

Abstract
Ethanol acts as a teratogen in developing fetuses causing abnormalities of the brain, heart, craniofacial bones, and limb skeletal elements. To assess whether some teratogenic actions of ethanol might occur via dysregulation of msx2 expression, we examined msx2 expression in developing mouse embryos exposed to ethanol on embryonic day (E) 8 of gestation and subjected to whole mount in situ hybridization on E11–11.5 using a riboprobe for mouse msx2. Control mice exhibited expression of msx2 in developing brain, the developing limb buds and apical ectodermal ridge, the lateral and nasal processes, olfactory pit, palatal shelf of the maxilla, the eye, the lens of the eye, otic vesicle, prevertebral bodies (notochord), and endocardial cushion. Embryos exposed to ethanol in utero were significantly smaller than their normal counterparts and did not exhibit expression of msx2 in any structures. Similarly, msx2 expression, as determined by reverse transcription–PCR and Northern blot hybridization, was reduced ≈40–50% in fetal mouse calvarial osteoblastic cells exposed to 1% ethanol for 48 hr while alkaline phosphatase was increased by 2-fold and bone morphogenetic protein showed essentially no change. Transcriptional activity of the msx2 promoter was specifically suppressed by alcohol in MC3T3-E1 osteoblasts. Taken together, these data demonstrate that fetal alcohol exposure decreases msx2 expression, a known regulator of osteoblast and myoblast differentiation, and suggest that one of the “putative” mechanisms for fetal alcohol syndrome is the inhibition of msx2 expression during key developmental periods leading to developmental retardation, altered craniofacial morphogenesis, and cardiac defects.
Keywords: development, craniofacial, homeodomain, fetal alcohol syndrome
Maternal consumption of ethanol severely affects the mental ability, facial appearance, limbs, and hearts of at least 3 in 1,000 infants born in the United States (1, 2) and 1 in 300 infants born in the western world (3). Fetal alcohol syndrome (FAS), a phrase coined by Jones et al. in 1973 (4, 5), describes the fetal consequence of maternal ethanol during pregnancy, and the associated array of physical dysmorphias, intrauterine growth retardation, and intellectual impairment postnatally (6–9). The altered morphogenesis of the craniofacial region includes micropthalmia, midfacial and maxillary hypoplasia, cleft palate, poorly developed philtrums, thin upper lips, short noses, epicanthal folds, and microcephaly (4, 10–15). Dysmorphogenesis of the limbs has also been observed (4, 11, 13) in addition to a variety of neural tube, cardiac and renal defects (11, 12, 14). Although the critical dosage of alcohol responsible for this wide range of abnormalities in FAS is still unknown, results from animal studies indicate that the fetotoxic effects of alcohol are more dependent on peak blood alcohol levels—often acquired by “binge drinking”—than upon the total amount of alcohol consumed throughout an entire pregnancy (16, 17).
Homologs of the Drosophila muscle segment homeobox gene (msh) have been identified in vertebrates and are classed into four subclasses [msh a, b, c, and d (18)]. In the mouse, three homologs have been identified, msx1, msx2 [formerly designated Hox-7 and Hox-8, respectively, (19, 20)], and msx3 (21), which are genetically unlinked. Similar homologs have been identified in the human and are denoted MSX1 and MSX2 (22–24). In the mouse, transcripts of the msx2 gene are localized in premigratory and migratory cephalic neural crest, neural crest derived mesenchyme of the first through fourth branchial arches, osteogenic tissue of the mandible and maxilla, eye, ear, developing teeth, apical ectodermal ridge and underlying mesenchyme of the limb bud, myoblast, endocardial cushion, and membranous bone and sutures of the calvaria (22, 25–29). Verification of the critical role of the msx2 gene in craniofacial development has come from examination of MSX2 mutants and mouse transgenic experiments. A mutation in the homeodomain of the human MSX2 gene was recently detected in a family affected with autosomal dominant craniosynostosis [the premature closure of one or more sutures (22)]. Likewise, transgenic mice expressing a mutated form of the msx2 gene exhibited premature closure of the cranial sutures, akin to Boston-type craniosynostosis (30). Since msx2-regulated morphogenetic fields overlap tissues altered in ethanol teratogenesis, we have examined the hypothesis that fetal exposure to alcohol disrupts the ordered progressive expression of msx2 and have examined the effect of alcohol on the msx2 promoter in cultured mouse calvarial derived osteoblasts to explore the possible mechanism of action. We now report that alcohol disrupts the earliest patterns of msx2 expression in fetal mice and inhibits the expression of msx2 in cranial osteoblasts in vitro.
MATERIALS AND METHODS
The protocol previously described by Kotch and Sulik (31) for induction of craniofacial anomalies in pregnant female C57BL/6J mice by intraperitoneal injection of ethanol was followed as described with some modification. Mice were maintained on Purina Mouse Breeder Chow and water ad libitum. Prior to mating, females were superovulated with pregnant mare serum and human chorionic gonadotrophin (Sigma). Female C57BL/6J mice were placed with males for a 1-hr period and then examined for the presence of a copulation plug. The time of plug detection was considered gestational day 0.0 hr. Two intraperitoneal injections of 25% (vol/vol) ethanol in lactated Ringer’s solution were administered at a dosing volume of 0.015 ml/g maternal body weight. The injections were given 4 hr apart, with the first administered at 8 days, 0 hr, and the second at 8 days, 4 hr. Control animals were injected with lactated Ringer’s solution according to the regimen for ethanol injections.
Hybridization Probes.
Hybridization probes for msx2 (32, 33) were prepared using a Genius 4 digoxigenin RNA labeling in vitro transcription kit (Boehringer Mannheim) according to the manufacturers instructions. A plasmid containing exon 2 of mouse msx2 was constructed as follows: oligonucleotide hox3 [5′-CCTCGGTACCATATGAGCCCCACCACCTGCACCCTG-3′ (KpnI site)] and oligonucleotide hox4 [5′-GGGGGATCCTTAGGATAGATGGTACATGCCATATCC-3′ (BamHI site)] were utilized to PCR amplify from mouse genomic DNA the portion of exon 2 of mouse msx2 as described (32). This fragment was subcloned into the EcoRV site of bluescript pKS(+) (Stratagene) and sequenced to verify orientation. The plasmid thus contains exon 2 of the murine msx2 gene with sense orientation directed by the T3 promoter and antisense directed by the T7 promoter. After linearization with HindIII, an antisense riboprobe was generated from the T7 promoter.
Whole-Mount in Situ Hybridization.
Pregnant females were killed by CO2 asphyxiation on embryonic day (E) 11. Embryos were harvested, fixed, and subjected to whole-mount in situ hybridization as described by Wilkinson and Nieto (34). Embryos were prehybridized for 1 hr at 70°C in hybridization buffer [50% formamide/5× standard saline citrate (SSC), pH 4.5/50 μg/ml yeast RNA/1% SDS/50 μg/ml heparin). The hybridization buffer was replaced, single-stranded RNA probes labeled with digoxigenin-dUTP were added to 1 μg/ml, and the embryos hybridized overnight at 70°C. The embryos were washed through two changes of solution 1 (50% formamide/5× SSC, pH 4.5/1% SDS) for 30 min at 70°C followed by one wash with solution 1 diluted 1:1 with solution 2 (0.5 M NaCl/10 mM Tris⋅HCl, pH 7.5/0.1% Tween-20) for 10 min at 70°C and three washes with solution 2. The wash buffer was replaced by two changes of RNase buffer (100 μg/ml RNase A in solution 2) for 30 min at 37°C, followed by one wash with solution 2 then one wash with solution 3 (50% formamide/2× SSC, pH 4.5) at room temperature, two washes with solution 3 for 30 min at 65°C, and three washes with TBST (136 mM NaCl/2.7 mM KCl/25 mM Tris⋅HCl, pH 7.5/0.1% Tween-20).
The embryos were preblocked with 10% sheep serum (heat inactivated just before use at 70°C for 30 min) in TBST containing 2 mM levamisole for 2 hr. Sufficient embryo acetone powder [prepared from E13.5 embryos as described by Harlow and Lane (35)] was heat-inactivated just before use in a small volume of TBST at 70°C for 30 min. Anti-digoxigenin antibody coupled to alkaline phosphatase (Boehringer Mannheim) at 1:2,000 dilution was preabsorbed for 30 min at 4°C with 1% (wt/vol) embryo acetone powder in TBST containing 1% heat-inactivated goat serum and 2 mM levamisole, then cleared by centrifugation at 10,000 × g for 10 min. Embryos were incubated with the preabsorbed antibody overnight at 4°C with gentle rocking, then washed three times with TBST for 5 min, five times with TBST for 1 hr, and three times with NTMT (100 mM NaCl/100 mM Tris⋅HCl, pH 9.5/50 mM MgCl2/1% Tween-20/2 mM levamisole) for 10 min. The color reaction was initiated by washing the embryos into NTMT containing 2 mM levamisole, 4.5 μl/ml NBT (75 mg/ml nitroblue tetrazolium salt in 70% dimethylformamide), and 3.5 μl/ml BCIP (x-phosphate, 50 mg/ml 5-bromo-4-chloro-3-indolyl phosphate toluidine salt in 100% dimethylformamide), and the embryos were incubated in the dark for 3 hr to develop the color. After color development, the embryos were washed three times with PBT (PBS plus 0.1% Tween 20). Photomicrographs were taken with an Olympus Stereo Zoom Microscope equipped with an Olympus automatic photosystem.
Reverse Transcription–PCR (RT-PCR).
Cultures of 18 day fetal mouse calvaria were isolated essentially as described for neonatal mice calvaria (36, 37). Cells were grown to confluence (4 days) then treated for 48 hr with either nothing or medium containing 1% ethanol. Total RNA was extracted from each cell layer as described (38). RT-PCR was carried out according to described methods (38, 39). Amplimer pairs for bone morphogenetic protein 2 [BMP-2 (40)] and alkaline phosphatase [AP (41)] have been described.
Preparation of RNA and Northern Blot Hybridization.
Fetal mouse calvarial osteoblasts were seeded into culture at 4 × 104 cells/cm2 and incubated for 48 hr. The cells were then treated with 1% ethanol and incubated for 24 hr prior to isolation of total RNA. Total RNA was isolated and electrophoresed as described (32, 38). The gels were stained with ethidium bromide, and 28S and 18S rRNA bands were photographed. The mRNAs were blotted onto a nitrocellulose membrane and probed with a 32P-labeled cDNA to msx2 as described (32). The membranes were stripped and reprobed with a 32P-labeled cDNA to AP (kindly provided by Paula Henthorn, University of Pennsylvania, Philadelphia).
Transient Transfection Assays and Promoter–Luciferase Construct.
The mouse msx2 promoter–luciferase construct (MSXLUC) containing the proximal 1 kb of the mouse msx2 promoter and the cytomegalovirus promoter–luciferase construct (CMVLUC) containing a cytomegalovirus promoter were synthesized as described (32). Transfection was performed in MC3T3-E1 cells by DEAE dextran as described (42). Transfection efficiency was normalized to a pGL2 promoter vector (SV40LUC; Promega) promoter construct as described (42). Cellular luciferase activity was measured as previously detailed using a Berthold AutoLumat 953 luminometer (EG & G Instruments, Oak Ridge, TN). Data are presented as the mean (±SD) luciferase activity of three independent transfections. Reagents were from Promega.
Statistical Analysis.
Data were analyzed by one-way ANOVA using Microsoft excel, version 4.0.
RESULTS
Alcohol Interrupts the Normal Developmental Progression of Mouse Embryos.
Mouse embryos were collected from control and ethanol-treated mother mice at E11–11.5, 3 days posttreatment. Beating embryonic hearts were observed in all embryos from control and ethanol-treated mother mice. Embryos harvested from mothers exposed to ethanol were ≈50% smaller, as compared with embryos harvested from control mothers, indicating a delay in development (Fig. 1). The embryos exposed to ethanol in utero had reached the approximately 16 somite stage in contrast to control embryos which had developed to the 25–40 somite stage.
Figure 1.

Comparison of mouse embryos of E11–11.5 from either control mother mice (A) or mother mice treated with ethanol on E8 (B) and harvested on E11–11.5. The embryos have all been subjected to in situ hybridization for msx2 expression as described. Both photomicrographs represent the same magnification and show that the embryos exposed in utero to ethanol are ≈50% smaller than the equivalent gestationally aged controls. (×10.)
Msx2 Expression in Ethanol Exposed Embryos Is Suppressed.
Alcohol could retard the development of the primitive brain as well as the craniofacial bones and other target organs such as the heart by inhibiting the expression of msx2, thus altering a cascade of events that are necessary for normal development. If the effect of alcohol were to be localized to specific sites of msx2 expression, one would expect expression in certain sites and none in sensitive sites. Alternatively, if alcohol inhibits the expression of msx2 globally, one would expect that all the organs known to exhibit expression of msx2 would show no hybridization with an msx2 riboprobe. To test these hypotheses, embryos were submitted to whole mount in situ hybridization. Examination of the control embryos revealed that msx2 was expressed in: the maxillary process of the first branchial arch, mandibular process of the first branchial arch, second branchial arch, third branchial arch, fourth branchial arch, the olfactory pit of the developing nasal region, regions above the developing brain, the developing limb buds and apical ectodermal ridge, the lateral and nasal processes, olfactory pit, palatal shelf of the maxilla, the eye, the lens of the eye, pre-vertebral bodies (notochord) (Fig. 2), otic vesicle (Fig. 3), and the A-V region of the developing heart (Fig. 4). In contrast, multiple embryos exposed to ethanol in vivo were also examined by whole mount in situ hybridization for msx2 expression and none exhibited a positive reaction for msx2 (Fig. 5) in any of the developing structures noted for control embryos. Despite this fact, embryo development did proceed as several structures formed not present on E8, such as the otic vesicle, forelimb bud, and maxillary and mandibular processes of the first branchial arch. While the embryos from control mothers exhibited features typical of E11–11.5 embryos including developing forelimb and hindlimb buds, these structures were absent in the embryos from alcohol-treated mothers.
Figure 2.

Various views of E11–11.5 mouse embryos from the control group subjected to in situ hybridization for msx2. A positive reaction is represented by the purple color that is the product of specific binding of the antisense digoxigenin-labeled riboprobe to msx2 and subsequent visualization with an alkaline phosphatase conjugated anti-digoxigenin antibody. Only the specific binding resulted in color production, whereas the absence of gene expression resulted in no color reaction. (A) Lateral view of left side of embryo. (×15.) (B) This figure shows that while the (1) maxillary process of the first branchial arch exhibits a positive reaction, the lateral surfaces of (2) the mandibular process of the first branchial arch and (3) the second branchial arch are negative while the aboral surfaces of those structures (not visible) are positive. (×30.) (C) A representative whole mount in situ hybridization of the head of an E11.5 mouse embryo using the msx2 anti-sense riboprobe. The photomicrograph was taken of the facial region with first and second branchial arches removed to reveal structures located below the removed anatomical structures. (×50.) Numbers on each figure refer to the following: 1, maxillary process of first branchial arch; 2, mandibular process of first branchial arch; 3, second branchial arch; 4, third branchial arch; 5, fourth branchial arch; 6, proximal region of forelimb bud; 7, distal region of forelimb bud; 8, apical ectodermal ridge; 9), proximal region of hindlimb bud; 10, lateral nasal process; 11, medial nasal process; 12, olfactory pit; 13, palatal shelf of the maxilla; 14, eye; 15, lens of the eye; 16, dorsal cephalic mesenchyme over telencephalon; 17, dorsal cephalic mesenchyme over midbrain; 18, dorsal cephalic mesenchyme over rhombencephalon (developing hindbrain); 19, prevertebral bodies and notochord.
Figure 3.

A representative whole mount in situ hybridization of the head of an E11–11.5 mouse embryo using the msx2 antisense riboprobe. The photomicrograph was taken of the dorsal aspect of the developing head region in a control mouse embryo. Msx2 expression is shown in the otic vesicle (OV) and leading edges of the neural fold. (×50.)
Figure 4.
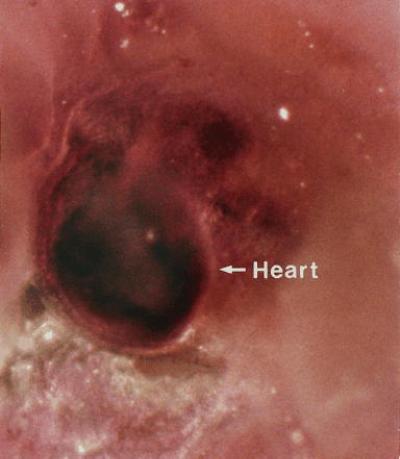
A representative whole mount in situ hybridization of the developing heart in an E11–11.5 control mouse embryo. Msx2 expression is found throughout the heart region but is most notable in the A-V cushion. (×60.)
Figure 5.

Mouse embryo treated in utero on E8.0 and harvested on E11–11.5. This embryo has been subjected to in situ hybridization using the msx2 antisense riboprobe. Note the complete absence of any signal for msx2 and the retarded development of the fore limb bud, heart, optic eminence (OE), maxillary process of first branchial arch (Max), mandibular process of first branchial arch (Mand), and otic vesicle (OV). (×50.)
Alcohol Suppresses msx2 Expression in Osteoblastic Cells.
To define the mechanism of action of ethanol on the suppression of msx2 we analyzed total RNA from cultured fetal mouse calvaria cells either exposed to control medium or to medium containing 1% ethanol for 48 hr prior to RNA isolation. RNA was subjected to RT-PCR for msx2, BMP-2, and AP. The results showed that ethanol was a potent inhibitor of msx2 expression in developing osteoblasts. Cells exposed to ethanol showed a 50% reduction in msx2 message content as compared with control cultures (Fig. 6). By contrast AP mRNA was up-regulated, while essentially no change occurred in BMP-2 mRNA accumulation. Thus ethanol decreases msx2 mRNA accumulation in fetal mouse calvarial osteoblasts. The results of the RT-PCR analysis of msx2 and AP regulation by ethanol were confirmed by Northern blot hybridization. After only 24-hr exposure, ethanol reduced msx2 mRNA steady state by 37% while stimulating AP mRNA by nearly 4-fold (Fig. 7).
Figure 6.

RT-PCR of msx2, BMP-2, and AP mRNA expression in fetal mouse calvarial osteoblast cultures treated with 1% ethanol (ETOH). Calvarial osteoblasts were treated and mRNA isolated as described. Cells exposed to ethanol showed a 50% reduction in msx2 message (441 bp), a 2-fold increase in AP message (313 bp), and essentially no change in BMP-2 message (720 bp). RT-PCRs are representative of duplicate determinations.
Figure 7.

Northern blot analysis of the effect of alcohol on AP and msx2 expression in fetal mouse calvarial osteoblasts. Fetal mouse calvarial osteoblasts were seeded into culture at 4 × 104 cells/cm2 and incubated for 48 hr. The cells were then treated with 1% ethanol (ETOH) and incubated for 24 hr prior to isolation of total RNA. Total RNA (20 μg) were loaded into each lane. The two msx2 transcripts observed reflect differences in processing of the 3′-untranslated region (32). Since the probe hybridizes to the homeodomain region of the msx2 mRNA, these two bands are detected while after RT-PCR (Fig. 6) only one band is observed since it represents the sum of the two transcripts. Cells exposed to ethanol showed a 37% reduction in msx2 message and a 3.6-fold increase in AP message. Data represents typical Northern blots of three independent analyses.
To further analyze the mechanism of the effect of alcohol on the regulation of msx2 gene expression, MC3T3-E1 mouse osteoblasts were transfected with either MSXLUC or CMVLUC and exposed to a dose response (0–3%) of ethanol for 48 hr prior to luciferase activity measurements. Ethanol treatment reduced msx2 promoter activity by 30% at an alcohol concentration of 1% and by 50% at an alcohol concentration of 2.0% (Fig. 8). This effect was specific for the msx2 promoter since a CMVLUC was unaffected by alcohol treatment (Fig. 8).
Figure 8.

Alcohol suppresses msx2 promoter activity in MC3T3-E1 cells. Cells transfected with either MSXLUC or CMVLUC were incubated in the absence or presence of a dose range of ethanol from 1–3%. The cells were harvested and examined for luciferase activity. Data are presented as the mean ± SD luciferase activity of three independent transfections. ANOVA analysis showed that MSXLUC activity varied significantly with ethanol dose (P = 0.011), while CMVLUC activity did not (P = 0.609).
DISCUSSION
Studies designed to examine ethanol embryo toxicity in humans and rodents have failed to uncover a single underlying mechanism for the teratogenic action of ethanol (43), suggesting that ethanol embryo toxicity may proceed by several mechanisms. Numerous studies have been performed, variably implicating abnormal prostaglandin metabolism, chromosomal alterations, placental dysfunction, hypoxia, altered protein synthesis, altered growth signaling, interference with neurotransmitter production, and alterations of enzymes that regulate glycogen metabolism (31, 44–51). However, to our knowledge no prior study has examined the effects of alcohol on the temporo-spatial regulation of homeodomain transcription factors known to be crucial in normal craniofacial morphogenesis.
Homeodomain factors are DNA binding proteins, which regulate transcription via protein–protein and protein–DNA interactions (52, 53). As a group, its members are transcription factors that regulate the timing, patterning, and tissue-specific expression of developmentally regulated genes (52, 54). The 60-amino acid homeodomain, which defines this group, mediates such DNA–protein and protein–protein interactions (53). Homeodomain factors mediate both positive and negative transcriptional regulation (53). The expression of msx2, a homeobox gene that regulates the process of development of embryonic structures which give rise to the craniofacial bones (25, 26, 55–57), limb (26, 29, 58), vertebral processes (59), eye (26) and its lens (26, 28), ear (26, 28), and heart (26, 60), has been examined in mouse embryos transiently exposed to ethanol. Our results demonstrate that at day 11 of gestation, this gene is highly expressed in control embryos in structures known to be effected by ethanol toxicity. However, in embryos exposed to ethanol for a short period of time, this important developmental gene is not expressed. As a result, the embryos exposed to ethanol are developmentally retarded.
We have confirmed that the msx2 gene is suppressed by ethanol in cranial osteoblasts in in vitro experiments. Isolated fetal mouse calvarial osteoblasts exposed to ethanol for 48 hr had reduced mRNA for msx2 as compared with controls. Furthermore, in experiments in which an MSXLUC was transiently transfected into osteoblastic cultures, exposure to ethanol significantly reduced promoter activity. Ethanol has been reported to selectively effect mRNA transcription in various tissues including the inhibition of immunoglobulin κ chain in B lymphocytes while not effecting total RNA production or mRNA for β actin (61) and the up-regulation of the molecular chaperonin HSC70 in neuroblastoma × glioma cell hybrids (62, 63). The mechanism of ethanol regulation of HSC70 has been found to be at the level of the promoter, involving an ethanol-responsive cis-acting element in the proximal region of the promoter. Thus, the effects of ethanol that we have observed on the msx2 promoter are consistent with such a mechanism although this remains to be determined. However, alcohol has many affects including alteration of signal transduction (64). Indeed, msx2 expression is regulated by molecules such as BMP-2 and BMP-4, which convey epithelial-mesenchymal inductive events (65–67). Of interest in this study is the finding that alcohol did not affect BMP-2 mRNA in ethanol treated calvarial cells. However, we cannot exclude the possibility that ethanol may regulate the expression of both BMP-2 and BMP-4 in the embryo. The mechanism(s) whereby alcohol impacts msx2 expression in vivo needs to be carefully detailed.
Epidemiologic studies suggest that there exists a window of enhanced susceptibility of the fetus to the dysmorphic effects of maternal alcohol ingestion in the first trimester. These data suggested that episodes of “binge drinking”—ingestion of large amounts of alcohol in a short time period, with associated higher maternal-fetal ethanol levels within this window of enhanced susceptibility—may combine to greatly increase the risk for fetal malformations. This notion has been supported by animal studies. Single intraperitoneal injections of ethanol given to pregnant C57BL/6J mice between E7 and E11 resulted in craniofacial abnormalities, including exencephaly and hypoplasia of the midfacial region and anomalies of the DiGeorge sequence (68–70). The onset of expression of msx2 in mouse embryos has been reported to occur as early as E9 and is still expressed as late as E17 (26). Using closely spaced (4 hr) dual intraperitoneal injections of alcohol to mimic “binge” drinking, abnormalities with facial features similar to those noted in human FAS infants have been induced in fetal mice only when alcohol is present on or before E8 (31, 70–73). By contrast, similar alcohol exposures at later stages of murine gestation could not induce murine craniofacial abnormalities. Thus, as evident in the human disorder, the rodent model of FAS reveals both a window of susceptibility and an alcohol concentration threshold for effects upon the developing embryo. Our data now show that at least one gene responsible for the development of craniofacial bones, msx2, is suppressed under these conditions and may represent a “putative” target for the effects of alcohol, subject to further confirmation.
Finally, with regards to expression, it is intriguing to note the reciprocal changes in msx2 and alkaline phosphatase expression in response to alcohol. Preliminary studies using MC3T3-E1 osteoblastic cells constitutively expressing msx2 indicate that msx2 suppresses both alkaline phosphatase mRNA accumulation and alkaline phosphatase enzyme activity (D.A.T., unpublished observations). Since alkaline phosphatase plays a role in the initiation and maintenance of mineralization, dysregulated timing of msx2 expression and alkaline phosphatase activity in response to alcohol may explain craniofacial abnormalities associated with FAS.
Acknowledgments
We wish to acknowledge the excellent technical assistance of Ms. Linda R. Halstead, Mrs. Chunyue Zhang, and Mr. Sean McCaul. This work was supported by a grant from the Barnes–Jewish Hospital Foundation (to L.R.). D.A.T. is a 1996 Charles E. Culpeper Foundation Medical Science Scholar.
ABBREVIATIONS
- FAS
fetal alcohol syndrome
- E
embryonic day
- RT-PCR
reverse transcription–PCR
- BMP-2
bone morphogenetic protein 2
- AP
alkaline phosphatase
- MSXLUC
msx2 promoter–luciferase construct
- CMVLUC
cytomegalovirus promoter–luciferase construct
References
- 1.Hanson J W, Streissguth A P, Smith D W. J Pediatr. 1978;92:457–460. doi: 10.1016/s0022-3476(78)80449-1. [DOI] [PubMed] [Google Scholar]
- 2.Little B B, Snell L M, Rosenfeld C R, Gilstrap L C, III, Gant N F. Am J Dis Child. 1990;144:1142–1146. doi: 10.1001/archpedi.1990.02150340088030. [DOI] [PubMed] [Google Scholar]
- 3.Abel E L, Sokol R J. Alcohol Clin Exp Res. 1991;15:514–524. doi: 10.1111/j.1530-0277.1991.tb00553.x. [DOI] [PubMed] [Google Scholar]
- 4.Jones K L, Smith D W, Ulleland C N, Streissguth P. Lancet. 1973;i:1267–1271. doi: 10.1016/s0140-6736(73)91291-9. [DOI] [PubMed] [Google Scholar]
- 5.Jones K L, Smith D W. Lancet. 1973;ii:999–1001. doi: 10.1016/s0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- 6.Coles C D. Clin Obstet Gynecol. 1993;36:255–266. doi: 10.1097/00003081-199306000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Escobar L F, Bixler D, Padilla L M. Am J Med Genet. 1993;45:25–29. doi: 10.1002/ajmg.1320450109. [DOI] [PubMed] [Google Scholar]
- 8.Beattie, J. O. (1992) Eur. J. Clin. Nutr. 46, Suppl. 1, S7–S17. [PubMed]
- 9.Alpert J J, Zuckerman B. Pediatr Rev. 1991;12:375–379. [PubMed] [Google Scholar]
- 10.Altman B. J Pediatr Ophthalmol. 1976;13:255–258. [PubMed] [Google Scholar]
- 11.Palmer R H, Oullette E M, Warner L, Leichtman S R. Pediatrics. 1974;53:490–494. [PubMed] [Google Scholar]
- 12.Christoffel K K, Salafsky I. J Pediatr. 1975;87:963–967. doi: 10.1016/s0022-3476(75)80919-x. [DOI] [PubMed] [Google Scholar]
- 13.Collins E, Turner G. Med J Aust. 1978;2:606–608. doi: 10.5694/j.1326-5377.1978.tb131776.x. [DOI] [PubMed] [Google Scholar]
- 14.DeBeukelaer M M, Randall C L, Stroud D R. J Pediatr. 1977;91:759–760. doi: 10.1016/s0022-3476(77)81033-0. [DOI] [PubMed] [Google Scholar]
- 15.Ouellette E M, Rosett H L, Rosman N P, Weiner L. N Engl J Med. 1977;297:528–530. doi: 10.1056/NEJM197709082971003. [DOI] [PubMed] [Google Scholar]
- 16.Ernhart C B, Sokol R J, Martier S, Moron P, Nadler D, Ager J W, Wolf A. Am J Obstet Gynecol. 1987;156:33–39. doi: 10.1016/0002-9378(87)90199-2. [DOI] [PubMed] [Google Scholar]
- 17.Schenker S, Becker H C, Randall C L, Phillips D K, Baskin G S, Henderson G I. Alcohol Clin Exp Res. 1990;14:635–647. doi: 10.1111/j.1530-0277.1990.tb01220.x. [DOI] [PubMed] [Google Scholar]
- 18.Akimenko M A, Johnson S L, Westerfield M, Ekker M. Development (Cambridge, UK) 1995;121:347–357. doi: 10.1242/dev.121.2.347. [DOI] [PubMed] [Google Scholar]
- 19.Scott M P. Cell. 1992;71:551–553. doi: 10.1016/0092-8674(92)90588-4. [DOI] [PubMed] [Google Scholar]
- 20.Scott M P. Nucleic Acids Res. 1993;21:1687–1688. doi: 10.1093/nar/21.8.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimeld S M, McKay I J, Sharpe P T. Mech Dev. 1996;55:201–210. doi: 10.1016/0925-4773(96)00505-9. [DOI] [PubMed] [Google Scholar]
- 22.Jabs E W, Muller U, Li X, Ma L, Luo W, Haworth I S, Klisak I, Sparkes R, Warman M L, Mulliken J B. Cell. 1993;75:443–450. doi: 10.1016/0092-8674(93)90379-5. [DOI] [PubMed] [Google Scholar]
- 23.Ivens A, Flavin N, Williamson R, Dixon M, Bates G, Buckingham M, Robert B. Hum Genet. 1990;84:473–476. doi: 10.1007/BF00195823. [DOI] [PubMed] [Google Scholar]
- 24.Hewitt J E, Clark L N, Ivens A, Williamson R. Genomics. 1991;11:670–678. doi: 10.1016/0888-7543(91)90074-o. [DOI] [PubMed] [Google Scholar]
- 25.Hill R E, Jones P F, Rees A R, Sime C M, Justice M J, Copeland N G, Jenkins N A, Graham E, Davidson D R. Genes Dev. 1989;3:26–37. doi: 10.1101/gad.3.1.26. [DOI] [PubMed] [Google Scholar]
- 26.MacKenzie A, Ferguson M W, Sharpe P T. Development (Cambridge, UK) 1992;115:403–420. doi: 10.1242/dev.115.2.403. [DOI] [PubMed] [Google Scholar]
- 27.Woloshin P, Song K, Degnin C, Killary A M, Goldhamer D J, Sassoon D, Thayer M J. Cell. 1995;82:611–620. doi: 10.1016/0092-8674(95)90033-0. [DOI] [PubMed] [Google Scholar]
- 28.Monaghan A P, Davidson D R, Sime C, Graham E, Baldock R, Bhattacharya S S, Hill R E. Development (Cambridge, UK) 1991;112:1053–1061. doi: 10.1242/dev.112.4.1053. [DOI] [PubMed] [Google Scholar]
- 29.Davidson D R, Crawley A, Hill R E, Tickle C. Nature (London) 1991;352:429–431. doi: 10.1038/352429a0. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y H, Kundu R, Wu L, Luo W, Ignelzi M A, Jr, Snead M L, Maxson R E., Jr Proc Natl Acad Sci USA. 1995;92:6137–6141. doi: 10.1073/pnas.92.13.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kotch L E, Sulik K K. Am J Med Genet. 1992;44:168–176. doi: 10.1002/ajmg.1320440210. [DOI] [PubMed] [Google Scholar]
- 32.Towler D A, Rutledge S J, Rodan G A. Mol Endocrinol. 1994;8:1484–1493. doi: 10.1210/mend.8.11.7877617. [DOI] [PubMed] [Google Scholar]
- 33.Hoffmann H M, Catron K M, van Wijnen A J, McCabe L R, Lian J B, Stein G S, Stein J L. Proc Natl Acad Sci USA. 1994;91:12887–12891. doi: 10.1073/pnas.91.26.12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilkinson D G, Nieto M A. Methods Enzymol. 1993;225:361–373. doi: 10.1016/0076-6879(93)25025-w. [DOI] [PubMed] [Google Scholar]
- 35.Harlow E, Lane D. Antibodies: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1988. [Google Scholar]
- 36.Rifas L, Dawson L L, Halstead L R, Roberts M, Avioli L V. Calcif Tissue Int. 1994;54:505–510. doi: 10.1007/BF00334333. [DOI] [PubMed] [Google Scholar]
- 37.Rifas L, Gupta A, Hruska K A, Avioli L V. Calcif Tissue Int. 1995;57:60–63. doi: 10.1007/BF00298998. [DOI] [PubMed] [Google Scholar]
- 38.Rifas L, Kenney J S, Marcelli M, Pacifici R, Cheng S L, Dawson L L, Avioli L V. Endocrinology. 1995;136:4056–4067. doi: 10.1210/endo.136.9.7649114. [DOI] [PubMed] [Google Scholar]
- 39.Estus S, Zaks W J, Freeman R S, Gruda M, Bravo R, Johnson E M., Jr J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johansson B M, Wiles M V. Mol Cell Biol. 1995;15:141–151. doi: 10.1128/mcb.15.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tong H S, Sakai D D, Sims S M, Dixon S J, Yamin M, Goldring S R, Snead M L, Minkin C. J Bone Miner Res. 1994;9:577–584. doi: 10.1002/jbmr.5650090418. [DOI] [PubMed] [Google Scholar]
- 42.Towler D A, Bennett C D, Rodan G A. Mol Endocrinol. 1994;8:614–624. doi: 10.1210/mend.8.5.7914673. [DOI] [PubMed] [Google Scholar]
- 43.Randall C L, Ekblad U, Anton R F. Alcoholism. 1990;14:807–812. doi: 10.1111/j.1530-0277.1990.tb01818.x. [DOI] [PubMed] [Google Scholar]
- 44.Spagnolo A. Ann Ist Super Sanita. 1993;29:89–96. [PubMed] [Google Scholar]
- 45.Edwards H G, Dow-Edwards D L. Teratology. 1991;44:373–378. doi: 10.1002/tera.1420440403. [DOI] [PubMed] [Google Scholar]
- 46.Pennington S N. Alcoholism. 1990;14:832–837. doi: 10.1111/j.1530-0277.1990.tb01823.x. [DOI] [PubMed] [Google Scholar]
- 47.Randall C L, Anton R F, Becker H C, Hale R L, Ekblad U. Teratology. 1991;44:521–529. doi: 10.1002/tera.1420440506. [DOI] [PubMed] [Google Scholar]
- 48.Randall C L, Becker H C, Anton R F. Alcohol Clin Exp Res. 1991;15:673–677. doi: 10.1111/j.1530-0277.1991.tb00577.x. [DOI] [PubMed] [Google Scholar]
- 49.Grummer M A, Langhough R E, Zachman R D. Alcoholism. 1993;17:592–597. doi: 10.1111/j.1530-0277.1993.tb00805.x. [DOI] [PubMed] [Google Scholar]
- 50.Mauceri H J, Lee W H, Conway S. Alcoholism. 1994;18:35–41. doi: 10.1111/j.1530-0277.1994.tb00877.x. [DOI] [PubMed] [Google Scholar]
- 51.Weston W M, Greene R M, Uberti M, Pisano M M. Alcoholism. 1994;18:177–182. doi: 10.1111/j.1530-0277.1994.tb00900.x. [DOI] [PubMed] [Google Scholar]
- 52.Scott M P, Tamkun J W, Hartzell G W., III Biochim Biophys Acta. 1989;989:25–48. doi: 10.1016/0304-419x(89)90033-4. [DOI] [PubMed] [Google Scholar]
- 53.Krumlauf R. Cell. 1994;78:191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 54.Akam M. Development (Cambridge, UK) 1987;101:1–22. [PubMed] [Google Scholar]
- 55.Takahashi Y, Le Douarin N. Proc Natl Acad Sci USA. 1990;87:7482–7486. doi: 10.1073/pnas.87.19.7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nishikawa K, Nakanishi T, Aoki C, Hattori T, Takahashi K, Taniguchi S. Biochem Mol Biol Int. 1994;32:763–771. [PubMed] [Google Scholar]
- 57.Jowett A K, Vainio S, Ferguson M W, Sharpe P T, Thesleff I. Development (Cambridge, UK) 1993;117:461–470. doi: 10.1242/dev.117.2.461. [DOI] [PubMed] [Google Scholar]
- 58.Robert B, Lyons G, Simandl B K, Kuroiwa A, Buckingham M. Genes Dev. 1991;5:2363–2374. doi: 10.1101/gad.5.12b.2363. [DOI] [PubMed] [Google Scholar]
- 59.Monsoro-Burq A H, Bontoux M, Teillet M A, Le Douarin N M. Proc Natl Acad Sci USA. 1994;91:10435–10439. doi: 10.1073/pnas.91.22.10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan-Thomas P S, Thompson R P, Robert B, Yacoub M H, Barton P J. Dev Dyn. 1993;197:203–216. doi: 10.1002/aja.1001970305. [DOI] [PubMed] [Google Scholar]
- 61.Aldo-Benson M, Kluve-Beckerman B, Hardwick J, Lockwood M. J Lab Clin Med. 1992;119:32–37. [PubMed] [Google Scholar]
- 62.Wilke N, Sganga M, Barhite S, Miles M F. EXS. 1994;71:49–59. doi: 10.1007/978-3-0348-7330-7_6. [DOI] [PubMed] [Google Scholar]
- 63.Miles M F, Diaz J E, DeGuzman V S. J Biol Chem. 1991;266:2409–2414. [PubMed] [Google Scholar]
- 64.Soszynski P A, Frohman L A. Endocrinology. 1992;131:173–180. doi: 10.1210/endo.131.1.1351838. [DOI] [PubMed] [Google Scholar]
- 65.Vainio S, Karavanova I, Jowett A, Thesleff I. Cell. 1993;75:45–58. [PubMed] [Google Scholar]
- 66.Francis P H, Richardson M K, Brickell P M, Tickle C. Development (Cambridge, UK) 1994;120:209–218. doi: 10.1242/dev.120.1.209. [DOI] [PubMed] [Google Scholar]
- 67.Francis-West P H, Tatla T, Brickell P M. Dev Dyn. 1994;201:168–178. doi: 10.1002/aja.1002010207. [DOI] [PubMed] [Google Scholar]
- 68.Webster W S, Walsh D A, McEwen S E, Lipson A H. Teratology. 1983;27:231–243. doi: 10.1002/tera.1420270211. [DOI] [PubMed] [Google Scholar]
- 69.Webster W S, Walsh D A, Lipson A H, McEwen S E. Neurobehav Toxicol. 1980;2:227–234. [Google Scholar]
- 70.Sulik K K, Johnston M C, Daft P A, Russell W E, Dehart D B. Am J Med Genet. 1986;2:97–112. doi: 10.1002/ajmg.1320250614. [DOI] [PubMed] [Google Scholar]
- 71.Sulik K K. Prog Clin Biol Res. 1985;163C:399–403. [PubMed] [Google Scholar]
- 72.Kotch L E, Dehart D B, Alles A J, Chernoff N, Sulik K K. Teratology. 1992;46:323–332. doi: 10.1002/tera.1420460403. [DOI] [PubMed] [Google Scholar]
- 73.Sulik K K, Johnston M C, Webb M A. Science. 1981;214:936–938. doi: 10.1126/science.6795717. [DOI] [PubMed] [Google Scholar]