

Abstract
A new class of matrix metalloproteinase (MMP) inhibitors has been identified by screening a collection of compounds against stromelysin. The inhibitors, 2,4,6-pyrimidine triones, have proven to be potent inhibitors of gelatinases A and B. An X-ray crystal structure of one representative compound bound to the catalytic domain of stromelysin shows that the compounds bind at the active site and ligand the active-site zinc. The pyrimidine triones mimic substrates in forming hydrogen bonds to key residues in the active site, and provide opportunities for placing appropriately chosen groups into the S1′ specificity pocket of MMPs. A number of compounds have been synthesized and assayed against stromelysin, and the variations in potency are explained in terms of the binding mode revealed in the X-ray crystal structure.
Keywords: Stromelysin, MMP, X-ray structure, protease inhibitor
Matrix metalloproteinases (MMPs) are normally involved in the breakdown of protein in connective tissue, which is important for growth and development and for wound healing. They are also implicated in several human diseases, where their overexpression or unregulated activation leads to aberrant degradation of connective tissue. Such breakdown of connective tissue can be directly associated with disease symptoms: for example, cartilage degradation in rheumatoid arthritis or osteoarthritis. MMPs are also key players in at least three events in malignancy: primary tumor growth, metastasis, and angiogenesis (Stetler-Stevenson et al. 1993; Nagase and Woessner 1999). In an oncology program to identify new, nonhydroxamate inhibitors of MMP-2 and MMP-9 (gelatinases A and B), we reinvestigated a compound that had shown weak activity against MMP-3 (stromelysin) in a high throughput screen. The 2,4,6-pyrimidine trione 1 has an IC50 of 30 μM against stromelysin; however, when compound 1 was assayed against the oncology targets (gelatinases A and B), it was found to be >30-fold more active against these enzymes than against stromelysin.
The mode of binding of the new class of MMP inhibitors was established via X-ray crystallography. Compound 2, a more potent analog of compound 1, was cocrystallized with stromelysin, and the crystal structure determined at 2.0 Å resolution. Because the catalytic mechanism and structure of the active site of MMPs are known to be conserved, the binding mode of the pyrimidine triones is expected to be similar across the MMP family. A number of the compounds prepared in the oncology program have been assayed against stromelysin, and the observed structure–activity relationships are consistent with the mode of binding revealed by the X-ray crystal structure, as described in the following section.
Results and Discussion
Compound 2, with an IC50 against stromelysin-1 of 2.0 μM, was cocrystallized with the catalytic domain of the enzyme. X-ray data to a resolution limit of 2.0 Å was collected and the crystal structure was solved by molecular replacement methods. The crystals represent a new crystal form with three stromelysin molecules in the asymmetric unit. The interactions between the inhibitor and the three crystallographically independent molecules appear to be identical. Because of differences in crystal packing, the conformation of the loop residues 224–233 bordering the S1′ specificity pocket differs in the three molecules. This region is, however, particularly well defined in molecule B as a result of stabilizing contacts with a neighboring molecule in the crystal lattice. A bound glycerol molecule, soaked into the crystal as a cryoprotectant, is present at the base of the S1′ specificity pocket and is also involved in several hydrogen-bonding interactions with this loop in molecule B.
As shown in Figure 1A ▶, the inhibitor is bound in an orientation that places the hydrophobic 4-phenoxyphenyl moiety in the deep S1′ pocket. The N3 nitrogen atom in the pyrimidine trione ring approaches the active-site zinc most closely, at 2.17 Å, and completes a tetrahedral arrangement of nitrogen ligands about the zinc. Two oxygen atoms of the pyrimidine trione are at distances of 3.00 and 3.12 Å from the zinc, respectively, and possibly contribute additional energetically favorable interactions with the central metal ion. The geometry of the ligands about the active-site zinc is reminiscent of that seen in the small molecule X-ray structures of complexes between another pyrimidine trione and Zn2+ (Wang and Craven 1971; Nassimbeni and Rodgers 1974). The N3–Zn bond vector is essentially coplanar with the imide moiety that spans positions 2–4 in the pyrimidine trione. This is consistent with the enol form of the inhibitor, as shown in Figure 1B ▶, where the deprotonated nitrogen N3 has the requisite in-plane lone pair for liganding to the zinc. The enol form is also favored because it places a proton on the O2 oxygen atom, which can be donated in a bidentate hydrogen bond to the oxygen atoms of the Glu 202 carboxylate group. O2 occupies the same position as the catalytic water molecule that is thought to protonate Glu 202 concomitant with attack on the carbon atom of the scissile bond of peptide substrates. Continuing around the 2,4,6-pyrimidine trione ring in a clockwise sense with reference to Figure 1 ▶, the nitrogen atom N1 donates a hydrogen bond to the backbone carbonyl group of Ala 165, and the oxygen atom O6 accepts a hydrogen bond from the backbone amide group of Leu 164. These hydrogen bonds are normally formed by bound substrates lying in an extended conformation across the active-site cleft.
Fig. 1.

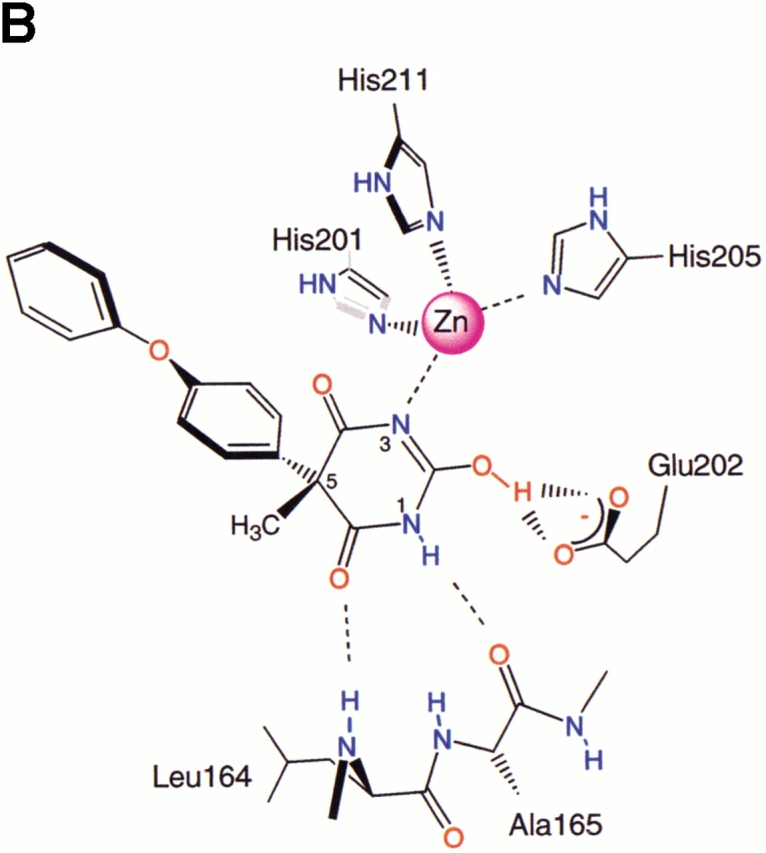
(A) Stereo view of the inhibitor bound to the active site of stromelysin. Coloring of heteroatoms is by atom type, with the remainder of the protein shown in green and the inhibitor in blue. The secondary structure elements of the protein are also shown in ribbon form to help orient the viewer. In this view, the S1′ pocket extends toward the upper left of the figure. The bound glycerol molecule can be seen at the back of the S1′ pocket. The figure was prepared by using the Ribbons program (Carson 1997). (B) Schematic drawing of the interactions between the inhibitor and the active site, in the same orientation as in A. Hydrogen bonds discussed in the text are indicated. The numbering scheme for the inhibitor is also shown. (Reprinted from Foley et al. 2001, with permission of Elsevier Science.)
The decreased potency of the modified compounds shown in Table 1 confirms the binding mode established via the X-ray analysis. Compound 3, the (racemic) N-methyl derivative of compound 2, was inactive. Neither methylation of the nitrogen, which is liganded to the active-site zinc, nor the nitrogen, which is hydrogen bonded to the backbone carbonyl of Ala 165, is tolerated. Substitution of the O2 oxygen with sulfur, as in compound 4, also abolishes inhibitory activity against stromelysin. A number of subtle factors could be involved in reducing the activity of this compound, including the reduced tendency of thioamides to enolize to the imidothiol form (Kjellin and Sandström 1973). The enol tautomer of the pyrimidine trione with N3 deprotonated and O2 donating hydrogen bonds to the carboxylate oxygen atoms of Glu 202 is most likely the bound form of the inhibitor, and the thioamide of compound 4 is not expected to be able to participate in hydrogen bonding to the side chain of Glu 202. Compounds 3 and 4 were similarly diminished in their activity against gelatinases A and B, supporting the assertion of a common binding mode for this inhibitor class against an assortment of MMPs (Foley et al. 2001).
Table 1.
Inhibition of stromelysin activity
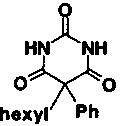
| Compound number | Structure | IC50 | |
| 1 |
|
30 μM | |
| 2 |
|
2.0 μM | |
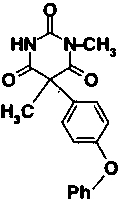
| 3 |
|
>40 μM | |
| 4 |
|
>40 μM | |
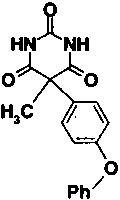
| 5 |
|
33 μM | |
| 6 |
|
9 μM |
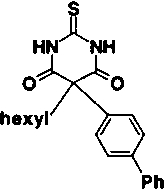
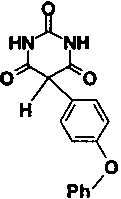
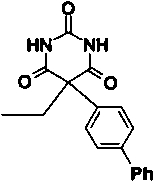
Some modifications to the alkyl and aryl substituents on the pyrimidine trione ring were also tested. Removal of the methyl group of compound 2, giving compound 5, reduces potency by a factor of 16. The methyl is solvent exposed in the X-ray structure and does not make a significant number of van der Waals contacts with the enzyme. However, replacement of the methyl by hydrogen allows enolization to place the aryl group in the same plane as C4, C5, and C6 of the pyrimidine trione ring system, as shown in Figure 2 ▶. Nuclear magnetic resonance studies of compound 5 in solution detected both the keto and enol forms shown in Figure 2 ▶, in a ratio of 3:2. In the X-ray complex with compound 2, the phenoxyphenyl substituent is in an axial position nearly perpendicular to the pyrimidine trione ring, whereas in the enol form of compound 5, the bridging phenyl group is coplanar with the pyrimidine trione. The enol form of compound 5 is thus not able to bind to stromelysin in the same fashion as compound 2, which simultaneously places the hydrophobic phenoxyphenyl group in the S1′ pocket and ligands the active-site zinc. Substitution of the phenoxyphenyl moiety by the linear, more rigid biphenyl group reduced potency by a factor of 4.5-fold (compare compound 6 to compound 2 in Table 1). The angle of entry to the S1′ pocket is not ideal for the linear biphenyl group, as it would clash with residues 197 and 223 on the wall of the S1′ pocket if it were to enter in the same fashion as the phenoxyphenyl moiety of compound 2. A wide variety of hydrophobic groups are accepted by the S1′ pocket of stromelysin (Hajduk et al. 1997), and the optimum choice of this group will be different for each class of inhibitor with a unique strategy for liganding the active-site zinc.
Fig. 2.

Tautomeric forms of compound 5 observed in solution. Replacement of the hydrogen on C5 by a methyl group, as in compound 2, prevents this enolization.
In summary, we have discovered a new class of zinc-liganding inhibitors of MMPs through high-throughput screening of a library of small molecules. In a parallel study, the structure of MMP-8 in complex with a pyrimidine trione has been determined (Brandstetter et al. 2001). Through a combination of medicinal chemistry and X-ray crystallography, we now understand the interactions between the 2,4,6-pyrimidine trione inhibitors and the active site of representative MMPs at the atomic level of detail. The two substituents at C5 of the pyrimidine trione ring provide separate opportunities for fine-tuning the selectivity and potency of this class of inhibitors. One substituent is ideally oriented for entry to the S1′ pocket of MMPs, and the other extends toward the S2′ site of these enzymes. This class of molecule provides unique opportunities for developing inhibitors of selected MMPs, with possible advantages over the previously known classes of zinc-liganding inhibitors such as the hydroxamic acids.
Materials and methods
Synthesis of the inhibitors is described in Foley et al. (2001). The IC50 values against stromelysin were determined as described in Bickett et al. (1993); however, we used MES buffer at pH 6.5. The recombinant stromelysin used for the crystallographic work was purified as described in Marcy et al. (1991) and includes residues 83–250 after activation with (4-aminophenyl)mercuric acetate. For crystallization, stromelysin (10 mg/mL in 20 mM Tris-HCl, 5 mM CaCl2, 0.02% NaN3 at pH 7.5) was preincubated overnight at 277K with 2.5 mM inhibitor. Crystals were grown via the hanging-drop method at 293K from 2 μL of the protein–inhibitor complex mixed with 1 μL of well solution. The well solution contained 0.2 M (NH4)2SO4, 30% PEG-MME 5000, and 0.1 M HEPES buffer at pH 7.5. Before collecting X-ray data, crystals were transferred to a cryoprotectant consisting of 8% glycerol, 0.2 M (NH4)2SO4, 30% PEG-MME 5000, and 0.1 M HEPES buffer at pH 7.5. Crystals were frozen in situ in a stream of nitrogen gas at 110K. A data set extending to 2.8 Å was collected on a Mar imaging plate with Cu Kα radiation provided by a Rigaku rotating-anode generator operating at 50 kV, 90 mA. A second set of data extending to 2.0 Å was obtained with the Mar imaging plate at the Brookhaven NSLS beamline X8C by using radiation of wavelength 1.072 Å. All data were reduced by using the Denzo and Scalepack software (Otwinowski and Minor 1996). The crystals are of space-group P43212 with unit cell dimensions a = b = 103.8 Å, c = 147.5 Å.
Molecular replacement calculations were performed with a model consisting of residues 83–250 from another crystal form. Molecular replacement in AMoRe (Navaza 1994) gave a solution with two molecules per asymmetric unit. The R-factor for data in the 20.0 - 3.5 Å range after rigid-body refinement in AMoRe was 44.1%, and examination of the packing showed that the two molecules formed a connected three-dimensional lattice. Inspection of the electron-density map calculated with phases based on the first two molecules revealed the presence of a third molecule. Rigid-body refinement of all three molecules in AMoRe lowered the R-factor to 38.4%. With three molecules per asymmetric unit, the solvent content is 60%.
Noncrystallographic symmetry restraints were applied in Refmac (Murshudov et al. 1997) during the initial stages of refinement against the 2.8 Å data; the restraints were gradually released as the resolution limit was extended to 2.0 Å. Solvent molecules were added with the aid of ARPP (Lamzin and Wilson 1997). The conformation of the loop including residues 226–231 in the current structure differs from the conformation seen in the molecular replacement model, and this section of the model was rebuilt in O (Jones et al. 1991). Cycles of adjustments to the model with O and refinement in Refmac lowered the working R-factor to 24.4% for data in the range 20.0 - 2.0 Å (free R-factor 28.4% for a test set consisting of 10% of the reflections). The average B-factors for the three independent molecules in the asymmetric unit are 19.8, 23.5, and 43.4 Å2. Data quality and refinement statistics are given in Table 2. The structure has been deposited in the Protein Data Bank (Berman et al. 2000) as entry 1G4K.
Table 2.
Data and refinement statistics
| Resolution | 30.0 − 2.0 Å |
| No. of unique reflections | 55,161 |
| Redundancy | 8.6 fold |
| R-merge | 4.7% (12.1%)a |
| Completeness | 99.9% (99.9%)a |
| R-work | 24.4% |
| R-free | 28.4% |
| Resolution used in refinement | 20.0 − 2.0 Å |
| No. of reflections used in refinement | 54,951 |
| No. of atoms in model | 4294 |
| No. of water molecules | 187 |
a Values in parentheses are for the highest resolution shell (2.07 − 2.00 Å).
Acknowledgments
We thank Kwei Lan Tsao and Alejandro Lugo for performing the IC50 assays, Leon Flaks for assistance at beamline X8C, Jens Birktoft for encouragement, and Hans Brandstetter, Rick Engh, and Frank Grams for providing data on their crystal structure prior to publication.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.48401.
References
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The protein data bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickett, D.M., Green, M.D., Berman, J., Dezube, M., Howe, A.S., Brown, P.J., Roth, J.T., and Mcgeehan, G.M. 1993. A high throughput fluorogenic substrate for interstitial collagenase (MMP-1) and gelatinase (MMP-9). Anal. Biochem. 212 58–64. [DOI] [PubMed] [Google Scholar]
- Brandstetter, H., Grams, F., Glitz, D., Lang, A., Huber, R., Bode, W., Krell, H.-W., and Engh, R.A. 2001. The 1.8 angstrom crystal structure of an MMP8-barbiturate inhibitor complex reveals a previously unobserved mechanism for collagenase substrate recognition. J. Biol. Chem. (in press). [DOI] [PubMed]
- Carson, M. 1997. Ribbons. Methods Enzymol. 277 493–505. [PubMed] [Google Scholar]
- Foley, L.H., Palermo, R., Dunten, P., and Wang, P. 2001. Novel 5,5-disubstituted pyrimidine-2,4,6-triones as selective MMP inhibitors. Bioorg. Med. Chem. Lett. 11 969–972. [DOI] [PubMed] [Google Scholar]
- Hajduk, P.J., Sheppard, G., Nettesheim, D.G., Olejniczak, E.T., Shuker, S.B., Meadows, R.P., Steinman, D.H., Carrera, G.M., Marcotte, P.A., Severin, J., et al. 1997. Discovery of potent nonpeptide inhibitors of stromelysin using SAR by NMR. J. Am. Chem. Soc. 119 5818–5827. [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A A47 110–119. [DOI] [PubMed] [Google Scholar]
- Kjellin, G. and Sandström, J. 1973. The thione-thiol tautomerism in simple thioamides. Acta Chem. Scand. 27 209–217. [Google Scholar]
- Lamzin, V.S. and Wilson, K.S. 1997. Automated refinement for protein crystallography. Methods Enzymol. 277 269–305. [DOI] [PubMed] [Google Scholar]
- Marcy, A.I., Eiberger, L.L., Harrison, R., Chan, H.K., Hutchinson, N.I., Hagmann, W.K., Cameron, P.M., Boulton, D.A., and Hermes, J.D. 1991. Human fibroblast stromelysin catalytic domain: Expression, purification, and characterization of a C-terminally truncated form. Biochemistry 30 6476–6483. [DOI] [PubMed] [Google Scholar]
- Murshudov, G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-liklihood method. Acta Crystallogr. D D53 240–255. [DOI] [PubMed] [Google Scholar]
- Nagase, N. and Woessner, J.F. 1999. Matrix metalloproteinases. J. Biol. Chem. 274 21491–21494. [DOI] [PubMed] [Google Scholar]
- Nassimbeni, L.R. and Rodgers, A. 1974. The crystal structure of the bis(5,5′-diethylbarbiturato) bispicoline complex of zinc(II). Acta Crystallogr. B B30 1953–1961. [Google Scholar]
- Otwinowski, Z. and Minor, W. 1996. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Stetler-Stevenson, W.G., Aznavoorian, S., and Liotta, L.A. 1993. Tumor cell interactions with extracellular matrix during invasion and metastasis. Annu. Rev. Cell Biol. 9 941–973. [DOI] [PubMed] [Google Scholar]
- Wang, B.C. and Craven, B.M. 1971. Synthesis and crystal structure determinations of the bis-(5,5-diethylbarbiturato)-bisimidazole complexes of cobalt(II) and zinc(II). J. Chem. Soc. (D) Chem. Commun. 290–291.