

Abstract
Coeliac disease is characterized by immunoglobulin-A (IgA)-class autoantibodies targeted against transglutaminase 2 (TG2), a multi-functional protein also with a role in angiogenesis. These antibodies are present in patient serum but are also found bound to TG2 below the epithelial basement membrane and around capillaries in the small intestinal mucosa. Based on these facts and the information that the mucosal vasculature of coeliac patients on a gluten-containing diet is disorganized, we studied whether the coeliac disease-specific autoantibodies targeted against TG2 would disturb angiogenesis. The effects of coeliac disease-specific autoantibodies on in vitro angiogenesis were studied in angiogenic cell cultures. The binding of the antibodies to cells, endothelial sprouting, migration of both endothelial and vascular mesenchymal cells, the integrity of the actin cytoskeleton in both cell types and the differentiation of vascular mesenchymal cells were recorded. In vitro, IgA derived from coeliac disease patients on a gluten-containing diet binds to surface TG2 on endothelial and vascular mesenchymal cells and this binding can be inhibited by the removal of TG2. In addition, coeliac disease-specific autoantibodies targeting TG2 disturb several steps of angiogenesis: endothelial sprouting and the migration of both endothelial and vascular mesenchymal cells. Furthermore, the autoantibodies cause disorganization of the actin cytoskeleton in both capillary cell types that account most probably for the defective cellular migration. We conclude that coeliac disease-specific autoantibodies recognizing TG2 inhibit angiogenesis in vitro. This disturbance of the angiogenic process could lead in vivo to the disruption of the mucosal vasculature seen in coeliac disease patients on a gluten-containing diet.
Keywords: angiogenesis, coeliac disease, disease-specific autoantibodies, transglutaminase 2
Introduction
Coeliac disease is an intestinal disorder triggered by dietary gluten from wheat, rye and barley and is characterized by small-intestinal lesions with villous atrophy and crypt hyperplasia. At cellular level, classical hallmarks include increased epithelial cell proliferation and decreased differentiation as well as massive T cell infiltration into the mucosa, which is thought to be the focal driving force in the disease pathogenesis [1,2]. Another characteristic feature for the disease is the presence of specific immunoglobulin-A (IgA) class autoantibodies in the serum of patients on a gluten-containing diet. These antibodies are targeted against transglutaminase 2 (TG2) [3], a multi-functional enzyme involved in processes such as angiogenesis [4,5] and wound healing [6]. TG2 regulates distinct cellular functions, including organization of the cytoskeleton, cell adhesion and cell death [7]. Although present in the serum of coeliac disease patients not on a diet, the antibodies are produced locally in the small-intestinal mucosa [8], where they are deposited below the epithelial basement membrane as well as around mucosal blood vessels [9–12]. On a gluten-free diet the mucosal antibody deposits, similar to the serum antibodies, disappear slowly as the mucosa begins to heal [10]. The mucosa heals without scarring within 1–2 years on a strict gluten-free diet and deteriorates again if gluten is reintroduced into the diet.
Contrary to the general view that coeliac disease-specific gluten-induced IgA class autoantibodies targeting TG2 are only bystanders in the disease pathogenesis [1,2], several facts suggest that TG2 autoantibodies might themselves be pathogenic. These autoantibodies are functional, because the IgA deposits in the small bowel, i.e. the antibodies produced locally and targeting in vivo TG2 in the gut can bind recombinant TG2 [12] and because the serum antibodies also inhibit intestinal epithelial cell differentiation [13]. Moreover, these gluten-triggered autoantibodies induce intestinal epithelial cell proliferation [14] and increase epithelial permeability and activate monocytes [15].
The small-intestinal vasculature in the lamina propria is composed of a single arteriole which traverses the long axis of the villi before branching at the tip of the villus to form a capillary tuft. In addition to providing mechanical support to the villi, the mucosal microvasculature plays an important role in the digestion of nutrients, nutrient absorption and barrier function [16]. The small-intestinal vasculature undergoes continuous remodelling throughout life, because vessels are constantly lost and gained at an equal rate [17,18]. This ongoing angiogenesis involves two cell types, the endothelial cells and vascular mesenchymal cells, and several phases: endothelial cell sprouting, migration and the formation of the endothelial tube, followed by the recruitment and migration of vascular mesenchymal cells from the surrounding connective tissue to the endothelial tube, and finally the maturation of the nascent vessel [19]. Thus, proper capillary development and function requires controlled behaviour of the endothelial and the vascular mesenchymal cells as well as their precise movement and coordination of differentiation. The mucosal vasculature in the small intestine of a coeliac disease patient on a gluten-containing diet differs considerably from the above-described normal vasculature in the healthy small intestine. As described by Cooke and Holmes as far back as 1984, in coeliac disease-affected mucosa the capillary tufts are totally missing and the entire vasculature is disorganized [20]. In addition to the gross changes in the appearance of the vasculature network, even older data describe changes in the structure of the capillary endothelial cells following alteration in gluten intake [21,22]. On the basis of the previously mentioned reports, the fact that the coeliac patient IgA class antibodies are functional [13–15], the presence of TG2-targeted antibody deposits around blood vessels in vivo[9] and the role of TG2 in angiogenesis [4,5], we hypothesized that the coeliac disease-specific autoantibodies have a biological effect in interfering with angiogenesis.
Materials and methods
Purification of IgA and IgG autoantibodies
Total IgA fractions from serum samples from eight coeliac disease patients on a gluten-containing diet positive for anti-TG2 antibodies (median titre > 100 U/ml, minimum 56·4, maximum > 100 U/ml) and three non-coeliac controls negative for anti-TG2 antibodies (< 5 U/ml) were purified using cyanogen bromide (CNBr)-activated Sepharose 4B (Pharmacia Upjohn, Uppsala, Sweden) coupled with 7 mg/ml rabbit anti-human IgA antibodies (Sigma Aldrich, St Louis, MO, USA). The serum sample was passed through the column, after which the column was washed prior to elution of the IgA with 0·1 M glycine-HCl in 0·5 M NaCl, pH 2·5. The collected IgA fractions were neutralized with 1 M Tris-HCl pH 8·0 before removal of glycine by passing the samples through PD-10 columns (Pharmacia Upjohn). The concentrations were determined by enzyme-linked immunosorbent assay (ELISA) (Celikey®; Pharmacia Diagnostics GmbH, Freiburg, Germany) using affinity-purified human IgA (Dako, Copenhagen, Denmark) as standard. The IgA samples were lyophilized and resolubilized in Hank's balanced salt solution to a final concentration of 100 μg/ml.
Affinity purification of TG2-specific IgA class autoantibodies was not performed because of the high content of oligosaccharide side chains in IgA molecules, which leads to technical difficulties. In order to show that the effects of coeliac disease patient IgA are mediated by antibodies targeting TG2, we purified TG2-specific IgG class autoantibodies from IgA-deficient coeliac disease patients on a gluten-containing diet for comparison. Total IgG fractions from one IgA-deficient coeliac patient not on a diet and one non-coeliac control subject were purified using Protein G Sepharose 4 fast flow (Pharmacia Biotech AB, Uppsala, Sweden), similarly to IgA samples. The collected IgG fractions were neutralized and glycine removed as described above. The TG2-specific antibodies of the pooled total IgG fractions were affinity-purified using CNBr-activated Sepharose 4B coupled with guinea pig TG2 (Sigma-Aldrich), also as described above. The concentrations were determined by ELISA. The total IgG sample from the healthy subject and the TG2-specific IgG from the coeliac disease patient were lyophilized and resolubilized in Hank's balanced salt solution to a final concentration of 100 μg/ml.
Cell lines and cultures
Human umbilical vein endothelial cells (HUVECs) were cultured in endothelial growth medium-2 (EGM-2) (Clonetics, San Diego, CA, USA), 20% fetal bovine serum (FBS), 2 mM glutamine (Gibco brl, Paisley, Scotland, UK), 100 U penicillin, 100 μg/ml streptomycin (Gibco brl) and 25 μg/ml endothelial cell growth supplement (Clonetics).
C3H/10T1/2 cells (abbreviated in the text as 10T1/2) (CCL-226) were purchased from the American Type Culture Collection (LGC Promochem, Borås, Sweden) and were maintained in Dulbecco's modified Eagle's medium (Gibco brl) with high glucose, 10% FBS, 2 mM glutamine, 100 U penicillin and 100 μg/ml of streptomycin.
In vitro angiogenic culture
The in vitro angiogenic cultures were performed as described previously [23]. Briefly, 24-well plates were coated withrat-tail tendon-derived type I native collagen (1·6 mg/ml) for 1 h. 10T1/2 cells and HUVECs were plated on the collagen-coated wells at a ratio of 1:2 and cultured in EGM-2 medium. To test the effects of different antibodies, the culture media were supplemented with 1·2 μg of coeliac disease patient or healthy control IgA, 1·2 μg of anti-TG2-specific coeliac disease patient IgG or control IgG, or with 60 ng of the mouse monoclonal IgG class TG2 antibody, CUB7402 [24] (NeoMarkers, Fremont, CA, USA) or negative mouse IgG1 (Dako). Cultures were maintained at 5% CO2 in a temperature of 37°C and the branching of the cells was analysed by an inverted-phase contrast microscope (Carl Zeiss Vision GmbH, Munchen-Hallbergmoos, Germany) after 3 days of culture. The length of the branches from four longest tubes in four images taken from randomly selected microscopic fields was measured by Analysis-software (Soft Imaging Systems GmbH, Munster, Germany). All experiments were performed in duplicate and repeated twice. The experiments with IgA were performed with three different coeliac disease and non-coeliac patient IgAs.
Western blotting
Proteins were isolated from HUVEC and 10T1/2 co-cultures using RIPA buffer containing Complete Mini Protease inhibitors (Boehringer Mannheim, Indianapolis, IN, USA). Protein lysates were electrophored on NuPAGE Novex 10% Bis-Tris Gels (Invitrogen, San Diego, CA, USA) and transferred electrophoretically to Hybond-C Extra membranes (Amersham Life Sciences, Arlington Heights, IL, USA). The membranes were blocked and then incubated overnight with α-smooth muscle actin antibody [25] (Sigma-Aldrich) (1:1000). After washing, secondary horseradish peroxidase-conjugated anti-mouse antibody (1:3000) (Dako) was incubated for 1 h before the signal was detected using the enhanced chemiluminescence detection system (Amersham Life Sciences). Quantification of the bands was performed using the Kodak 1D image analysis software (Kodak, New Haven, CT, USA).
Scratch wound assay
HUVECs and 10T1/2 cells (25 000 cells/well) were plated on type I collagen-coated 24-well plates (Nunc, Roskilde, Denmark). When the cells reached confluence, the monolayers were wounded and the antibodies under study were added. To distinguish migration from proliferation, proliferation was inhibited by mitomycin C (0·02 mg/ml) (Sigma-Aldrich). After a 24-h culture period the cells were washed, fixed in 4% paraformaldehyde and stained with 0·5% crystal violet (Sigma-Aldrich). The migration of the cells was assessed by counting the number of nuclei observed in the denuded area of preselected microscopic fields. The experiments were carried out in duplicate and repeated three times.
Immunofluorescent stainings
For co-localization studies of coeliac disease-specific type IgA and TG2 both HUVECs and 10T1/2 cells were plated separately on type I collagen-coated four-chamber polystyrene vessel culture slides. At approximately 80% confluence both cultures were exposed to either coeliac disease or non-coeliac patient IgA for 24 h. Subsequently, the cells were fixed in 4% paraformaldehyde. After washing, the cells were incubated for 30 min with 0·1 M sodium citrate buffer (pH 5) or with 1 M potassium thiocyanate (KSCN). Consequently, after blocking with 0·5% bovine serum albumin, the cells were stained with mouse CUB7402 antibody (1:200), which was detected with Alexa Fluor 568 conjugated secondary anti-mouse antibody (1:1000) (Molecular Probes, Leiden, the Netherlands) and finally with fluorescein isothiocyanate (FITC)-conjugated anti-human IgA antibody (1:40) (Dako) prior to mounting.
In further experiments, extracellular TG2 was removed from the cells using 0·25% chloroacetic acid (Fluka Chemie AG, Buchs, Switzerland) in 0·2 M NaCl, pH 2·7, after treatment with KSCN according to the protocol described by Salmi et al. [12]. Chloroacetic acid disrupts the tight binding of TG2 to fibronectin and removes TG2 from cell surfaces. Thereafter, the cells were stained similarly for TG2 and IgA.
The co-localization of IgA with TG2 on the cell surface was analysed from images taken using the Axiovision 3·0 program (Carl Zeiss Vision GmbH) by ImageJ [26,27] using ImageJ Colocalization Finder-PlugIn to show Pearson's correlation and the overlap coefficient of co-localization.
For intracellular actin stainings the cells were fixed as above and permeabilized with 0·2% Triton X-100 (Sigma-Aldrich). After blocking, the cells were incubated with Alexa FITC-conjugated mixed isomers of phalloidin (Molecular Probes) 1:200 for 30 min at room temperature and washed three times in PBS prior to mounting. Samples were viewed with a confocal laser microscope.
Statistical analysis
Statistical analysis was performed using the non-parametric two-tailed Mann–Whitney U-test. The data are presented as mean ± standard error of the mean (s.e.m.). A P-value < 0·05 was considered significant.
Ethical considerations
The study protocol was approved by the Ethics Committee of Tampere University Hospital, Tampere, Finland and of Heim Pal Children's Hospital, Budapest, Hungary. All subjects gave written informed consent.
Results
Coeliac disease patient IgA binding to capillary cell types in culture
Double immunofluorescence stainings and co-localization analyses were performed in order to verify the binding of coeliac disease patient IgA to cell surface TG2. The staining showed that IgA derived from a coeliac disease patient on a gluten-containing diet binds to the cell surfaces of both HUVECs and 10 T1/2 cells, where it co-localizes with TG2 (co-localization percentage > 90) (Fig. 1). When cell surface TG2 was removed from the surface of both cell types, the binding of IgA to the cell surface was abolished completely (data not shown).
Fig. 1.
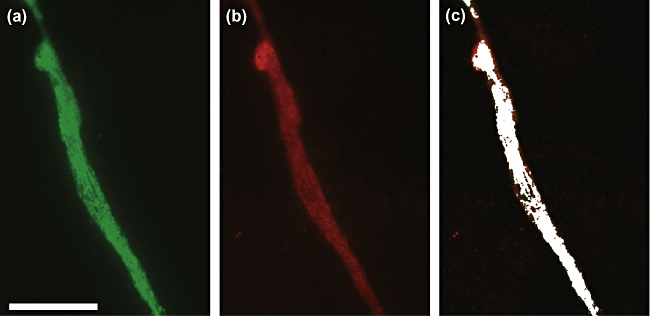
Affinity-purified coeliac disease immunoglobulin-A from a patient on a gluten-containing diet (a) and transglutaminase 2 (b) are co-localized (c) on the surfaces of capillaries formed in in vitro angiogenic cultures, as described in Material and methods. Scale bar 200 μm; co-localization 95%.
The effects of coeliac disease patient autoantibodies on in vitro behaviour of endothelial cells
We first tested the effects of all the antibodies on endothelial cell sprouting on collagen I, where control cells aggregate and form sprouts before fusing into tube-like structures [28]. IgA derived from coeliac disease patients on a gluten-containing diet reduced the length of endothelial sprouts significantly, by approximately 50%, compared with sprouts formed in the presence of non-coeliac IgA (mean 439 μm, s.e.m. = 32 μm versus mean 863 μm, s.e.m. = 82 μm, P < 0·001) (Fig. 2). Similarly, affinity purified patient TG2-specific IgG reduced endothelial sprouting significantly when compared with non-coeliac IgG (mean 409 μm, s.e.m. = 66 μm versus mean 949 μm, s.e.m. = 154 μm, P = 0·0028) (Fig. 2). In contrast to coeliac disease patient autoantibodies, commercially available monoclonal anti-TG2 antibody, CUB7402, failed to exhibit such an effect (mean 759 μm, s.e.m. = 40 μm versus mean 853 μm, s.e.m. = 59 μm) (Fig. 2).
Fig. 2.
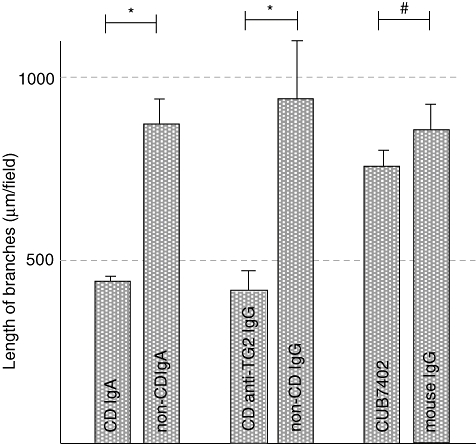
The length of endothelial cell branches in cultures supplemented with coeliac disease patient and non-coeliac subject immunoglobulin-A (IgA), coeliac disease patient anti-transglutaminase 2 (TG2) IgG from an IgA-deficient patient and healthy control IgG as well as CUB7402 and irrelevant mouse IgG. *P < 0·001; #not significant; CD: coeliac disease.
We next sought to determine whether coeliac disease patient autoantibodies also affect the migration of endothelial cells. To this end, we performed scratch wound assays with endothelial cells in the presence of coeliac disease patient IgA, IgA-deficient coeliac disease patient anti-TG2-IgG and CUB7402 as well as their corresponding controls. Both types of coeliac disease-specific autoantibodies, total IgA and anti-TG2-specific IgG, reduced significantly the number of migrating endothelial cells when compared with their controls (mean 26, s.e.m. = 1 versus mean 101, s.e.m. = 2, P < 0·001, mean 23 s.e.m. = 2 versus mean 108, s.e.m. = 3, P < 0·001 respectively) (Fig. 3). Commercial anti-TG2-specific antibody, CUB7402, exerted an effect similar to that of the coeliac patient antibodies (mean 36, s.e.m. = 1 versus mean 77, s.e.m. = 3, P < 0·001) (Fig. 3).
Fig. 3.
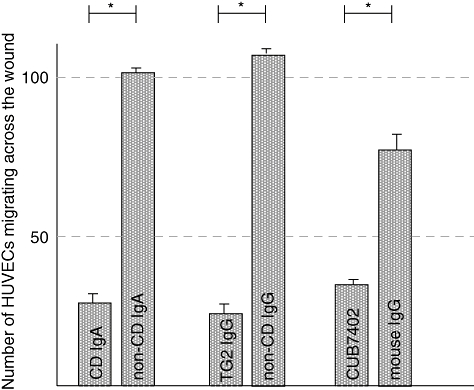
The effects of coeliac disease patient and healthy control subject immunoglobulin-A (IgA), coeliac patient affinity-purified anti-transglutaminase 2 (TG2) IgG and healthy control IgG as well as CUB7402 and control mouse IgG on the number of migrating endothelial cells. *P < 0·001; CD, coeliac disease.
We performed phalloidin–FITC staining to visualize the actin cytoskeleton in endothelial cells in order to study whether the inhibited endothelial cell migration could be due to a disarranged actin cytoskeleton. The staining showed clearly a disorganization of the actin stress fibres in cells treated with coeliac disease patient IgA and anti-TG2-IgG as well as CUB7402, but not in cells treated with control antibodies (Fig. 4).
Fig. 4.
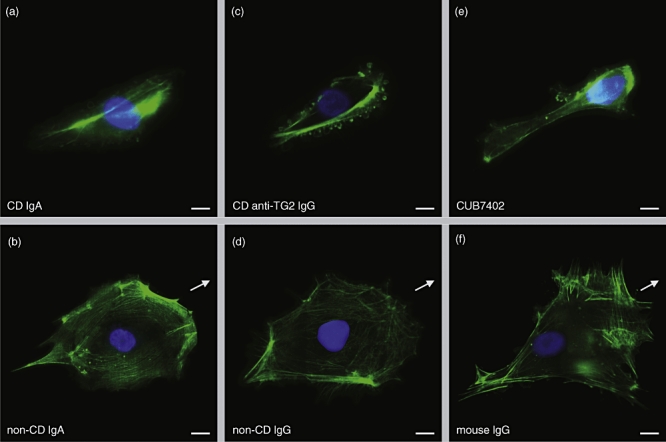
The organization of actin stress fibres in endothelial cells cultured in medium containing coeliac disease patient (a) or non-coeliac patient immunoglobulin-A (IgA) (b), coeliac patient affinity-purified anti-transglutaminase 2 (TG2) IgG (c) and healthy control IgG (d) as well as CUB7402 (e) or control mouse IgG (f). Arrows point in the direction of the movement. Scale bar 20 μm; CD, coeliac disease.
The in vitro effects of coeliac disease patient autoantibodies on vascular mesenchymal cells
We investigated whether coeliac disease patient IgA or anti-TG2-IgG also disrupts in vitro capillary formation in an angiogenic cell culture composed of both endothelial and mesenchymal cells. IgA derived from coeliac disease patients on a gluten-containing diet also inhibited branching significantly in co-culture conditions, reducing the length of branches by approximately 50% when compared with cultures treated with control IgA (mean 860 μm, s.e.m. = 42 μm versus mean 1503 μm, s.e.m. = 118 μm, P < 0·001), similarly to coeliac patient anti-TG2-specific IgG (mean 780 μm, s.e.m. = 70 μm versus mean 1606 μm, s.e.m. = 109 μm, P < 0·001) (Fig. 5). The commercial monoclonal TG2 antibody, CUB7402, had an inhibiting effect on capillary branches similar to that of coeliac disease patient autoantibodies (mean 1041 μm, s.e.m. = 84 μm versus mean 1517 μm, s.e.m. = 101 μm, P < 0·001) (Fig. 5).
Fig. 5.
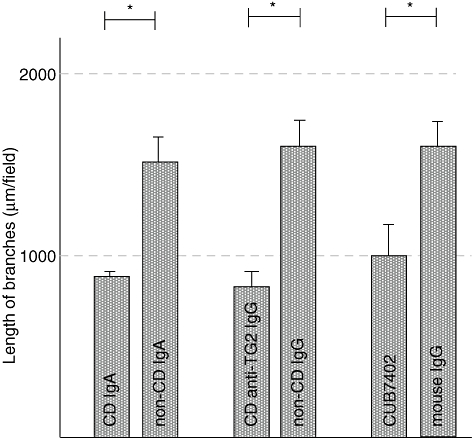
The length of the capillary branches in cocultures of human umbilical vein endothelial cells and 10T1/2 cells cultured in the presence of coeliac disease patient and non-coeliac patient immunoglobulin-A (IgA), IgA-deficient coeliac disease patient anti-transglutaminase 2 (TG2) IgG and healthy control IgG as well as commercial TG2 antibody CUB7402 and its relevant control. *P < 0·001; CD, coeliac disease.
Because coeliac disease patient IgA and anti-TG2-IgG inhibited the migration of endothelial cells, we studied whether vascular mesenchymal cell migration was also affected. The wound closure assays showed that coeliac disease IgA inhibited significantly the migration of vascular mesenchymal cells, by 67% when compared with control IgA (mean 99 cells, s.e.m. = 3 versus mean 300 cells, s.e.m. = 3, P < 0·001) (Fig. 6). Coeliac disease anti-TG2-IgG (mean 93 cells, s.e.m. = 3 versus mean 311 cells, s.e.m. = 3, P < 0·001) and CUB7402 had a similar effect (mean 136 cells, s.e.m. = 3 versus mean 313 cells, s.e.m. = 5, P < 0·001) (Fig. 6).
Fig. 6.
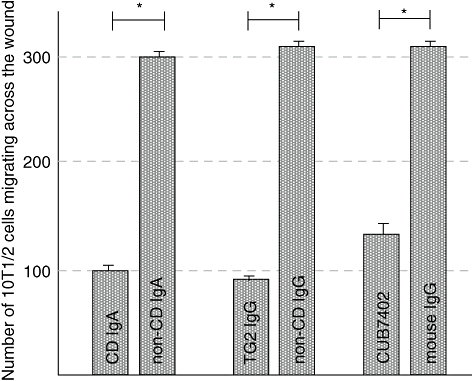
The number of migrating 10T1/2 cells in the presence of coeliac patient immunoglobulin-A (IgA), IgA-deficient coeliac patient anti-transglutaminase 2 (TG2) IgG and CUB7402 or their corresponding controls. *P < 0·001; CD, coeliac disease.
As in the case of endothelial cells, there was a complete disorganization of the vascular mesenchymal cell actin cytoskeleton in the presence of coeliac disease patient IgA and anti-TG2-IgG or CUB7402 (Fig. 7).
Fig. 7.
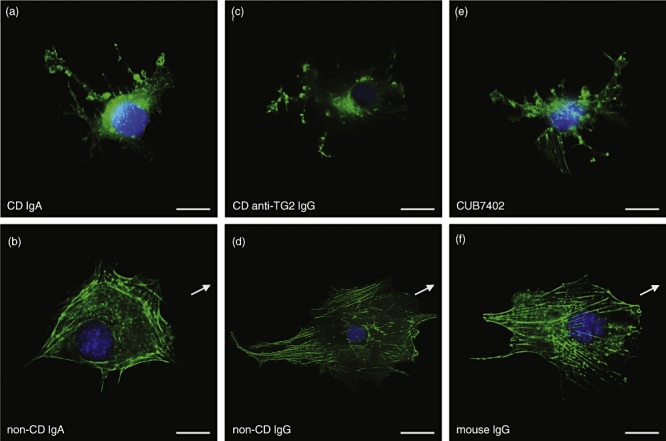
Actin stress fibres organization in vascular mesenchymal cells cultured in a medium containing coeliac patient immunoglobulin-A (IgA) (a), non-coeliac patient IgA (b), coeliac patient anti-transglutaminase 2 (TG2) IgG (c), control IgG (d), CUB7402 (e) or control mouse IgG (f). Arrows indicate the direction of movement. Scale bar 20 μm; CD, coeliac disease.
We also analysed the effects of the coeliac disease-specific autoantibodies on the expression level of vascular mesenchymal cell differentiation marker α-smooth muscle actin in mesenchymal 10T1/2 cells which differentiate when co-cultured with endothelial cells [29]. Western blotting experiments showed that none of the tested antibodies affected the expression of α-smooth muscle actin in 10T1/2 cells (Fig. 8).
Fig. 8.

Coeliac disease patient immunoglobulin-A (IgA) or CUB7402 do not affect the differentiation of vascular smooth muscle cells/pericytes, as indicated by the expression of α-smooth muscle actin (α-sma).
Discussion
In this study we provide evidence that coeliac disease-specific autoantibodies have a role in the disease pathogenesis by hindering angiogenesis. Angiogenesis commences with sprouting of endothelial cells, which is followed by migration of endothelial cells along the formed sprout and the formation of the endothelial tube [30]. Subsequently, mesenchymal cells migrate from the surrounding connective tissue to surround the endothelial tubes [19], after which they differentiate into vascular smooth muscle cells or pericytes [31]. Our findings show that coeliac disease-specific autoantibodies inhibit all the previously mentioned steps of angiogenesis except vascular mesenchymal cell differentiation. In addition, we show that the autoantibodies caused the disorganization of the actin cytoskeleton in both capillary cells types. As proper actin dynamics is a prerequisite for the migration of cells [32] we believe that the coeliac disease-specific autoantibody-induced cytoskeletal disarrangement might explain the inhibited cell migration. The deranged behaviour of the endothelial cells induced by the autoantibodies could lead to the abnormal appearance of the entire vasculature network seen in the small-intestinal mucosa if left without treatment in coeliac disease patients.
The present results show that the inhibited angiogenesis observed is due specifically to coeliac disease patient-specific anti-TG2 antibodies. Removal of TG2 from the cell surface of both endothelial and vascular mesenchymal cells strengthened this conception further by abolishing the binding of coeliac disease patient IgA to these cells. In addition, the effects exerted by the patient IgA class autoantibodies were strikingly similar to those seen using anti-TG2-specific IgG autoantibody derived from the serum of an IgA-deficient coeliac disease patient who had similar intestinal lesions or using commercial monoclonal anti-TG2 antibody CUB7402. These findings are thus in line with our other recent results, showing the gluten-dependent TG2-specific autoantibodies to be specifically of primary importance, in spite of the presence of antibodies targeted against other molecules [33]. Recently it has been shown that a fraction of autoantibodies of coeliac patients on gluten-containing diet cross-reacts with TG2 and deamidated gliadin peptides [34]. Based on this novel finding, it is theoretically possible that patient autoantibodies recognizing deamidated gliadin peptides modulate the function of TG2 and thereby anti-gliadin antibodies could also play a role in vivo. Furthermore, in coeliac disease-affected mucosa TG2 might be complexed to hitherto unknown antigenic structures and thus antibodies targeting these complexes might also be involved in inhibiting angiogenesis.
The effects of coeliac disease patient autoantibodies on angiogenesis can be explained by the altered function of TG2 in response to autoantibody binding. It has not been shown conclusively whether the coeliac disease-specific IgA bound in vivo to TG2 represses [35] or enhances [36] the target enzyme activity. However, in the light of recent results reporting that blocking the cross-linking function of TG2 does not inhibit angiogenesis [5], it seems unlikely that the inhibitory effects of coeliac patient autoantibodies on angiogenesis could be due to decreased enzymatic activity as a result of antibody binding. In contrast, exogenous addition of active TG2 to angiogenic cultures resulted in a dose-dependent reduction in vessel formation [5], suggesting that the effects exerted by coeliac disease patient autoantibodies might be mediated by increased TG2 activity.
On the other hand, overexpression of either catalytically active or inactive TG2 in cells leads to reduced cell migration [37] and altered overall organization of the actin cytoskeleton [38], the same phenomena that we see in cells treated with coeliac disease autoantibodies. Thus coeliac disease patient autoantibodies may also inhibit angiogenesis by interfering with the non-enzymatic functions of TG2. However, further experiments are needed to clarify the mechanism behind the effects exerted by the autoantibodies.
In addition to being present in the serum and small intestinal mucosa of coeliac disease patients on a gluten-containing diet, the disease-specific autoantibodies can also be found in other organs. The autoantibody deposits have been identified around blood vessels in the liver [9] and interestingly, mild liver abnormalities are common in patients with coeliac disease, but they usually resolve with a gluten-free diet [39]. Furthermore, there are data suggesting that hepatic failure in coeliac disease patient with severe liver disease can be reversed with dietary treatment [39]. Moreover, the autoantibodies are also present in the brain vessels of coeliac disease patients suffering from neurological symptoms [40]. It is of interest that the coeliac patient autoantibodies have been shown to induce neuroblast apoptosis [41] and also cause gluten ataxia, a neurological manifestation of coeliac disease, when injected into mouse brain [42]. Taken together, it might be possible that the disease-specific autoantibodies are involved in the extraintestinal manifestations of coeliac disease by hindering local angiogenesis or otherwise affecting the function of capillaries.
Based on the results presented in this paper and on earlier studies showing a role for the coeliac disease-specific autoantibodies in the disease pathogenesis, coeliac disease might thus be included in the group of other autoimmune diseases, such as myasthenia gravis [43] or pemphigus vulgaris [44], in whose pathogenesis the disease-specific autoantibodies have been shown experimentally to play a fundamental role. We present here new evidence to indicate that the gluten-induced disease-specific autoantibodies might constitute an important contributor in the development and persistence of the flat mucosal lesion seen in coeliac disease, in addition to activated T cell-driven mechanisms.
Acknowledgments
The authors wish to thank Mr Jorma Kulmala for technical assistance. The contribution of Taneli Tani MD, PhD (Tampere University Hospital, Tampere, Finland) is acknowledged in providing the HUVECs. The Coeliac Disease Study Group has been supported financially by the Research Council for Health, the Academy of Finland, the Pediatric Research Foundation, the Competitive Research Funding of the Pirkanmaa Hospital District, the Yrjö Jahnsson Foundation, the Finnish Medical Foundation, the Research Fund of the Finnish Coeliac Society and the European Commission (contract number MRTN-CT-2006-036032).
References
- 1.Sollid LM, Jabri B. Is celiac disease an autoimmune disorder? Curr Opin Immunol. 2005;17:595–600. doi: 10.1016/j.coi.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Koning F, Schuppan D, Cerf-Bensussan N, Sollid LM. Pathomechanisms in celiac disease. Best Pract Res Clin Gastroenterol. 2005;19:373–87. doi: 10.1016/j.bpg.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Dieterich W, Ehnis T, Bauer M, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- 4.Haroon ZA, Hettasch JM, Lai TS, Dewhirst MW, Greenberg CS. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999;13:1787–95. doi: 10.1096/fasebj.13.13.1787. [DOI] [PubMed] [Google Scholar]
- 5.Jones RA, Kotsakis P, Johnson TS, et al. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006;13:1442–53. doi: 10.1038/sj.cdd.4401816. [DOI] [PubMed] [Google Scholar]
- 6.Telci D, Griffin M. Tissue transglutaminase (TG2) − a wound response enzyme. Front Biosci. 2006;11:867–82. doi: 10.2741/1843. [DOI] [PubMed] [Google Scholar]
- 7.Fésüs L, Piacentini M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci. 2002;27:534–9. doi: 10.1016/s0968-0004(02)02182-5. [DOI] [PubMed] [Google Scholar]
- 8.Marzari R, Sblattero D, Florian F, et al. Molecular dissection of the tissue transglutaminase autoantibody response in celiac disease. J Immunol. 2001;166:4170–6. doi: 10.4049/jimmunol.166.6.4170. [DOI] [PubMed] [Google Scholar]
- 9.Korponay-Szabo IR, Halttunen T, Szalai Z, et al. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut. 2004;53:641–8. doi: 10.1136/gut.2003.024836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaukinen K, Peräaho M, Collin P, et al. Small-bowel mucosal transglutaminase 2-specific IgA deposits in coeliac disease without villous atrophy: a prospective and randomized clinical study. Scand J Gastroenterol. 2005;40:564–72. doi: 10.1080/00365520510023422. [DOI] [PubMed] [Google Scholar]
- 11.Salmi TT, Collin P, Järvinen O, et al. Immunoglobulin A autoantibodies against transglutaminase 2 in the small-intestinal mucosa predict forthcoming coeliac disease. Aliment Pharmacol Ther. 2006;24:541–52. doi: 10.1111/j.1365-2036.2006.02997.x. [DOI] [PubMed] [Google Scholar]
- 12.Salmi TT, Collin P, Korponay-Szabó IR, et al. Endomysial antibody-negative coeliac disease: clinical characteristics and intestinal autoantibody deposits. Gut. 2006;55:1746–53. doi: 10.1136/gut.2005.071514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halttunen T, Mäki M. Serum immunoglobulin A from patients with celiac disease inhibits human T84 intestinal crypt epithelial cell differentiation. Gastroenterology. 1999;116:566–72. doi: 10.1016/s0016-5085(99)70178-2. [DOI] [PubMed] [Google Scholar]
- 14.Barone MV, Caputo I, Ribecco MT, et al. Humoral immune response to tissue transglutaminase is related to epithelial cell proliferation in celiac disease. Gastroenterology. 2007;132:1245–53. doi: 10.1053/j.gastro.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 15.Zanoni G, Navone R, Lunardi C, et al. In celiac disease, a subset of autoantibodies against transglutaminase binds Toll-like receptor 4 and induces activation of monocytes. PLoS Med. 2006;3:e358. doi: 10.1371/journal.pmed.0030358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matheson PJ, Wilson MA, Garrison RN. Regulation of intestinal blood flow. J Surg Res. 2000;93:182–96. doi: 10.1006/jsre.2000.5862. [DOI] [PubMed] [Google Scholar]
- 17.Unthank JL, Lash JM, Bohlen HG. Maturation of the rat intestinal microvasculature from juvenile to early adult life. Am J Phys. 1990;259:G282–9. doi: 10.1152/ajpgi.1990.259.2.G282. [DOI] [PubMed] [Google Scholar]
- 18.Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci USA. 2002;99:15451–5. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–64. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooke WT, Holmes GKT. Coeliac disease. Edinburgh: Churchill Livingstone; 1984. [Google Scholar]
- 21.Shiner M. Ultrastructural changes suggestive of immune reactions in the jejunal mucosa of coeliac children following gluten challenge. Gut. 1973;14:1–12. doi: 10.1136/gut.14.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shiner M, Ballard J. Antigen–antibody reactions in jejunal mucosa in childhood coeliac disease after gluten challenge. Lancet. 1972;1:1202–5. doi: 10.1016/s0140-6736(72)90924-5. [DOI] [PubMed] [Google Scholar]
- 23.Kurzen H, Manns S, Dandekar G, Schmidt T, Prätzel S, Kräling BM. Tightening of endothelial cell contacts: a physiologic response to cocultures with smooth-muscle-like 10T1/2 cells. J Invest Dermatol. 2002;119:143–53. doi: 10.1046/j.1523-1747.2002.01792.x. [DOI] [PubMed] [Google Scholar]
- 24.Birckbichler PJ, Upchurch HF, Patterson MK, Conway E. A monoclonal antibody to cellular transglutaminase. Hybridoma. 1985;4:179–86. doi: 10.1089/hyb.1985.4.179. [DOI] [PubMed] [Google Scholar]
- 25.Skalli O, Ropraz P, Trzeciak A, Benzonana G, Gillessen D, Gabbiani G. A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol. 1986;103:2787–96. doi: 10.1083/jcb.103.6.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 27.Rasband WS. ImageJ U.S. Bethesda, MD: National Institutes of Health; [2 January 2007]. Available at: http://rsb.info.nih.gov/ij/ 1997–2005. [Google Scholar]
- 28.Montesano R, Orci L, Vassalli P. In vitro rapid organization of endothelial cells into capillary-like networks is promoted by collagen matrices. J Cell Biol. 1983;97:1648–52. doi: 10.1083/jcb.97.5.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirschi KK, Rohovsky SA, D'Amore PA. PDGF, TGF-beta, and heterotypic cell–cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141:805–14. doi: 10.1083/jcb.141.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weihua Z, Tsan R, Schroit AJ, Fidler IJ. Apoptotic cells initiate endothelial cell sprouting via electrostatic signaling. Cancer Res. 2005;65:11529–35. doi: 10.1158/0008-5472.CAN-05-2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kale S, Hanai J, Chan B, et al. Microarray analysis of in vitro pericyte differentiation reveals an angiogenic program of gene expression. FASEB J. 2005;19:270–1. doi: 10.1096/fj.04-1604fje. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi M, Nishita M, Mishima T, Ohashi K, Mizuno K. MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. EMBO J. 2006;25:713–26. doi: 10.1038/sj.emboj.7600973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korponay-Szabó IR, Laurila K, Szondy Z, et al. Missing endomysial and reticulin binding of coeliac antibodies in transglutaminase 2 knockout tissues. Gut. 2003;52:199–204. doi: 10.1136/gut.52.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korponay-Szabó IR, Vecsei Z, Király R, et al. Deamidated gliadin peptides form epitopes that transglutaminase antibodies recognize. J Pediatr Gastroenterol Nutr. in press. [DOI] [PubMed]
- 35.Esposito C, Paparo F, Caputo I, et al. Anti-tissue transglutaminase antibodies from coeliac patients inhibit transglutaminase activity both in vitro and in situ. Gut. 2002;51:177–81. doi: 10.1136/gut.51.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Király R, Vecsei Z, Deményi T, Korponay-Szabó IR, Fésüs L. Coeliac autoantibodies can enhance transamidating and inhibit GTPase activity of tissue transglutaminase: dependence on reaction environment and enzyme fitness. J Autoimmun. 2006;26:278–87. doi: 10.1016/j.jaut.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 37.Balklava Z, Verderio E, Collighan R, Gross S, Adams J, Griffin M. Analysis of tissue transglutaminase function in the migration of Swiss 3T3 fibroblasts: the active-state conformation of the enzyme does not affect cell motility but is important for its secretion. J Biol Chem. 2002;277:16567–75. doi: 10.1074/jbc.M109836200. [DOI] [PubMed] [Google Scholar]
- 38.Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol. 2000;148:825–38. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaukinen K, Halme L, Collin PF, et al. Celiac disease in patients with severe liver disease: gluten-free diet may reverse hepatic failure. Gastroenterology. 2002;122:881–8. doi: 10.1053/gast.2002.32416. [DOI] [PubMed] [Google Scholar]
- 40.Hadjivassiliou M, Mäki M, Sanders DS, et al. Autoantibody targeting of brain and intestinal transglutaminase in gluten ataxia. Neurology. 2006;66:373–7. doi: 10.1212/01.wnl.0000196480.55601.3a. [DOI] [PubMed] [Google Scholar]
- 41.Cervio E, Volta U, Verri M, et al. Sera of patients with celiac disease and neurologic disorders evoke a mitochondrial-dependent apoptosis in vitro. Gastroenterology. 2007;133:195–206. doi: 10.1053/j.gastro.2007.04.070. [DOI] [PubMed] [Google Scholar]
- 42.Boscolo S, Sarich A, Lorenzon A, et al. Gluten ataxia: passive transfer in a mouse model. Ann NY Acad Sci. 2007;1107:319–28. doi: 10.1196/annals.1381.034. [DOI] [PubMed] [Google Scholar]
- 43.Vincent A, Willcox N, Hill M, Curnow J, MacLennan C, Beeson D. Determinant spreading and immune responses to acetylcholine receptors in myasthenia gravis. Immunol Rev. 1998;164:157–68. doi: 10.1111/j.1600-065x.1998.tb01217.x. [DOI] [PubMed] [Google Scholar]
- 44.Aoki-Ota M, Tsunoda K, Ota T, et al. A mouse model of pemphigus vulgaris by adoptive transfer of naïve splenocytes from desmoglein 3 knockout mice. Br J Dermatol. 2004;151:346–54. doi: 10.1111/j.1365-2133.2004.06056.x. [DOI] [PubMed] [Google Scholar]