

A complex of human carbonic anhydrase II with its inhibitor acetate is described where the ligand is found to bind in an orientation different from that previously described.
Keywords: carbonic anhydrase II, acetate binding
Abstract
The enzyme human carbonic anhydrase II (hCAII) crystallized in an acetate-bound complex belonging to space group P212121, with unit-cell parameters a = 42.3, b = 71.8, c = 74.0 Å. The structure was solved by the molecular-replacement method and refined to an R value of 0.18 and an R free of 0.21. The acetate molecule replaced the zinc-bound water molecule in the structure, differing from previous reports regarding the site of acetate binding. This mode of binding disrupts the hydrogen-bonded solvent network required for activity of the enzyme. This mode of inhibitor binding is a novel one that has not been observed previously.
1. Introduction
Human carbonic anhydrase II (hCAII) is a zinc metalloenzyme that is abundant in erythrocytes (Lindskog, 1997 ▶). The enzymatic reaction, the hydration of carbon dioxide to bicarbonate or the reverse dehydration of bicarbonate, occurs at the zinc ion.
The enzyme is roughly spherical in shape and the active site lies within a conical cleft about 15 Å deep; the zinc ion lies at the bottom of this cleft and is tetrahedrally liganded by His94, His96, His119 and a hydroxide ion at physiological pH (Håkansson et al., 1992 ▶; Stams & Christianson, 2000 ▶). The zinc-bound hydroxide forms a hydrogen bond to the hydroxyl group of Thr199, which in turn forms a hydrogen bond to Glu106. This hydrogen-bond network, along with the so-called ‘deep water’ molecule located at the mouth of the substrate-binding pocket, orients the zinc-bound nucleophilic hydroxide for optimal attack on the substrate. A second hydrogen-bond network engages the zinc-bound hydroxide and the imidazole side chain of His64 through two intervening solvent molecules and this network is likewise important for catalysis. His64 acts as a proton shuttle in catalysis, in which it accepts the proton product (via the bridging solvent molecules) from zinc-bound water as the zinc-bound hydroxide is regenerated; subsequently, the proton product is passed along to buffers. Conformational mobility has been noted for the imidazole ring of the His64 side chain. It has been noted in either an ‘in’ (facing towards zinc) or an ‘out’ (facing away from zinc) position. This facilitates proton transfer between the active-site waters and solvent water at the mouth of the active-site cavity (Tripp et al., 2001 ▶).
Inhibitors such as inorganic anions and aromatic/heterocyclic sulfonamides bind within the carbonic anhydrase (CA) active site by coordinating to the metal ion either in a tetrahedral geometry, replacing the zinc-bound water/hydroxide, or in a pentacoordinated mode, adding to the coordination sphere without displacing the zinc-bound water molecule (Pastorekova et al., 2004 ▶; Supuran & Scozzafava, 2007 ▶). The crystal structures of the acetate complexes of wild-type hCAII (PDB code 1cay) and E106Q mutant hCAII (PDB code 1caz) have previously been solved to 2.1 and 1.9 Å resolution, respectively (Håkansson et al., 1994 ▶). Both crystallized in a typical manner in space group P21, with unit-cell parameters a = 42.7, b = 41.7, c = 73.0 Å, β = 104.6°. The altered active-site hydrogen-bond network caused by the mutation resulted in a different binding mode of the inhibitor in the two complexes.
2. Materials and methods
2.1. Cloning, expression and purification of hCAII
Human carbonic anhydrase II (hCAII) cDNA was overexpressed in Escherichia coli strain BL21(DE3)pLysS from the plasmid pACA/hCAII (a kind gift from Professor Sven Lindskog, Umea University, Sweden) and purified following a previously published procedure (Jackman et al., 1996 ▶). Briefly, the protein was induced by the addition of 0.25 mM IPTG to mid-log E. coli BL21(DE3)pLysS cells followed by the addition of 0.45 mM ZnSO4 and incubation at 303 K for 6 h. The cells were pelleted and resuspended in buffer containing 50 mM Tris–SO4 pH 8.0 (at 277 K); they were then lysed by sonication and the cellular debris removed by centrifugation. The protein was purified by sequential chromatography on DEAE Sephacel, Mono-S and Sephadex G-75 media. The hCAII obtained is ≥98% pure as shown by SDS–PAGE.
2.2. Crystallization
The purified protein (in 50 mM Tris–HCl pH 7.7–7.8) was concentrated to a final concentration of 10 mg ml−1 (the enzyme concentrations were determined at 280 nm spectrophotometrically using a molar absorptivity of 54 mM −1 cm−1). The protein was crystallized using the hanging-drop vapour-diffusion method from a precipitant solution containing 0.2 M sodium acetate trihydrate, 0.1 M Tris pH 8.5, 30% PEG 4000; crystals grew to maximum dimensions of 0.2 × 0.2 × 1.0 mm in about 5 d.
2.3. Data collection
Data were collected at beamline X29 of the National Synchrotron Light Source, Brookhaven National Laboratory. An ADSC Quantum 315 detector was used to collect data using a crystal-to-detector distance of 260 mm (λ = 0.979 Å). The crystal was flash-cooled in liquid nitrogen after transferring it to mother liquor containing 15% glycerol for 20 s. A single crystal was used to collect 180 frames of data with an exposure time of 3 s per 1° oscillation. Data were collected to 1.8 Å resolution and were processed and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▶). Autoindexing followed by integration of diffraction maxima determined the crystal unit cell to be orthorhombic with space group P212121. The Matthews coefficient was calculated to be 1.9 Å3 Da−1, which corresponds to one molecule in the asymmetric unit with a solvent content of 33.8%. The data-collection statistics are given in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Crystal parameters | |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 42.3, b = 71.8, c = 74.0 |
| Data processing | |
| Resolution (Å) | 50–1.8 (1.86–1.80) |
| No. of measured reflections | 434736 |
| No. of unique reflections | 21002 |
| Completeness (%) | 98.6 (95.6) |
| Redundancy (%) | 6.8 (6.7) |
| I/σ(I) | 13.5 (4.1) |
| Rmerge† | 0.050 (0.205) |
| Refinement parameters | |
| Resolution range (F > 0) (Å) | 50–1.8 |
| No. of reflections | 20675 |
| R‡ | 0.187 |
| Rfree | 0.215 |
| No. of atoms | |
| Protein | 2059 |
| Zn | 1 |
| No. of water molecules | 244 |
| No. of ligands | 1 (acetate) |
| R.m.s.d. | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.4 |
| B factors (Å2) | |
| Average, main-chain atoms | 11.7 |
| Average, side-chain atoms | 14.4 |
| Solvent | 20.9 |
R
merge = 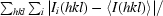
 , where Ii(hkl) is the intensity measurement for a given reflection and 〈I(hkl)〉 is the average intensity for multiple measurements of I(hkl).
, where Ii(hkl) is the intensity measurement for a given reflection and 〈I(hkl)〉 is the average intensity for multiple measurements of I(hkl).
R = 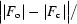
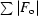 .
.
2.4. Structure solution and refinement
The structure was determined by the molecular-replacement method using MOLREP (Vagin & Teplyakov, 1997 ▶) incorporated in the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶). The starting search model for molecular replacement included the atomic coordinates of one of the subunits of native hCAII (PDB code 1xev) from which all water molecules and heteroatoms, including metal ions, had been excluded. Intensity data in the resolution range 50–3.0 Å were used in a combined cross-rotation and translation search and yielded a solution with a peak height of 12.94σ. A random 5% (1004) of the reflections were set aside for use as a test set for cross-validation in order to remove model bias. Models were refined with CNS v.1.1 (Brünger et al., 1998 ▶) until convergence, combined with inspection of the 2|F o| − |F c| and |F o| − |F c| electron-density maps at all stages. Initial rigid-body refinement to optimize the orientation and translation position of the model was followed by refinement cycles involving Cartesian molecular dynamics by heating to 2500 K and slow-cooling followed by energy minimization and B-factor refinement. A region of extra electron density connected to the zinc was modelled as an acetate ion since sodium acetate had been used in the crystallization solution. A total of 244 water molecules were included using the automatic water-picking function of CNS in combination with visual inspection. After the final cycle of refinement the values of R and R free were 0.187 and 0.215, respectively. PROCHECK (Laskowski et al., 1993 ▶) validation of the final model showed 88% of the residues to be in the most favourable region of the Ramachandran plot. Details of the refinement are given in Table 1 ▶.
3. Results
3.1. General shape of the molecule
The overall structure of the protein is similar to the previously reported structures of hCAII with an additional novel acetate-binding site. The protein has three α-helices, of which one starts and ends with a 310-helical conformation, five 310-helices, four γ-turns, 26 β-turns, five β-bulges, four β-hairpins and 15 β-strands forming two sheets.
3.2. Hydrogen-bonded solvent network
The acetate molecule is directly bound to the zinc ion, replacing the native hydroxide anion in the metal-coordination sphere. One of the carboxylate O atoms (B factor 19.5 Å2) is hydrogen bonded to Thr199 N and the other (B factor 18.9 Å2) is coordinated to zinc at a distance of 2.11 Å and hydrogen bonded to Thr199 Oγ1, which in turn is hydrogen bonded to Glu106 O∊1 (Fig. 1 ▶). The molecule extends into the hydrophobic region made up of residues Val121, Val143, Leu198 and Thr200 (within a distance of 5 Å) of the active-site cavity, displacing the deep water molecule which is usually found to occupy the most remote part of the hydrophobic cavity in the structures of hCAII (Fig. 2 ▶). In the present structure the side chain of His64 appears to be well defined and oriented away from Zn towards the outside, although it has almost always been found to be disordered both in the native and in many hCAII complexes.
Figure 1.

(a) Stereoview of the active site generated with |2F o| − |F c| Fourier coefficients and contoured at 1.2σ. (b) Stereoview of the OMIT |F o| − |F c| electron-density map contoured at the 3σ level. The final refined acetate molecule is superimposed upon the electron-density map. This figure was generated using PyMOL (DeLano, 2002 ▶).
Figure 2.

Active-site solvent network. The zinc ion and water molecules are indicated by gold and red spheres, respectively. Interactions between atoms are indicated by pink dotted lines. Atom colours are black, carbon; red, oxygen; blue, nitrogen; gold, zinc. This figure was prepared using MOLSCRIPT (Kraulis, 1991 ▶) and RASTER3D (Merritt & Bacon, 1997 ▶).
3.3. Comparison with other acetate-complexed hCAII structures
The protein crystallized in space group P212121, in contrast to the P21 space group of previous acetate-complexed structures. The previously reported complexes had been obtained by soaking crystals of hCAII (initially grown in Tris–SO4, ammonium sulfate and HgCl2, with the Hg then being removed using ammonium sulfate, Tris pH 7.8 and β-mercaptoethanol) in 0.3 M acetate and 80 mM citrate pH 6 as opposed to the present crystal form, which was obtained at pH 8.5. Overall, the structure of the complex is similar to that of wild-type hCAII complexed with acetate (Håkansson et al., 1994 ▶), with an r.m.s.d. of 0.47 Å over all Cα atoms. However, the mode of binding of the acetate molecule is different. Contrary to the previously reported structure, the acetate molecule replaces the zinc-bound water in this case, binding directly to the zinc ion. In the previously reported structure the methyl group is oriented towards the hydrophobic side of the active site, in contrast to the structure under discussion in which the methyl group is directed in between the hydrophobic and hydrophilic regions. The B factors of the various atoms of the acetate moiety are comparable in the wild-type and mutant acetate-complexed hCAII structures: 19.9, 23.8 and 18.4 Å2 for CH3, 19.5, 23.2 and 20.0 Å2 for OXT, 18.9, 23.7 and 18.8 Å2 for O, and 21.3, 23.4 and 18.6 Å2 for C for 1xeg, 1cay and 1caz, respectively.
4. Discussion
The complex of hCAII with acetate crystallized in space group P212121. The structure was solved at 1.8 Å resolution by the molecular-replacement method and refined to a final R value of 0.18. The acetate molecule is bound to the zinc, replacing a water/hydroxide moiety in tetrahedral coordination, in contrast to the known acetate-bound structure (Fig. 3 ▶). The overall structure is similar to the known hCAII structures. (This difference in the space group is not unknown for hCAII crystals and may be a consequence of the absence of Hg, which is generally present in the crystallization of the enzymes, from the setup; another possibility is the presence of extra chemical compounds that have been shown to be activators of the enzyme in the crystallization solution with which attempts were made at cocrystallization.) Normally (as in 1cay), acetate is found to interact with Thr199 with the methyl group facing towards the hydrophobic patch formed by Val121, Val143 and Leu198. However, the present structure of the complex shows that acetate has replaced the zinc-bound water, forming a tetrahedral coordination, and thus disrupts the hydrogen-bonded solvent network. Both the carboxylate O atoms interacted with Thr199, with the methyl group directed away from the hydrophobic patch similar to that in the E106Q mutant (PDB code 1caz). The mode of binding of the acetate is similar to that found in the case of the binding of the substrate bicarbonate to mutated T200H hCAII. The bicarbonate group also replaces zinc-bound water, with one of the carboxylate O atoms coordinating to Zn and Thr199 Oγ1, while another is coordinated to Thr199 N. However, the third O atom is coordinated to His94 in the hydrophilic region, whereas the methyl group is directed away from the hydrophilic patch. Calculations made using the CASTP server (Binkowski et al., 2003 ▶) showed that the cavity has a solvent-accessible surface area of 303 Å2 and a volume of 377 Å3, compared with values of 250 Å2 and 285 Å3 for 1cay and 248 Å2 and 279 Å3 for 1caz. In the case of native uncomplexed hCAII, the area of the same pocket is 260 Å2 with a volume of 282 Å3. The volume of the cavity in which the acetate is bound is larger than the volume in the case of the other acetate-bound structures (PDB codes 1cay or 1caz). The strength of ligand binding will be less in this case.
Figure 3.

Superposition of acetate-bound human carbonic anhydrase II structures. 1xeg is in marine, 1cay is in yellow and 1caz is in orange. The alternate conformations of His64 in 1cay are shown. This figure was generated using PyMOL.
Supplementary Material
PDB reference: acetate-bound complex of human carbonic anhydrase II, 1xeg, r1xegsf
Acknowledgments
The authors acknowledge the financial assistance from the Department of Science and Technology, Government of India. PAM acknowledges the Council of Scientific and Industrial Research (India) for the award of a Senior Research Fellowship. We thank Dr Howard Robinson for providing data-collection time at beamline X29 of NSLS, BNL.
References
- Binkowski, T. A., Naghibzadeg, S. & Liang, J. (2003). Nucleic Acids Res.31, 3352–3355. [DOI] [PMC free article] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, California, USA.
- Håkansson, K., Briand, C., Zaitsev, V., Xue, Y. & Liljas, A. (1994). Acta Cryst. D50, 101–104. [DOI] [PubMed]
- Håkansson, K., Carlsson, M., Svensson, L. A. & Liljas, A. (1992). J. Mol. Biol.227, 1192–1204. [DOI] [PubMed]
- Jackman, J. E., Merz, K. M. Jr & Fierke, C. A. (1996). Biochemistry, 35, 16421–16428. [DOI] [PubMed]
- Kraulis, P. J. (1991). J. Appl. Cryst.24, 946–950.
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lindskog, S. (1997). Pharmacol. Ther.74, 1–20. [DOI] [PubMed]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol.277, 505–524. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pastorekova, S., Parkkila, S., Pastorek, J. & Supuran, C. T. (2004). J. Enzyme Inhib. Med. Chem.19, 199–229. [DOI] [PubMed]
- Stams, T. & Christianson, D. W. (2000). EXS, 90, 159–174. [DOI] [PubMed]
- Supuran, C. T. & Scozzafava, A. (2007). Bioorg. Med. Chem.15, 4336–4350. [DOI] [PubMed]
- Tripp, B. C., Smith, K. S. & Ferry, J. G. (2001). J. Biol. Chem.276, 48615–48618. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: acetate-bound complex of human carbonic anhydrase II, 1xeg, r1xegsf