% sequence identity (Peretz et al. 1997) but differ greatly (30°C) in thermostability (Bogin et al. 1998a). TbADH reversibly catalyses the oxidation of secondary alcohols to the corresponding ketones with a T1/260min (temperature at which 50% of the enzymatic activity is lost after 60 min) of 93.8°C, whereas in CbADH the T1/260min is 63.8°C. Both TbADH and CbADH are medium-chain, NADP(H)-linked, zinc-containing enzymes. Both ADHs are homotetramers composed of ∼38-kD subunits and have similar substrate specificity.
In previous studies, we isolated and characterized TbADH (Peretz and Burstein 1989; Peretz et al. 1993, 1997; Bogin et al. 1997), and cloned, sequenced, and overexpressed the structural adh genes from T. brockii and C. beijerinckii in Escherichia coli (Peretz et al. 1997). We also crystallized both TbADH (Zhang et al. 1993; Korkhin et al. 1996) and CbADH (Korkhin et al. 1996) and determined their respective structures at resolutions of 2.5 Å and 2.05 Å (Korkhin et al. 1998). Comparison of the very similar crystal structures of holo-TbADH (PDB entry 1YKF) and holo-CbADH (PDB entry 1KEV) (r.m.s. difference in Cα = 0.8 Å) revealed several features that might account for the higher thermal stability of TbADH. These include additional ion pairs, `charged-neutral' hydrogen bonds, and prolines as well as improved stability of α helices and tighter molecular packing. Using site-directed mutagenesis, we confirmed that proline, positioned at β-turns and at terminating external loops, is one element that increases the thermal stability of TbADH (Bogin et al. 1998b). A deeper structural insight, however, suggests that the most significant structural differences between the enzymes are the following nonconserved ion pairs of TbADH: (1) an intrasubunit ion pair, E224-K254, and (2) a short ion-pair network (Lys257-Asp237-Arg304-Glu165) at the interface between subunits A and D (and B–C; Korkhin et al. 1998, 1999). Hence, another explanation for the higher thermostability of TbADH could be superior inter- and intrasubunit salt bridging.
Here we report that using both single and multiple site-directed mutations to replace certain strategic residues in CbADH with the corresponding amino acids from TbADH enhanced the thermostability of the mesophilic enzyme. To investigate how such mutations alter the molecular structure to achieve enhanced thermostability, we crystallized the quadruple mutant and solved, refined, and analyzed its structure. The results of our structural analysis confirm our prediction that the increased thermal stability of the CbADH mutant is primarily due to the formation of an extended network of ion pairs and salt bridges, mediated by water molecules, and to the formation of a new intrasubunit salt bridge.
Results
Multiple sequence alignment
Amino acid sequence alignment of TbADH and CbADH with those of ADHs from two other members of this enzyme family, Mycoplasma pneumoniae ADH (MpADH) and Entamoeba histolytica ADH (EhADH) is presented in Figure 1 ▶. A definite pattern of high sequence conservation can be seen: 75% identity between TbADH and CbADH, 66% identity between TbADH and EhADH, 62% identity between TbADH and MpADH and between CbADH and either EhADH or MpADH, and 60% identity between MpADH and EhADH.
Fig. 1.

Amino acid sequence alignment of alcohol dehydrogenases from Thermoanaerobacter brockii (TbADH; TB; Peretz and Burstein 1989), Clostridium beijerinckii (CbADH; CB; Peretz et al. 1997), Mycoplasma pneumoniae (MpADH; MP; Himmelreich et al. 1996), and Entamoeba histolytica (EhADH; EH; Samuelson et al. 1992). Every tenth residue and the coenzyme-binding domain (residues 154–293) are marked above the sequences. Amino acid residues of TbADH are in the one-letter code, and identical residues in the other ADHs are marked with dashes. ⋄, intersubunit ion pair network between Lys257A-Asp237A-Arg304D-Glu165A; ⊗, intrasubunit ion pair network between Glu224 (of α-helix D) and Lys254 (of α-helix E); Glu280 (in the loop between β-strands S and F) is also marked. Secondary structure elements of β-strand (bbbbb) and α-helix (aaaaa) of CbADH are shown below the aligned sequences. Data from Korkhin et al. (1998).
Molecular and enzymatic properties of the mutant enzymes
We used site-directed mutagenesis (single, double, or multiple mutations) to replace the following amino acid residues in CbADH with the analogous residues from the thermophilic counterpart TbADH: V224E-, S254K-, V224E/S254K-, Q165E-, M304R-, Q165E/M304R-, V224E/S254K/Q165E/M304R-, and V224E/S254K/Q165E/M304R/R238ACbADH. Characterization of the highly purified recombinant mutants of CbADH by SDS-PAGE and nondenaturing gel filtration (Superdex S-200) revealed that all of the recombinant ADHs were intact tetramers having an electrophoretic mobility similar to that of the wild-type enzyme (data not shown).
All recombinant mutants displayed wild-type or near-wild-type levels of enzymatic activity and substrate specificity. Table 1 shows the kinetic properties of the enzymatic activity of TbADH, CbADH, and the CbADH mutants at 40°C, using 2-propanol as substrate. At 40°C, the values of Kcat and Kcat/Km in the thermophilic TbADH were significantly lower than those in the mesophilic CbADH. At elevated temperatures, however, the situation was reversed (Bogin et al. 1998b). The Km values of all mutants and the enzymatic efficiency (Kcat/Km) of most mutants were very similar to those of native CbADH, indicating that neither binding nor catalytic efficiency of the enzyme was severely affected by the mutations. Only the double mutant Q165E/M304R, the quadruple-mutant Q165E/M304R/V224E/S254K, and the penta-mutant R238A/Q165E/M304R/V224E/S254K of CbADH (Table 1) demonstrated a somewhat higher rate of catalysis. Indeed, all of the mutations were far from the active site of the enzyme (Korkhin et al. 1998), and any structural changes that might have resulted from these mutations did not appear to perturb the active site.
Table 1.
Kinetic parameters of CbADH and its mutantsa
| ADH | Km [mM] | Kcat [min−1] | Kcat/Km [min−1 • mM−1] |
| CbADHb | 7.2 | 8703 | 1209 |
| TbADHb | 5.8 | 2619 | 452 |
| CbADH mutants | |||
| V224E- | 7.1 | 7844 | 1105 |
| S254K- | 4.8 | 6069 | 1264 |
| V224E/S254K- | 6.2 | 5152 | 831 |
| Q165E- | 7.4 | 8144 | 1101 |
| M304R- | 6.3 | 7345 | 1166 |
| Q165E/M304R- | 7.1 | 15840 | 2231 |
| Q165E/M304R/V224E/S254K | 5.75 | 12542 | 2181 |
| R238A/Q165E/M304R/V224E/S254K | 7.47 | 15665 | 2097 |
a Enzymatic activity was measured by following the formation of NADPH at 340 nm. Km values for 2-propanol were determined with varying concentrations of 2-propanol (0.5–100 mM), enzyme (5–120 nM), and 0.5 mM NADP in 100 mM Tris-HCl (pH 8.8) in a total volume of 1 ml in triplicate. Values are the averages of three experiments, and the individual measurements were within 10% of the quoted mean.
b Values are from Bogin et al. (1998b).
Thermal stability parameters
The thermal stability of the CbADH and its mutants was determined by following their residual enzymatic activity after heat treatment and by thermal inactivation, followed by circular dichroism spectroscopy measurements. The temperature of 50% inactivation (T1/2) was determined by exposing the enzyme to temperatures of 30° to 100°C for 60 min, and measuring the residual enzymatic activity. T1/260min is the temperature at which 50% of the enzymatic activity is lost after 1-h incubation, interpolated from a plot of the residual enzymatic activity versus temperature (Fig. 2 ▶). T1/2CD is the temperature at which 50% of the original CD signal at 218 nm is lost upon heating the protein sample between 30° and 110°C, with an average increase in temperature of 1°C/min (as described in Materials and Methods). The T1/260min and T1/2CD values of the mutants are presented in Table 2 and discussed below.
Fig. 2.

The effect of the mutations on the thermal stability of C. beijerinckii ADH. Thermal stability of the enzymes was determined by monitoring the loss of their CD signal at 218 nm upon heating. The data (mdeg vs. wavelength) were collected every nm with an averaging time of 2 sec using quartz cells with a light path of 0.1 cm. Experiments were performed between 30°C and 110°C, with an average increase in temperature of 1°C/min. Scans were performed every 4°C. The protein concentration was about 0.5 mg/mL in 50 mM Na/K phosphate buffer (pH 6.8). The background for each was determined in a single scan with buffer alone at 25°C and subtracted from each scan. The background CD signal was temperature-independent.
Table 2.
Thermal parameters of CbADH and its mutants
| ADH | T1/260min [°C]1 | ΔT1/260min [°C]2 | T1/2CD [°C]1 | ΔT1/2CD [°C]2 |
| CbADH | 63.8 | 65.1 | ||
| TbADH | 93.8 | +30 | 92.1 | +25.9 |
| CbADH mutants | ||||
| V224E- | 63.6 | −0.2 | ||
| S254K- | 69.6 | +5.8 | ||
| V224E/S254K- | 69.2 | +5.4 | 70.5 | +5.4 |
| Q165E- | 66.0 | +2.2 | ||
| M304R- | 66.2 | +2.4 | ||
| Q165E/M304R- | 68.4 | +4.6 | 70.4 | +5.3 |
| V224E/S254K/Q165E/M304R- | 69.5 | +5.7 | 73.7 | +8.6 |
| V224E/S254K/Q165E/M304R/R238A- | 68.3 | +4.5 | 73.6 | +8.5 |
1T1/260min is the temperature at which 50% of the enzymatic activity is lost after 1-h incubation, interpolated from a plot of the residual enzymatic activity vs. temperature. T1/2CD is the temperature at which 50% of the original CD signal at 218 nm is lost upon heating the protein sample between 30° and 110°C, with an average increase in temperature at 1°C/min (as described in Experimental Conditions).
2 Relative to CbADH ±0.3°C of the quoted mean.
Structure of the quadruple mutant Q165E/M304R/V224E/S254K-CbADH (4mu-CbADH)
To study the structural features of the above mutations, we crystallized the quadruple mutant Q165E/M304R/V224E/S254K-CbADH (4mu-CbADH). Crystals of this mutant were isomorphous to those of wild-type CbADH. No significant changes were observed in the C-α chain of the mutant compared with that of the wild-type protein. Taking into account that the sites of the two ion pairs (Glu165-Arg304 and Glu224-Lys254) are separated both in space and via backbone (considering also the symmetry mates), the effect of each of the two double mutations can be considered local and independent.
The intersubunit ion pair network Lys257-Asp237-Arg304-Glu165
In both TbADH and CbADH, Lys257 and Asp237 form an intrasubunit ion pair (Korkhin et al. 1998, 1999). In TbADH, however, Asp237 is also involved in an ion pair bridge with Arg304 of a second subunit. The Arg304 in turn forms an additional intersubunit salt bridge (3.3 Å) with Glu165 of the first subunit. Thus, in TbADH a four-member ion-pair network involving Lys257A-Asp237A-Arg304D-Glu165A is present (the superscript refers to the subunit). All four side-chain functional groups are located on the surface of the respective subunits (Korkhin et al. 1998, 1999). In CbADH, both Arg304 and Glu165 are replaced by Met304 and Gln165, whose side chains do not interact electrostatically (Fig. 1 ▶; Korkhin et al. 1998); consequently, such an ion pair network does not exist in the mesophilic enzyme.
Replacing Gln165 with glutamic acid in CbADH increased its T1/260min by 2.2°C. Replacing Met304 with arginine increased the T1/260min value of the CbADH mutant by 2.4°C. In this experiment, the individual contributions of the two mutated charged residues, Q165E and M304R, of CbADH were additive. The thermal stability of the double mutant, Q165E/M304R-CbADH, is equal to the sums of those of the single mutants (ΔT1/260min = +4.6°C; Fig. 2 ▶, Table 2).
Figure 3A ▶ shows the structure of CbADH at the region of the Q165E/M304R mutation in the quadruple mutant. The structure of this region is very similar to that of TbADH, except that Arg238 of CbADH is alanine in TbADH. Consequently, Glu165A in the quadruple mutant does not form a single salt bridge with Arg304D, as was deduced from the structure of TbADH (Korkhin et al. 1998, 1999). Instead, Figure 3A ▶ shows that Glu165A is placed between two arginines—Arg304D and Arg238A, forming ion pairs with both residues (3.4 Å and 3.5 Å, respectively). Furthermore, Glu165A is involved in an extensive hydrogen-bond network connecting three subunits. In this network the central water molecule (crystallographically well resolved, temperature factor B = 28 Å2) is coordinated by the two positively charged guanidinium groups of Arg238A and Arg304D, by a negatively charged carboxyl group of Glu165A and by a hydroxyl of Ser168A. The hydroxyl hydrogen of this Ser168A residue forms a hydrogen bond with the oxygen of the main chain carbonyl group of Glu165A. Arg304D is also involved in a hydrogen-bond network: via a second solvent molecule (B = 28 Å2) it binds Asp237A, which in turn is involved in an intrasubunit salt bridge with Lys257A. Via a third solvent molecule (B = 37 Å2), Arg304 binds the main chain carbonyl group of subunit B (Ala97B). Thus, this seven-element hydrogen-bond/salt-bridge system connects the three subunits of the quadruple mutant of CbADH (Figs. 3B, 4 ▶ ▶).
Fig. 3.
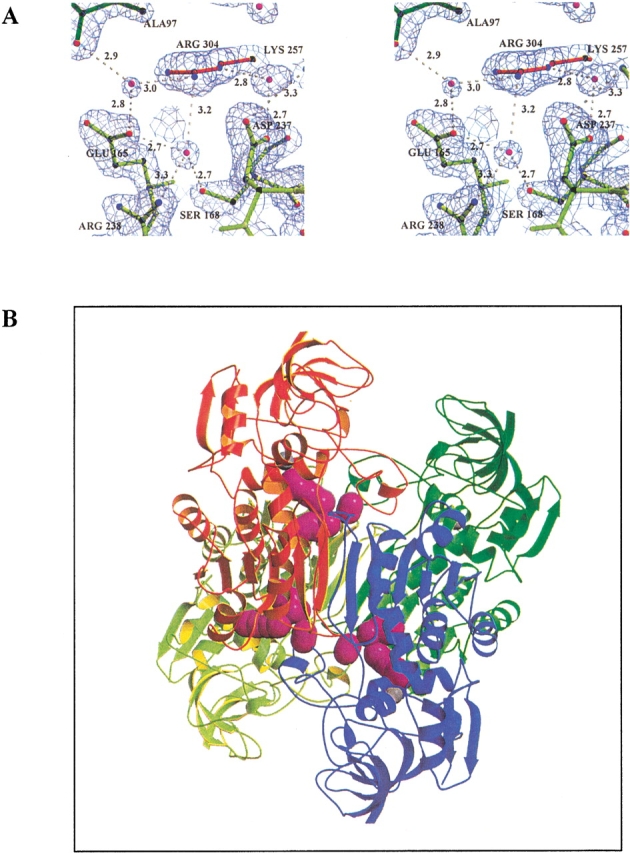
Three-dimensional structure of the tetramer of the quadruple mutant of CbADH. The four subunits are: A (yellow-light green), B (red), C (blue), and D (dark green). (A) Stereoview of the hydrogen-bond network at the interface of three subunits of the quadruple mutant of CbADH. Residues Asp237, Lys257, Arg238, Ser108, and Glu165 from Subunit A, Arg304 from Subunit B, and Ala97 of subunit D (via its main-chain carbonyl group) participate in this network. The water molecules are shown in magenta, hydrogen bond in white dashed lines, and distances are in Å. ′2Fo-Fc′ electron density map of the quadruple mutant is contoured at 1.7σ level. (B) Extra salt bridges (magenta) in subunit A (the yellow coil) in the Q165E/M304R mutant.
Fig. 4.

Schematic representation of the interactions at the interface of three subunits of the quadruple mutant of CbADH. Distances are given in Angstroms; electrostatic interactions are shown by solid lines.
In view of the above findings, and to investigate a possible contribution of the side chain of Ala238 to the thermal stability of the thermophilic ADH, we further mutated the quadruple mutant of CbADH by replacing Arg238 of CbADH with Ala of TbADH. The penta-mutant R238A/Q165E/M304R/V224E/S254K-CbADH was formed and isolated. The thermal stability of the penta-mutant, however, was not greater than that of the parent quadruple mutant (Table 2).
The intrasubunit ion pair of Glu224 and Lys254
In the hyperthermophilic TbADH, Glu224, located in α-helix D, forms an ion pair (4.3 Å) with Lys254, located in a neighboring α-helix E (Korkhin et al. 1998, 1999). In CbADH, Val and Ser replace Glu224 and Lys254 of TbADH, located at the coenzyme-binding domain (Fig. 1 ▶). Neither Val224 nor Ser254 of CbADH participates in any stabilizing interaction (Korkhin et al. 1998, 1999).
Replacing Val224 of CbADH with glutamic acid of TbADH had almost no effect on the thermal stability of the enzyme (ΔT1/260min = −0.2°C; Fig. 2 ▶, Table 2). On the other hand, replacing Ser254 of CbADH with lysine of TbADH, significantly stabilized the mutant (ΔT1/260min = +5.8°C; Fig. 2 ▶, Table 2).
Examining the high-resolution structure of the quadruple mutant of CbADH showed that this region varied significantly from subunit to subunit. The different structures of subunits A and D are shown in Figure 5A and B ▶, respectively. In subunit D of CbADH, Lys254 forms a hydrogen bond, via an ordered water molecule (temp. factor B = 38 Å2), with Glu224. It also forms ion pairs with both Glu280 and Glu224 (distances are 4.4 Å and 3.1 Å, respectively; Fig. 5B ▶). This result was surprising because in TbADH, the distance between Lys254 and Glu280 is 4.8 Å (Korkhin et al. 1998, 1999). Identical interactions were present in both subunits B and C of CbADH, and the water molecules were fixed in identical positions.
Fig. 5.

Two alternative conformations of Lys254 of the quadruple mutant of CbADH. (A) In subunit A, the ɛ-amino group of Lys254 points towards Glu280 to form a salt bridge; however neither hydrogen-bond connection nor electrostatic interaction between Lys254 and Glu224 is observed. (B) In subunit D, Lys254 is equally distanced from both Glu280 and Glu224; it interacts electrostatically with both of them and forms an additional hydrogen bond via a water molecule with Glu224. ′2Fo-Fc′ electron density map is contoured at 1.2σ.
In subunit A, however, Lys254 assumed a slightly different conformation. Its ɛ-amino-group points away from Glu224 (5.5 Å apart, Fig. 5A ▶), thus preventing the formation of an ion pair (present in subunits B, C, and D). This ɛ-amino group points toward Glu280 and forms a salt bridge (3.0 Å apart, Fig. 5A ▶). Although the water molecule adjacent to Glu224 is preserved in all four subunits, it does not mediate a hydrogen bond between Glu224 and Lys254 of subunit A (Fig. 5A ▶).
Superpositions of the side chains of Glu224, Lys254, and Glu280 in the four subunits of both the extreme thermophilic TbADH and the quadruple mutant of the mesophilic CbADH are presented in Figure 6 ▶.
Fig. 6.

Superposition of residues 224, 254, and 280 of TbADH and of the quadruple mutant of CbADH. The side chains of Glu224, Lys254, and Glu280 of TbADH (in magenta) and of the quadruple mutant of CbADH (in green) are shown in an orientation similar to that presented in Figure 5 ▶. The figure was prepared using the program O (Jones and Kjeldgaard 1997).
Discussion
Despite their low contribution to protein stability at room temperature, salt bridges have been proposed to play a crucial role in promoting hyperthermostability in many proteins (e.g., Kawamura et al. 1997; Elcock 1998; Hunenberger and McCammon 1999; Grinberg and Bernhardt 2001; Ozawa et al. 2001; Knochel et al. 2002). Moreover, structural analyses have indicated that among the molecular interactions (hydrophobic effect, van der Waals interactions, hydrogen bonds, and ionic effects, including dipole interactions and salt bridges) that could explain the unusual properties of hyperthermophiles, the most prominent are salt-bridge networks (Hennig et al. 1995; Yip et al. 1995; Pappenberger et al. 1997), which are energetically more favorable than isolated ion pairs (Horovitz et al. 1990).
Although individual surface salt bridges often promote the stability of proteins (Dao-pin et al. 1991; Strop and Mayo 2000), interactive combinations of complex salt bridges may exert a stronger stabilizing effect (Horovitz et al. 1990; Musafia et al. 1995). The finding by Olson and coworkers (Olson et al. 2001) that triads of charged side chains, Arg(+)−Glu(−)−Arg(+) spaced at i,i+4 or i,i+3 intervals in a helical peptide stabilize α helix by more than the additive contribution of two single salt bridges supports the hypothesis that interactions of this kind between salt bridges can account for much of the stabilization of certain thermophilic proteins. The results of our site-directed mutagenesis experiments agree with this notion, because amino acid substitutions leading to the enhanced thermal stability of the CbADH mutants reinforce the quaternary structure of the enzyme, (1) by forming extended networks of ion pairs and salt bridges, mediated by water molecules, and (2) by forming a new intrasubunit salt bridge. Further support for this view is provided by a report that ion pair networks are a major stabilizing force in the structure of hyperthermophilic and extreme-thermophilic proteins from Archaea and Bacteria (Hennig et al. 1995; Korndorfer et al. 1995; Yip et al. 1995; Tanner et al. 1996; Wallon et al. 1997). Moreover, significant thermal stabilization of glutamate dehydrogenase (GDH) was achieved by introducing into an extremely thermophilic GDH (from Thermococcus litoralis) a `missing' ion pair network present in the hyperthermophilic Pyrococcus furiosus GDH (Vetriani et al. 1998). On the other hand, the report that introducing a different ion pair network from the hyperthermophilic Pyrococcus furiosus GDH into the extremely thermophilic GDH (from Thermotoga maritimae) had no effect on its stability (Lebbink et al. 1998).
In certain thermophilic proteins, the frequency of salt bridges is higher than that of their mesophilic counterparts (Perutz 1978; Pervushin et al. 1996; Korkhin et al. 1998, 1999; Kumar et al. 2000a). Previous research by our group revealed that both TbADH and CbADH contain nine conserved intrasubunit salt bridges (seven single salt bridges, and one salt-bridge triad; Korkhin et al. 1998, 1999). Although Asp163 and Lys291 are present in both TbADH and CbADH, an additional ion pair between these residues is formed only in TbADH (3.05 Å apart) because in CbADH, the bulky side chain of Arg238 (Ala238 in TbADH) hinders the approach of these two charged residues (distanced 4.8 Å). The conserved intrasubunit salt bridge Lys257A-D237A in CbADH and TbADH is extended in TbADH to a four-residue intersubunit salt-bridge network comprising Lys257A-Asp237A-Arg304D-Glu165A. Residues 165 and 304 are not conserved in CbADH and do not participate in an ion-pair network. In addition, the thermophilic TbADH contains an additional, nonconserved intrasubunit ion pair in the coenzyme-binding domain, involving Glu224 from α-helix D and Lys254, located 3.94 Å away in a neighboring α-helix E. Both Glu224 and Lys254 are absent in CbADH, whereas in the other two mesophilic enzymes, EhADH and MpADH, one-half of the ion pair (Lys254) is preserved and Glu224 (the other half of the ion pair in TbADH) has been replaced by valine (see Fig. 1 ▶).
Therefore, we assumed that incorporating Arg304 into CbADH would extend the ion pair Lys257A-Asp237A to an intersubunit salt-bridge triad Lys257A-Asp237A-Arg304D, and as "multiple salt bridges are energetically more favorable than the sum of single bridges" (Horovitz and Fersht 1990), we anticipated a substantial increase in the thermostability of the mutant M304R-CbADH. The result (ΔT1/260min = +2.4°C) confirmed that the formation of a salt-bridge triad in M304R-CbADH is possible. Molecular modeling of the other single mutant Q165E-CbADH predicted that Glu165 might form a nonconserved, intersubunit salt bridge Glu165A-Arg189D (∼3.4 Å apart) that would stabilize the mutant. As anticipated, this mutation enhanced the thermal stability of M304R-CbADH (ΔT1/260min = +2.2°C). In theory, introducing the double mutation Q165E/M304R should have further stabilized the enzyme by extending the intrasubunit salt bridge Lys257A-Asp237A of CbADH to an intersubunit four-residue ion-pair network Lys257A-Asp237A-Arg304D-Glu165A. The results clearly showed that the double mutant was stable. The individual contributions of the two single mutations, Q165E- and M304R-CbADH, showed simple additivity, but not cooperativity, in the thermal stabilization of the double mutant Q165E/M304R-CbADH.
Analysis of the crystal structure of the quadruple mutant of CbADH demonstrated the significant contribution of the double mutation to the thermal stability of the mutated protein. The two newly introduced charged amino acids, Glu165A and Arg304D, indeed form an ion pair (3.5 Å apart, Fig. 3A ▶), and these residues are connected by additional hydrogen bonds via water molecules. This hydrogen-bond network also includes the main chain carbonyl of Ala97B (of subunit B). An additional ion pair formed by Glu165A and Arg238A (3.4 Å) creates an ion pair `sandwich' of Arg238A-Glu165A-Arg304D.
We predicted that mimicking the structure of this region in the thermophilic TbADH by mutating Arg238 in the double mutant of CbADH to alanine of TbADH would allow Glu165A to further approach Arg304D, thereby forming a shorter and more stable salt bridge (instead of the sandwich ion pair Arg238-Glu165-Arg304). The finding that the thermal stability of the penta-mutant R238A/Q165E/M304R/V224E/S254K-CbADH was not greater than that of the quadruple mutant suggests that the contribution of an extensive intersubunit hydrogen-bond network via solvent molecules (as observed in the quadruple mutant) to the thermal stability of the protein could sometimes be as good as that of a single intersubunit salt bridge.
The structure of the quadruple mutant allows speculation about the nature of the stabilization by single mutants. As each single mutant, Glu165A and Arg304D, can form a very stable hydrogen-bond network—including residues from the three subunits A, B, and D—each mutation increases the thermostability of the protein without a requirement for further cooperativity.
Enzymes from extreme thermophiles are characterized by high enzymatic activity at elevated temperatures and low enzymatic activity at lower temperatures (e.g., the specific enzymatic activity of TbADH is much lower than that of CbADH at 37°C, whereas at their respective optimal temperatures, the activity of TbADH is fourfold higher than that of CbADH). The mechanism of the enzymatic activity of the ADHs has been shown to be associated with a hinge-type movement of the catalytic domain toward the cofactor-binding domain (Colonna-Cesari et al. 1986; Korkhin et al. 1998). Such movement could be restricted by an extra ion pair on the surface of the protein monomer (e.g., ion pair Glu165-Arg304, shown in Fig. 2 ▶), thus lowering the enzymatic activity of the ADH. Only at elevated temperatures would the enzymatic activity increase from the higher kinetic energy of the movement. Nevertheless, this type of resilient ion-pair bond on the surface of monomers is essential for the protein to survive at elevated temperatures. In its absence, the high kinetic energy of free and unrestricted movement of the protein domains would most probably destabilize the protein and lead to its denaturation.
The quadruple-mutant of CbADH seems to exploit two structural features to improve its thermal stability (Williams et al. 1994; Aguilar et al. 1997; Elcock 1998): the formation of hydrogen bonds via solvent molecules, and the formation of long-range ion pair interactions. Solvent molecules can move freely without steric interference from the protein side chains, thus allowing more conformational freedom to the side chains, while retaining the possibility of short-range H-bond interactions. Ion pairs, on the other hand, are long-range interactions and are preserved even during considerable movement of the protein side chains. Several investigators have suggested that these structural features do not necessarily serve to enhance the thermodynamic stability of the protein, but rather provide resistance to denaturation by increasing the barrier of unfolding (Aguilar et al. 1997; Elcock 1998).
Replacing Ser254 of CbADH with Lys254 of TbADH increased the ΔT1/260min value of the mutant by 5.8°C. Examination of the X-ray structure of the quadruple mutant provides an explanation for the changes in the thermostability of the single mutants V224E- and S254K-, as well as in the double mutant V224E/S254K-CbADH. In the single mutant S254K-CbADH, the formation of a new salt bridge between Lys254 and Glu280 stabilized the mutant protein. In the other single mutant, V224E-CbADH, no stabilization occurred (ΔT1/260min = −0.2°C), because the newly introduced γ-carboxyl group of Glu224 can only be stabilized by its main-chain N atom. As the stability of the double mutant V224E/S254K-CbADH (ΔT1/260min = +5.4°C) was not improved over that of the single mutant S254K-CbADH (ΔT1/260min = +5.8°C), we assumed that a salt-bridge triad was not formed between residues Glu224-Lys254-Glu280. The absence of a salt-bridge triad in the structure of the quadruple mutant of CbADH provides two options for the formation of salt bridges—namely, Glu280-Lys254 or Glu224-Lys254 (Fig. 5 ▶), suggesting another mechanism of thermal stabilization. A thermophilic enzyme, acting at elevated temperatures, can afford significant displacement of amino acid residues without losing a stabilizing link. Wherever it moves, Lys254 is trapped by electrostatic and/or hydrogen-bond interaction with either glutamate 224 or 280.
In conclusion, comparative structural studies of highly homologous mesophilic and thermophilic ADHs, backed by selective site-directed mutagenesis of the mesophile, and by crystal structure of the mutant, indicate that specific ion pairs and salt bridges are important for maintaining the stability of the thermophilic enzyme. Introducing such elements into the structure of the mesophilic enzyme markedly enhanced its thermal stability. The results indicate that only a few mutations may be necessary to thermostabilize a mesophilic enzyme or vice versa. Structural studies of the quadruple mutant demonstrated the precise manner by which the newly introduced ion pairs reinforced the quaternary structure of the enzyme: by introducing an extensive hydrogen-bond network including a large number of water molecules in the "meeting point" of three subunits, and by creating two alternative options for ion pairing.
Both mechanisms are suitable for promoting enzymatic activity at elevated temperatures because they reinforce the quaternary structure while allowing the conformational freedom required for the enzymatic reaction of the ADHs.
Materials and methods
Materials
The enzymes for DNA cloning, sequencing, and amplification were from Amersham, New England Biolabs, Fermentas MBI, and Promega. [γ32P]ATP was from Amersham. Oligonucleotides for cloning, sequencing, and site-directed mutagenesis of the C. beijerinckii adh gene were synthesized by the WIS Chemical Synthesis Laboratory. All other chemicals were of analytical grade.
Strategy for site-directed mutagenesis studies
In previous studies, we determined the crystal structures of CbADH and TbADH in their holoenzyme forms at resolutions of 2.05 Å and 2.5 Å, respectively (Korkhin et al. 1998), and then examined several features that could account for the higher thermal stability of TbADH. The present study focused on intersubunit ion pairing interactions and intrasubunit salt bridging. We performed site-directed mutagenesis (single, double, and multiple mutations) to replace the following amino acid residues in CbADH with their analogous residues in the thermophilic counterpart TbADH: V224E-, S254K-, V224E/S254K-, Q165E-, M304R-, Q165E/M304R-, V224E/S254K/Q165E/M304R-, and R238A/V224E/S254K/Q165E/M304R-CbADH.
Isolation of DNA and mutagenesis
DNA was extracted by the alkaline lysis procedure (Sambrook et al. 1989), separated by agarose gel (1%, w/v) electrophoresis, and then purified to homogeneity using a Geneclean II kit (Bio 101). Construction and expression of the gene encoding CbADH was performed according to Peretz et al. (1997). The plasmid pBS-P200CbADH was used as the template to generate the recombinant plasmids coding for the V224E-, S254K-, V224E/S254K-, Q165E-, M304R-, and Q165E/M304R-CbADH mutants. For the construction of the quadruple mutant, the plasmid with Q165E/M304R mutation was used as a template to introduce V224E/S254K- mutations. For the construction of the penta-mutant, the quadruple mutant plasmid was used as a template, and the R238A mutation was introduced. Site-directed mutagenesis was performed according to the method of Kunkel (1985), and the mutations were verified by DNA sequencing of the relevant plasmids. The oligonucleotides used for generating the mutants are listed below, with the exchanged bases underlined:
for V224E-CbADH: 5′-CATAACTTGATCTTCTATATGACCA TT-3′
for S254K-CbADH: 5′-CCTGGTTTAACCATCTTTACTGCTTGGG-3′
for Q165E-CbADH: 5′-CTTGAACCCATTTCAATATCTGCAAG-3′
for M304R-CbADH: 5′-CCATATCTCTTAACCTTTCTGCTCTCAAAC-3′
for R238A-CbADH: 5′-GGAAAAGGCGTTGACGCCGTAATTATGGCAGG-3′
Protein expression and purification
All recombinant plasmids harboring CbADH and its mutated variants were transformed into Escherichia coli strain TG-2 (Sambrook et al. 1989). The recombinant proteins were purified according to a modification of a procedure described by Peretz and Burstein (1989). Briefly, the transformed cells were cultured in 2YT with zinc chloride (50 μM) and ampicillin (100 mg/L) for 17 h at 37°C, harvested by centrifugation at 8,000 g for 20 min at 4°C, and resuspended in 25 mM Tris-HCl, 0.1 mM DTT, 0.1 mM EDTA, 0.1 mM benzamidine, 0.02% sodium azide, and 10% glycerol (pH 7.3) (buffer A). The cells were disrupted by pulsed sonication (Branson Sonifier 450) for 5 min, using a rosette cup immersed in an ice bath, and then centrifuged (23,000 g for 15 min) to remove cell debris. The supernatant was then heat-treated at 65°C for 3 min and centrifuged again for 15 min at 12,000 g. The clear supernatant was applied onto a DEAE-52-cellulose column (13 × 3 cm), preequilibrated with buffer A at 4°C. The column was extensively washed with buffer A until no protein was detected in the eluate, measured according to Bradford (1976). The recombinant proteins were eluted from the column with a solution of 0.1 M NaCl in buffer A. The enzymatically active fractions were pooled and applied onto a short Red-Sepharose column (7 × 3 cm) (Pharmacia). The recombinant enzyme was eluted from the column using a linear gradient of NaCl (0.1–2 M) in buffer A. Fractions containing enzymatic activity were collected and concentrated by ultrafiltration (Amicon YM-30), and then stored at 4°C. All mutant enzymes used in this study were obtained as intact tetramers and purified to homogeneity, as judged by Coomassie Brilliant Blue staining of SDS-polyacrylamide gel electrophoresis (PAGE) gels.
Enzyme assay and kinetic experiments
The catalytic activity of ADH was measured at 40°C by following the reduction of NADP+ (and monitoring the formation of NADPH) at 340 nm (ɛ340 = 6.2 mM−1cm−1). The standard assay mixture contained 150 mM 2-propanol and 0.5 mM NADP+ and 100 mM Tris-HCl (pH 8.8) in a total volume of 1 mL. One unit of ADH is defined as the amount of enzyme that catalyzes the oxidation of 1 μmole of 2-propanol/min under initial velocity at the above-mentioned conditions. Kinetic parameters were measured and calculated using a Beckman DU-7500 spectrophotometer, equipped with a Multicomponent/SCA/Kinetics Plus software package and a thermostated circulating water bath. The Km values for 2-propanol were determined using different concentrations of the alcohol (0.1–100 mM) and enzyme (5–120 nM) and 0.5 mM NADP (above saturation concentration of the coenzyme) in 100 mM Tris-HCl (pH 8.8). The reported values represent the average of three experiments, and the individual measurements were within 10% of the quoted mean.
Thermal inactivation ( T1/260min) analysis
The thermal stability of ADH was determined by monitoring the residual enzymatic activity after 60 min incubation in 25 mM Na/K phosphate buffer (pH 6.8) and 1M NaCl at different temperatures. Enzymatic activity was assayed by spectrophotometric analysis of NADPH formation at 340nm, under the standard conditions described above. Values are the means of three experiments, and the individual measurements were within 10% of the quoted mean.
Circular dichroism (CD)
CD measurements were made using an Aviv spectrophotometer, Model 202. The data (mdeg versus wavelength) were collected every nm and averaging time of 2 sec using quartz cells with a light path of 0.1 cm for far UV (200–260 nm). Experiments were performed between 30°C and 110°C, with an average increase in temperature of 1°C/min. Scans were performed every 4°C. The protein concentration was about 3 μM (0.5 mg/mL) in 50 mM Na/K phosphate buffer (pH 6.8). The background for each analysis was determined in a single scan with buffer alone at 25°C and subtracted from each scan. The background CD signal was temperature-independent. The decrease in CD signal with the increase of temperature was evaluated at the local minima of 218 nm and further normalized by subtracting from every wavelength the lowest signal observed at the particular wavelength. Finally, the change in the percent residual CD signal was plotted as a function of temperature, and the residual 50% CD signal (T1/2CD) was estimated.
Analytical procedures
DNA sequencing was performed on an Applied Biosystems Model 373A DNA sequencer using the dideoxy method (Sanger and Coulson 1975) and appropriate primers.
SDS-PAGE was performed on 12% slab gels and 5% stacking gels (Laemmli 1970), using a Bio-Rad MiniProtean II system. The gels were stained with Coomassie Brilliant Blue.
Protein concentrations were determined according to Bradford (1976) and Lowry (Peterson 1979), using bovine serum albumin as the standard. The estimated molecular weight of the recombinant enzymes was judged using a column of Superdex S-200, as described (Peretz and Burstein 1989).
Crystallization, data collection, and refinement
Crystals of the quadruple mutant Q165E/M304R/V224E/S254K-CbADH were grown in the hanging drop crystallization setup with 18% PEG 4000; 50 mM Tris-Cl pH 8.2; 50 mM NaCl; 2 mM NADP; 50 μM ZnCl2 at the protein concentration 12 mg/mL. For data collection at cryogenic conditions (T = 120K), ethylene glycol was gradually added to the mother liquor solution to a final concentration of 15% (v/v). A complete data set was collected from a single crystal on a Rigaku R-AXIS IV++ imaging plate area detector using a Rigaku RU-H3R rotating anode operated at 5kW and Osmic multilayer x-ray focusing mirrors. Diffraction data were integrated, scaled, and reduced using the HKL program package (Otwinowski and Minor 1997).
The crystals of the quadruple mutant and of the wild-type holo-CbADH were isomorphous. As starting model for the refinement, the coordinates of the holo-CbADH molecule were used (Korkhin et al. 1998), PDB code 1KEV. All refinement protocols and electron density calculations listed below were performed with the CNS package (Brunger et al. 1998), map display and model rebuilding using program O (Jones and Kjeldgaard 1997), and model inspection using WHATIF and PROCHECK (Laskowsky et al. 1993). Initially, five cycles of rigid body refinement were performed to optimize the model position in the asymmetric unit. Electron density 2Fo-Fc and Fo-Fc maps demonstrated excellent correspondence to the molecular model except for the rearranged α5 region in one of the subunits, which was rebuilt using the O program. To avoid model bias in the mutated regions, a simulated annealing omit electron density map was calculated using the standard CNS protocol in a 3.5 Å vicinity of the mutated residues. Side chains of the mutated amino acid residues were then built into the resulting 2Fo-Fc map. Simulated annealing refinement was carried out with all reflections available, employing a torsion angle dynamics algorithm by cooling the model from 5000K to 300K, with a 25K drop per step. Noncrystallographic symmetry restraints were not imposed in the refinement. The model was then inspected to correct all side chains in error. The cofactor was not included in the final model because its density could be resolved only in the adenine-moiety part and not in the regions of the nicotinamide and the pyrophosphate. Thirty cycles of group B-factor refinement were followed by 30 cycles of individual B-factor refinement. The addition of 411 solvent atoms was performed using peaks higher than 4σ on the Fo-Fc difference map; only solvent atoms involved in hydrogen bonding with the protein and with refined B-factor lower than 100 Å2 were included. Bulk solvent correction was applied on all stages of the refinement and electron density map calculations. Data collection and refinement statistics are presented in Table 3.
Table 3.
Data collection and refinement statistics
| Data collection | |
| Space Group | P212121 |
| Unit cell (Å) | 86.7; 147.8; 125.1 |
| Resolutiona | 20−1.97 (2.0 − 1.97) |
| N of unique reflections | 111587 |
| Completeness | 98.1 (65.1) |
| ≤F>/≤σ(F)> | 20.4 (2.0) |
| Rmerge (%) | 7.2 (46.4) |
| Refinement | |
| N of reflections in the test set | 11159 |
| Atoms used in refinement | 10593 |
| Water molecules | 411 |
| R-factor (%) | 21.9% |
| Rfree-factor (%) | 24.7% |
| Deviation from ideal geometry | |
| RMSD bond distance (Å) | 0.006 |
| RMSD bond angles (deg) | 1.3 |
| RMSD dihedral angles (deg) | 23.6 |
| RMSD improper angles (deg) | 0.76 |
a Numbers in parentheses represent the values for the outermost resolution shell.
Acknowledgments
This work was supported by the Israel Science Foundation (grant 296-00). Y.B. is the Maynard I. and Elaine Wishner Professor for Bioorganic Chemistry. We thank Dr. Linda Shimon, Dr. Haim Rozenberg, and Mr. Yaakov Halfon for their help in data collection, and Dr. Virginia Buchner for helpful remarks during manuscript preparation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ADH, alcohol dehydrogenase
CbADH, Clostridium beijerinckii ADH
TbADH, Thermoanaerobacter brockii ADH
EhADH, Entamoeba histolytica ADH
MpADH, Mycoplasma pneumoniae ADH
DTT, d,l dithiothreitol
Gdn-HCl, guanidine hydrochloride
PAGE, polyacrylamide gel electrophoresis
SDS, sodium dodecyl sulfate
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0222102.
The authors dedicate this paper to the memory of Professor Hans Neurath, founding editor of Protein Science.
References
- Aguilar, C.F., Sanderson, I., Moracci, M., Ciaramella, M., Nucci, R., Rossi, M., and Pearl, L.H. 1997. Crystal structure of the β-glycosidase from the hyperthermophilic archeon Sulfolobus solfataricus: Resilience as a key factor in thermostability. J. Mol. Biol. 271 789–802. [DOI] [PubMed] [Google Scholar]
- Argos, P., Rossmann, M.G., Grau, U.M., Suborn, H., Frank, G., and Tratschin, J.D. 1979. Thermal stability and protein structure. Biochemistry 18 5698–5703. [DOI] [PubMed] [Google Scholar]
- Barlow, D.J. and Thornton, J.M. 1983. Ion-pairs in proteins. J. Mol. Biol. 168 867–885. [DOI] [PubMed] [Google Scholar]
- Barril, X., Aleman, C., Orozco, M., and Luque, F.J. 1998. Salt bridge interactions: Stability of ionic and neutral complexes in the gas phase, in solution and in proteins. Proteins 32 67–79. [DOI] [PubMed] [Google Scholar]
- Bogin, O., Peretz, M., and Burstein, Y. 1997. Thermoanaerobacter brockii alcohol dehydrogenase: Characterization of the active site metal and its ligand amino acids. Protein Sci. 6 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1998a. Probing structural elements of thermal stability in bacterial alcohol dehydrogenase. I. Construction and characterization of chimeras consisting of secondary ADHs from Thermoanaerobacter brockii and Clostridium beijerinckii. Lett. Peptide Sci. 5 399–408. [Google Scholar]
- Bogin, O., Peretz, M., Hacham, Y., Korkhin, Y., Frolow, F., Kalb(Gilboa), A.J., and Burstein, Y. 1998b. Enhanced thermal stability of Clostridium beijerinckii alcohol dehydrogenase after strategic substitution of amino acid residues with prolines from the homologous thermophilic Thermoanaerobacter brockii alcohol dehydrogenase. Protein Sci. 7 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72 248–254. [DOI] [PubMed] [Google Scholar]
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T., and Warren, G.L. 1998. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystall. D Biol. Crystall. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Colonna-Cesari, F., Perahia, D., Karplus, M., Eklund, H., Brändén, C., and Tapia, O. 1986. Interdomain motion in liver alcohol dehydrogenase. Structural and energetic analysis of the hinge bending mode. J. Biol. Chem. 261 15273–15280. [PubMed] [Google Scholar]
- Dao-pin, S., Anderson, D.E., Baase, W.A., Dahlquist, F.W., and Matthews, B.W. 1991. Structural and thermodynamic consequences of burying a charged residue within the hydrophobic core of T4 lysozyme. Biochemistry 30 11521–11529. [DOI] [PubMed] [Google Scholar]
- Elcock, A.H. 1998. The stability of salt bridges at high temperatures: Implications for hyperthermophilic proteins. J. Mol. Biol. 284 489–502. [DOI] [PubMed] [Google Scholar]
- Fersht, A.R. and Serrano, L. 1993. Principles of protein stability derived from protein engineering experiments. Curr. Opin. Struct. Biol. 3 75–83. [Google Scholar]
- Grinberg, A.V. and Bernhardt, R. 2001. Contribution of a salt bridge to the thermostability of adrenodoxin determined by site-directed mutagenesis. Arch. Biochem. Biophys. 396 25–34. [DOI] [PubMed] [Google Scholar]
- Hendsch, Z.S. and Tidor, B. 1994. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 3 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig, M., Darimont, B., Sterner, R., Kirschner, K., and Jansonius, J.N. 1995. 2.0- Å structure of indole-3-glycerol phosphate synthase from the hyperthermophile Sulfolobus solfataricus—Possible determinants of protein stability. Structure 3 1295–1306. [DOI] [PubMed] [Google Scholar]
- Hennig, M., Sterner, R., Kirschner, K., and Jansonius, J.N. 1997. Crystal structure at 2.0 Å resolution of phosphoribosyl anthranilate isomerase from the hyperthermophile Thermotoga maritima: Possible determinants of protein stability. Biochemistry 36 6009–6016. [DOI] [PubMed] [Google Scholar]
- Himmelreich, R., Hilbert, H., Plagens, H., Pirkl, E., Li, B.C., and Herrmann, R. 1996. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 24 4420–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig, B. and Nicholls, A. 1995. Classical electrostatics in biology and chemistry. Science 268 1144–1149. [DOI] [PubMed] [Google Scholar]
- Horovitz, A. and Fersht, A.R. 1990. Strategy for analysing the cooperativity of intramolecular interactions in peptides and proteins. J. Mol. Biol. 214 613–617. [DOI] [PubMed] [Google Scholar]
- ———. 1992. Cooperative interactions during protein folding. J. Mol. Biol. 224 733–740. [DOI] [PubMed] [Google Scholar]
- Horovitz, A., Serrano, L., Avron, B., Bycroft, M., and Fersht, A.R. 1990. Strength and cooperativity of contributions of surface salt bridges to protein stability. J. Mol. Biol. 216 1031–1044. [DOI] [PubMed] [Google Scholar]
- Hunenberger, P.H. and McCammon, J.A. 1999. Molecular dynamics simulations of the hyperthermophilic protein sac7d from Sulfolobus acidocaldarius: Contribution of salt bridges to thermostability. J. Mol. Biol. 285 1811–1830. [DOI] [PubMed] [Google Scholar]
- Ismaiel, A.A., Zhu, C.X., Colby, G.D., and Chen, J.S. 1993. Purification and characterization of a primary-secondary alcohol dehydrogenase from two strains of Clostridium beijerinckii. J. Bacteriol. 175 5097–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, T.A. and Kjeldgaard, M. 1997. Electron-density map interpretation. Methods Enzymol. 277 173–208. [DOI] [PubMed] [Google Scholar]
- Kawamura, S., Tanaka, I., Yamasaki, N., and Kimura, M. 1997. Contribution of a salt bridge to the thermostability of DNA binding protein HU from Bacillus stearothermophilus determined by site-directed mutagenesis. J. Biochem. (Tokyo) 121 448–455. [DOI] [PubMed] [Google Scholar]
- Knochel, T., Pappenberger, A., Jansonius, J.N., and Kirschner, K. 2002. The crystal structure of indoleglycerol-phosphate synthase from Thermotoga maritima. Kinetic stabilization by salt bridges. J. Biol. Chem. 277 8626–8634. [DOI] [PubMed] [Google Scholar]
- Korkhin, Y., Frolow, F., Bogin, O., Peretz, M., Kalb(Gilboa), A.J., and Burstein, Y. 1996. Crystalline alcohol dehydrogenase from the mesophilic bacterium Clostridium beijerinckii and the thermophilic bacterium Thermoanaerobium brockii: Preparation, characterization and molecular symmetry. Acta Crystal. D52 882–886. [DOI] [PubMed] [Google Scholar]
- Korkhin, Y., Kalb (Gilboa), A.J., Peretz, M., Bogin, O., Burstein, Y., and Frolow, F. 1998. NADP-dependent bacterial alcohol dehydrogenases: Crystal structure, cofactor binding and cofactor specificity of the ADHs of Clostridium beijerinckii and Thermoanaerobacter brockii. J. Mol. Biol. 278 965–979. [DOI] [PubMed] [Google Scholar]
- Korkhin, Y., Bogin, O., Peretz, M., Kalb(Gilboa), A.J., Burstein, Y., and Frolow, F. 1999. Oligomeric integrity—The structural key to thermal stability in bacterial alcohol dehydrogenases. Protein Sci. 8 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korndorfer, I., Steipe, B., Huber, R., Tomschy, A., and Jaenicke, R. 1995. The crystal structure of holo-glyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima at 2.5 Å resolution. J. Mol. Biol. 246 511–521. [DOI] [PubMed] [Google Scholar]
- Kumar, S., Ma, B., Tsai, C.-J., and Nussinov, R. 2000a. Electrostatic strengths of salt bridges in thermophilic and mesophilic glutamate dehydrogenase monomers. Proteins 38 368–383. [DOI] [PubMed] [Google Scholar]
- Kumar, S., Tsai, C.-J., and Nussinov, R. 2000b. Factors enhancing protein thermostability. Protein Eng. 13 179–191. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Thermodynamic differences among homologous thermophilic and mesophilic proteins. Biochemistry 40 14152–14165. [DOI] [PubMed] [Google Scholar]
- Kunkel, T.A. 1985. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. 82 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kursula, I., Partanen, S., Lambeir, A.M., and Wierenga, R.K. 2002. The importance of the conserved Arg191-Asp227 salt bridge of triosephosphate isomerase for folding, stability, and catalysis. FEBS Lett. 518 39–42. [DOI] [PubMed] [Google Scholar]
- Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685. [DOI] [PubMed] [Google Scholar]
- Lamed, R.J. and Zeikus, J.G. 1981. Novel NADP-linked alcohol-aldehyde/ketone oxidoreductase in thermophilic ethanologenic bacteria. Biochem. J. 195 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowsky, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Lebbink, J.H., Knapp, S., van der Oost, J., Rice, D., Ladenstein, R., and de Vos, W.M. 1998. Engineering activity and stability of Thermotoga maritima glutamate dehydrogenase. I. Introduction of a six-residue ion-pair network in the hinge region. J. Mol. Biol. 280 287–296. [DOI] [PubMed] [Google Scholar]
- Lounnas, V. and Wade, R.C. 1997. Exceptionally stable salt bridges in cytochrome P450 can have functional roles. Biochemistry 36 5402–5417. [DOI] [PubMed] [Google Scholar]
- Marqusee, S. and Sauer, R.T. 1994. Contribution of a hydrogen bond/salt bridge network to the stability of secondary and tertiary structures in λ repressor. Protein Sci. 3 2217–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B.W. 1993. Structural and genetic analysis of protein stability. Annu. Rev. Biochem. 62 139–160. [DOI] [PubMed] [Google Scholar]
- Menéndez-Arias, L. and Argos, P. 1989. Engineering protein thermal stability. Sequence statistics point to residue substitutions in α-helices. J. Mol. Biol. 206 397–406. [DOI] [PubMed] [Google Scholar]
- Musafia, B., Buchner, V., and Arad, D. 1995. Complex salt bridges in proteins: Statistical analysis of structure and function. J. Mol. Biol. 254 761–770. [DOI] [PubMed] [Google Scholar]
- Olson, C.A., Spek, E.J., Shi, Z., Vologodskii, A., and Kallenbach, N.R. 2001. Cooperative helix stabilization by complex Arg-Glu salt bridges. Proteins 44 123–132. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Ozawa, T., Hakamada, Y., Hatada, Y., Kobayashi, T., Shirai, T., and Ito, S. 2001. Thermostabilization by replacement of specific residues with lysine in a Bacillus alkaline cellulase: Building a structural model and implications of newly formed double intrahelical salt bridges. Protein Eng. 14 501–504. [DOI] [PubMed] [Google Scholar]
- Pappenberger, G., Schurig, H., and Jaenicke, R. 1997. Disruption of an ionic network leads to accelerated thermal denaturation of d-glyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima. J. Mol. Biol. 274 676–683. [DOI] [PubMed] [Google Scholar]
- Peretz, M. and Burstein, Y. 1989. Amino acid sequence of alcohol dehydrogenase from the thermophilic bacterium Thermoanaerobium brockii. Biochemistry 28 6549–6555. [DOI] [PubMed] [Google Scholar]
- Peretz, M., Bogin, O., Keinan, E., and Burstein, Y. 1993. Stereospecificity of hydrogen transfer by the NADP-linked alcohol dehydrogenase from the thermophilic bacterium Thermoanaerobium brockii. Int. J. Pept. Protein Res. 42 490–495. [DOI] [PubMed] [Google Scholar]
- Peretz, M., Bogin, O., Tel-Or, S., Cohen, A., Li, G., Chen, J.-S., and Burstein, Y. 1997. Molecular cloning, nucleotide sequencing, and expression of genes encoding alcohol dehydrogenases from the thermophile Thermoanaerobacter brockii and the mesophile Clostridium beijerinckii. Anaerobe 3 259–270. [DOI] [PubMed] [Google Scholar]
- Perutz, M.F. 1970. Stereochemistry of cooperative effects in haemoglobin. Nature 228 726–739. [DOI] [PubMed] [Google Scholar]
- ———. 1978. Electrostatic effects in proteins. Science 201 1187–1191. [DOI] [PubMed] [Google Scholar]
- Perutz, M.F. and Raidt, H. 1975. Stereochemical basis of heat stability in bacterial ferredoxins and in haemoglobin A2. Nature 255 256–259. [DOI] [PubMed] [Google Scholar]
- Pervushin, K., Billeter, M., Siegal, G., and Wuthrich, K. 1996. Structural role of a buried salt bridge in the 434 repressor DNA-binding domain. J. Mol. Biol. 264 1002–1012. [DOI] [PubMed] [Google Scholar]
- Peterson, G.L. 1979. Review of the Folin phenol protein quantitation method of Lowry, Rosebrough, Farr and Randall. Anal. Biochem. 100 201–220. [DOI] [PubMed] [Google Scholar]
- Phelan, P., Gorfe, A.A., Jelesarov, I., Marti, D.N., Warwicker, J., and Bosshard, H.R. 2002. Salt bridges destabilize a leucine zipper designed for maximized ion pairing between helices. Biochemistry 41 2998–3008. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., Fritch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory manual, 2nd ed., pp. 125–128. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Samuelson, J., Zhang, W.W., Kumar, A., Descoteaux, S., Shen, P.S., and Bailey, G. 1992. Primary structures of alcohol and aldehyde dehydrogenase genes of Entamoeba histolytica. Arch. Med. Res. 23 31–33. [PubMed] [Google Scholar]
- Sanger, F. and Coulson, A.R. 1975. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 94 441–448. [DOI] [PubMed] [Google Scholar]
- Serrano, L., Horovitz, A., Avron, B., Bycroft, M., and Fersht, A.R. 1990. Estimating the contribution of engineered surface electrostatic interactions to protein stability by using double-mutant cycles. Biochemistry 29 9343–9352. [DOI] [PubMed] [Google Scholar]
- Singh, U.C. 1988. Probing the salt bridge in the dihydrofolate reductase-methotrexate complex by using the coordinate-coupled free energy perturbation method. Proc. Natl. Acad. Sci. 85 4280–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner, R. and Liebl, W. 2001. Thermophilic adaptation of proteins. Crit. Rev. Biochem. Mol. Biol. 36 39–106. [DOI] [PubMed] [Google Scholar]
- Strop, P. and Mayo, S.L. 2000. Contribution of surface salt bridges to protein stability. Biochemistry 39 1251–1255. [DOI] [PubMed] [Google Scholar]
- Sun, D.P., Sauer, U., Nicholson, H., and Matthews, B.W. 1991. Contributions of engineered surface salt bridges to the stability of T4 lysozyme determined by directed mutagenesis. Biochemistry 30 7142–7153. [DOI] [PubMed] [Google Scholar]
- Szilagyi, A. and Zavodszky, P. 2000. Structural differences between mesophilic, moderately thermophilic and extremely thermophilic protein subunits: Results of a comprehensive survey. Structure 8 493–504. [DOI] [PubMed] [Google Scholar]
- Tanner, J.J., Hecht, R.M., and Krause, K.L. 1996. Determinants of enzyme thermostability observed in the molecular structure of Thermus aquaticus D-glyceraldehyde-3-phosphate dehydrogenase at 2.5 Å resolution. Biochemistry 35 2597–2609. [DOI] [PubMed] [Google Scholar]
- Vetriani, C., Maeder, D.L., Tolliday, N., Yip, K.S.P., Stillman, T.J., Britton, K.L., Rice, D.W., Klump, H.H., and Robb, F.T. 1998. Protein thermostability above 100°C: A key role for ionic interactions. Proc. Natl. Acad. Sci. 95 12300–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldburger, C.D., Schildbach, J.F., and Sauer, R.T. 1995. Are buried salt bridges important for protein stability and conformational specificity? Nat. Struct. Biol. 2 122–128. [DOI] [PubMed] [Google Scholar]
- Wallon, G., Kryger, G., Lovett, S.T., Oshima, T., Ringe, D., and Petsko, G.A. 1997. Crystal structures of Escherichia coli and Salmonella typhimurium 3-isopropylmalate dehydrogenase and comparison with their thermophilic counterpart from Thermus thermophilus. J. Mol. Biol. 266 1016–1031. [DOI] [PubMed] [Google Scholar]
- Warren, G.L. and Petsko, G.A. 1995. Composition analysis of α-helices in thermophilic organisms. Protein Eng. 8 905–913. [DOI] [PubMed] [Google Scholar]
- Williams, M.A., Goodfellow, J.M., and Thornton, J.M. 1994. Buried waters and internal cavities in monomeric proteins. Protein Sci. 3 1224–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, D., Lin, S.L., and Nussinov, R. 1997a. Protein binding versus protein folding: The role of hydrophilic bridges in protein associations. J. Mol. Biol. 265 68–84. [DOI] [PubMed] [Google Scholar]
- Xu, D., Tsai, C.J., and Nussinov, R. 1997b. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 10 999–1012. [DOI] [PubMed] [Google Scholar]
- Yip, K.S.P., Stillman, T.J., Britton, K.L., Artymiuk, P.J., Baker, P.J., Sedelnikova, S.E., Engel, P.C., Pasquo, A., Chiaraluce, R., Consalvi, V., Scandurra, R., and Rice, D.W. 1995. The structure of Pyrococcus furiosus glutamate dehydrogenase reveals a key role for ion-pair networks in maintaining enzyme stability at extreme temperatures. Structure 3 1147–1158. [DOI] [PubMed] [Google Scholar]
- Zhang, Z., Djebli, A., Shoham, M., Frolow, F., Peretz, M., and Burstein, Y. 1993. Crystal parameters of an alcohol dehydrogenase from the extreme thermophile Thermoanaerobium brockii. J. Mol. Biol. 230 353–355. [DOI] [PubMed] [Google Scholar]