

Abstract
Airway surface liquid (ASL) absorption is initiated by Na+ entry via epithelial Na+ channels (ENaC), which establishes an osmotic gradient that drives fluid from the luminal to serosal airway surface. We and others have recently reported that a protease/anti-protease balance regulates ENaC in human airway epithelial cells (HAEC) and provides a mechanism for autoregulation of ASL volume. In cystic fibrosis (CF), this balance is disturbed, leading to constitutive proteolytic activation of ENaC and the pathological Na+ hyperabsorption characteristic of this airway disease. Prostasin is a glycosylphosphatidylinositol-anchored serine protease that activates ENaC and is expressed on the surface epithelium lining the airway. In this report we present evidence that prostasin expression is regulated by the ASL volume, allowing for increased proteolytic activation of ENaC when the ASL volume is high. Prostasin activity is further regulated by the cognate serpin protease nexin-1 (PN-1), which is expressed in HAEC and inhibits Na+ absorption by forming an inactive complex with prostasin and preventing the proteolytic processing of prostasin. Whereas these mechanisms regulate prostasin expression in response to ASL volume in non-CF epithelia, HAEC cultured from CF patients express >50% more prostasin on the epithelial surface. These findings suggest that a proteolytic cascade involving prostasin, an upstream prostasin-activating protease, and PN-1 regulate Na+ absorption in the airway and that abnormal prostasin expression contributes to excessive proteolytic activation of ENaC in CF patients.
Keywords: epithelial sodium channels, protease nexin 1, bronchial epithelial cultures, matriptase
Amiloride-sensitive epithelial sodium channels (ENaC) mediate Na+ absorption across the apical (luminal) epithelial surfaces lining the lung, colon, sweat duct, and kidney (15). In airway epithelium, Na+ absorption via ENaC in conjunction with a basolateral Na+-K+-ATPase establishes an osmotic gradient that drives airway surface liquid (ASL) absorption (29-31, 41). ENaC activity is increased in cystic fibrosis (CF), which, together with impaired CFTR-mediated anion secretion, leads to pathological ASL depletion and impaired mucus clearance from the lung (20, 23, 27, 29-31, 41). Previous work from our group (34) and Tarran et al. (42) has indicated that a balance between the protease activity of membrane-tethered channel-activating proteases (CAPs) and soluble protease inhibitors in the ASL modulates Na+ absorption in human airway epithelial cells (HAEC). When the ASL volume is low, as in the steady state, the soluble protease inhibitor concentration is sufficient to minimize constitutive activation of ENaC by CAPs in non-CF epithelium. However, when the ASL volume is expanded, the soluble protease inhibitors are diluted, and this allows for CAP-mediated ENaC activation. More importantly, recent evidence suggests that excessive activation of ENaC by CAPs contributes to Na+ hyperabsorption in CF airways (5, 34, 42). However, the soluble protease inhibitor concentration in the CF ASL would be predicted to be high, because the low ASL volume of CF airways would lead to concentration of the ASL proteins. To reconcile the apparent contradiction between a high degree of proteolytic ENaC activation and the predicted high level of protease inhibitor in the CF ASL, we questioned whether the protease/anti-protease balance present at the airway surface is modulated directly by changes in the expression of CAPs.
Prostasin, the human ortholog of CAP1, is a glycosylphosphatidylinositol (GPI)-anchored serine protease that activates ENaC (4, 8, 13, 44). Donaldson et al. (13) demonstrated that prostasin is expressed in airway epithelium and increases the Na+ current by ∼75% when coexpressed with ENaC in Xenopus oocytes. Furthermore, Tong et al. (43) reported an ∼75% reduction in the Na+ conductance of an immortalized airway epithelial cell line following small interference (si)RNA-mediated prostasin knockdown. Recent evidence from Bruns et al. (4) demonstrated that prostasin cleaves the extracellular loop of the γENaC subunit and thereby increases the open probability of the channel. Although the activating effects of prostasin on Na+ channels are well known, little is known regarding the regulation of prostasin expression and activity in the airway.
To investigate whether prostasin expression is regulated to maintain ASL volume homeostasis, we examined the expression of prostasin in primary HAEC cultured on an air-liquid interface with basal and expanded ASL volumes. We observed that prostasin expression and processing are regulated by changes in the ASL volume. Accordingly, the surface expression of prostasin increases under conditions when the ASL volume is expanded, presumably allowing for augmented Na+ and fluid absorption. In CF epithelia, these regulatory mechanisms are altered, leading to increased expression of processed prostasin on the luminal airway surface. Prostasin is synthesized as an inactive zymogen and has not been shown to be capable of autocatalysis (2, 39); activation requires its cleavage by an upstream protease, which is believed to be matriptase (10, 25, 26, 37). Protease nexin-1 (PN-1), an associated inhibitor of prostasin (9), inhibited the amiloride-sensitive Na+ current by formation of an inactive prostasin complex and by preventing the processing of prostasin zymogen to active enzyme. These results suggest that ENaC activity in airway epithelium is determined by a proteolytic cascade involving prostasin, an upstream prostasin-activating protease, and PN-1 and suggest that excessive Na+ absorption in CF airways is caused by abnormal prostasin regulation.
MATERIALS AND METHODS
Materials
All cell culture medium was obtained from GIBCO (Invitrogen, Carlsbad, CA), except bronchial epithelial growth medium (Clonetics, San Diego, CA) and Ultroser G (BioSepra, Cedex, France). Recombinant human PN-1 and recombinant human matriptase were purchased from R&D Systems (Minneapolis, MN). Sulfo-NHS-SS-biotin and streptavidin beads were obtained from Pierce Biotechnology (Rockford, IL). Unless otherwise specified, all other reagents were obtained from Sigma (St. Louis, MO).
Primary HAEC
HAEC were cultured from excess pathological tissue following lung transplantation and organ donation under a protocol approved by the University of Pittsburgh Investigational Review Board. HAEC were cultured on human placental collagen-coated Costar Transwell filters (catalog no. 3470; 0.33 cm2, 0.4-μm pore) as previously described (12, 35) and used for experimentation following 4–6 wk of culture at an air-liquid interface. Where indicated, 20–100 μl of PBS were gently pipetted onto the apical surface of differentiated HAEC to expand the ASL volume. Non-CF HAEC were obtained from donors with chronic obstructive pulmonary disease (11 patients), idiopathic pulmonary fibrosis (6 patients), scleroderma (3 patients), or primary pulmonary hypertension (3 patients). Qualitative differences due to disease state were not observed. CF HAEC were obtained from 12 donors with the following CFTR genotypes: ΔF508, G551D, G542X, N1303K, and two unknowns. Because the majority of the patients had at least one ΔF508 allele, there was insufficient power to assess for differences due to CFTR mutation severity.
Western blotting and surface biotinylation
Before experimental use, the apical surface of differentiated HAEC cultures was washed once with PBS plus 5 mM dithiothreitol and then three times with PBS to remove the accumulated cellular debris and mucus. After the indicated period of time, the apical secretions were collected in 100 μl of PBS and concentrated by acetone precipitation overnight; the basolateral medium was collected and similarly concentrated. After collection of the secretions, surface biotinylation and Western blotting were performed as previously described (34). Briefly, the HAEC cultures grown on filter supports were placed on ice, and the apical surface was washed with ice-cold PBS plus 1 mM CaCl2 to remove cellular debris. Subsequently, the apical or basolateral surface of the HAEC was incubated with 0.5 mg/ml sulfo-NHS-SS-biotin in borate buffer (85 mM NaCl, 4 mM KCl, and 15 mM Na2B4O7, pH 9). After 20 min, the biotinylation reaction was quenched with PBS plus 10% fetal bovine serum, and the cells were rinsed with ice-cold PBS plus Ca2+. The cells were then lysed in cell lysis buffer (10 mM Tris-Cl, 50 mM EGTA, 0.4% sodium deoxycholate, and 1% NP-40, pH 7.4). Cellular debris was removed by centrifugation, and protein concentration was determined using the Bradford method (Bio-Rad, Hercules, CA). The biotinylated proteins in 100 μg of cellular lysate were recovered by incubation with streptavidin beads overnight at 4°C. The proteins were resolved using standard SDS-PAGE under reducing conditions and transferred to nitrocellulose. The membrane was then immunoblotted using monoclonal anti-prostasin (1:500; BD Transduction Laboratories, San Jose, CA) or monoclonal anti-protease nexin antibodies (1:500; R&D Systems). Subsequently, the blots were stripped and reprobed with monoclonal β-actin antibodies (1:3,000) or monoclonal α1 Na+-K+-ATPase antibodies (1:1,000; Millipore, Billerica, MA). Band intensity was quantified by densitometry (Quantiscan; Biosoft, Cambridge, UK).
Human sputum collection and processing
Sputum samples were collected from adult patients with CF (mean age: 22 yr) under a protocol approved by the local Institutional Review Board. Sputum samples were processed using Sputolysin (Dade Behring, Deerfield, Illinois) as previously described (33). Briefly, 1 ml of 10% Sputolysin was added per 1 mg of sputum, and the sample was incubated for 5 min at 37°C with vigorous shaking and mixed with a transfer pipette. Samples were then centrifuged at 2,000 rpm for 5 min at 4°C, and the supernatants were used for Western blotting.
Biochemical characterization of prostasin in cultured human airway epithelium
To determine whether prostasin is attached to the cell surface via a GPI anchor, we exposed the cultured HAEC to 1 U/ml recombinant phosphatidylinositol-specific phospholipase C (PI-PLC; Molecular Probes, Carlsbad, CA) for 1 h before collection of the secreted proteins, surface biotinylation, and lysis (45). Enzymatic digestion of the N-glycans was performed using N-glycosidase F (PNGase F) per the manufacturer's instructions (New England Biolabs, Ipswich, MA).
Real-time PCR
TaqMan probes for human prostasin and β-actin were purchased from Applied Biosystems (Foster City, CA). The ASL of differentiated HAEC was expanded for the indicated period of time, and the total RNA was subsequently extracted using Trizol (Invitrogen). After purification (RNeasy; Qiagen, Valencia, CA), the RNA was reverse-transcribed to cDNA using the High Capacity cDNA reverse transcription kit (Applied Biosystems). Real-time PCR was performed with an ABI Prism 7900HT sequence detection system (Applied Biosystems), and the relative gene expression was determined using the ΔΔCt method, where Ct is the threshold cycle: relative expression = 2e − (ΔCt sample − ΔCt average of control), where the ΔCt for an individual sample is Ct prostasin − Ct actin.
Short-circuit current recordings
Before short-circuit current (Isc) recording, the apical surface of differentiated HAEC was exposed to increasing concentrations of recombinant human PN-1 or vehicle control in 20 μl of PBS for 4 h. The addition of 20 μl of PBS for 4 h to the apical surface caused the baseline amiloride-sensitive Isc to increase approximately twofold compared with control filters that remained at an air-liquid interface (data not shown). The Isc were measured as previously described (34). In brief, cells cultured on filter supports were mounted in modified Ussing chambers, and the cultures were continuously short-circuited with an automatic voltage clamp (Department of Bioengineering, University of Iowa, Iowa City, IA). Transepithelial resistance was measured by periodically applying a 2.5-mV bipolar voltage pulse and was calculated using Ohm's law. The bathing Ringer solution was composed of 120 mM NaCl, 25 mM NaHCO3, 3.3 mM KH2PO4, 0.8 mM K2HPO4, 1.2 mM MgCl2, 1.2 mM CaCl2, and 10 mM glucose. Chambers were constantly gassed with a mixture of 95% O2 and 5% CO2 at 37°C, which maintained the pH at 7.4. After a 5-min equilibration period, the baseline Isc was recorded. Subsequently, 1 μM trypsin was added to the apical surface for 5 min, providing a measure of protease-activatable channels (1, 5, 22, 34). The amiloride-sensitive Isc (INa) was determined by the addition of amiloride to the apical cell chamber to a concentration of 10 μM.
In vitro biochemical characterization of the PN-1 and matriptase interaction
As an initial test for matriptase inhibition, 10 nM recombinant human matriptase was incubated with a 10-fold molar excess of recombinant human PN-1, aprotinin, 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF), or ovalbumin in assay buffer (150 mM NaCl, 50 mM Tris, 10 mM CaCl2, and 0.05% Brij 35, pH 7.5) for 30 min at 25°C. Residual enzyme activity was measured by adding the substrate Boc-Gln-Ala-Arg-AMC (Bachem, King of Prussia, PA) and monitoring the change in relative fluorescence units over time with the FluorImager 575 (Molecular Dynamics, Sunnyvale, CA). To determine the stoichiometry of inhibition (SI), we incubated increasing molar ratios of PN-1 with 10 nM matriptase in assay buffer for 30 min at 25°C. The SI was determined by plotting the ratio of the velocity of the inhibited enzyme to the velocity of the uninhibited control versus the molar ratio of PN-1 to matriptase, and extrapolating to zero activity. PN-1 and matriptase complex formation was determined by incubating 250 nM PN-1 with 50 nM matriptase in assay buffer for 30 min at 25°C, resolving the product by SDS-PAGE, and immunoblotting for PN-1 and matriptase (R&D Systems).
Statistics
Results are means ± SE. Significance was determined using unpaired Student's t-tests unless otherwise indicated. A P value <0.05 was considered statistically different. For analysis of Western blotting data, the densitometry units from the blots containing both experimental and control conditions were normalized to the darkest band, and the provided n value indicates the number of tissue donors. For each tissue donor, the mean densitometry values were obtained from more than three individual cultures. The IC50 for PN-1 was estimated from the amiloride-sensitive current (normalized to values obtained from matched HAEC treated with vehicle control) plotted as a function of PN-1 concentration fitted to the Hill equation. All statistical analysis and data fitting were performed using SigmaPlot 10 (SPSS).
RESULTS
Prostasin is expressed in a nonpolarized distribution but only secreted and processed on the apical membrane of primary HAEC
To localize prostasin expression in primary HAEC, we performed Western blotting on the secreted proteins, surface biotinylated proteins, and whole cell lysate of HAEC. Before selective biotinylation of the apical and basolateral surfaces, the secretions into the apical and basolateral compartments were collected and concentrated by acetone precipitation. As shown in the Western blots in Fig. 1A, prostasin migrates as 37- and 40-kDa bands in the apical secretions; however, no prostasin was observed in the basolateral secretions. These results are consistent with previous reports of prostasin secretion from human prostate tissue (48) and from various immortalized mouse cell lines (36, 45) but differ from a study in the immortalized bronchial epithelial cell line JME/CF15, in which prostasin was not present in the secretions (43). To clarify whether prostasin is secreted in the airway, we also performed Western blotting on sputum from a CF patient (Fig. 1B). These data demonstrate that prostasin is apically secreted from HAEC in vivo and in culture.
Fig. 1.

Polarized apical expression of active prostasin in human airway epithelial cells (HAEC). A: the apical (AP) and basolateral (BL) secretions were collected from cultured HAEC for 24 h before AP or BL cell surface biotinylation. The concentrated secretions, recovered biotinylated proteins, and whole cell lysate were separated by SDS-PAGE and immunoblotted for prostasin. The blots were subsequently stripped and reprobed for β-actin (β) to ensure that no intracellular biotinylation had occurred and that protein loading was equal. B: prostasin Western blot of sputum from a cystic fibrosis (CF) patient; 10, 20, or 30 μl of processed sputum were analyzed. C: prostasin Western blot of AP and BL secretions, biotinylated proteins, and whole cell lysate of HAEC with or without treatment with 1 U/ml phosphatidylinositol-specific phospholipase C (PI-PLC) for 1 h at 37°C. D: Na+-K+-ATPase Western blot from the recovered biotinylated proteins and whole cell lysate of HAEC following selective AP or BL biotinylation. IB, immunoblot.
Because the GPI anchor acts as a strong signal for apical sorting (32), we anticipated that prostasin would only be present in the apically biotinylated proteins. As expected, both the 37- and 40-kDa prostasin molecular species were present in the biotinylated proteins from the apical membrane (Fig. 1A). Surprisingly, the 40-kDa prostasin molecular species was also present in the proteins recovered from the basolateral membrane, suggesting some degree of basolateral delivery. To determine whether the prostasin present on the basolateral surface is attached to the cell membrane via a GPI anchor, we treated the HAEC with PI-PLC. After PI-PLC treatment, the amount of cell surface prostasin decreased and prostasin correspondingly increased in the secretions (Fig. 1C), suggesting that prostasin is attached to both the apical and basolateral surfaces via a GPI anchor. To confirm that the selective apical and basolateral biotinylation reactions were segregated, we stripped the blots and reblotted for Na+-K+-ATPase, a basolaterally expressed membrane protein; as anticipated and as shown in Fig. 1D, Na+-K+-ATPase was only present in the basolateral biotinylated proteins of HAEC. In the whole cell lysate, prostasin migrated primarily as a 40-kDa band with a less prominent 37-kDa band. These results suggest that although prostasin is present on both the apical and basolateral membranes, it is only secreted and processed to a lower molecular mass species on the apical surface.
Next, we assessed whether differential posttranslational modification accounts for the difference in electrophoretic mobility between the 37- and 40-kDa prostasin bands. Because prostasin is known to be N-glycosylated, we examined the effects of glycan digestion in CF and control HAEC (Fig. 2A). After digestion with PNGase, both prostasin molecular species migrated faster, consistent with the previous reports of prostasin glycosylation (2, 38, 39, 45). However, differential glycosylation does not account for the difference between the 37- and 40-kDa prostasin bands, because both molecular species were present following N-glycan digestion.
Fig. 2.

Posttranslational modification of prostasin in control and CF HAEC.A: apical biotinylated proteins and whole cell lysates of control and CF HAEC were treated with and without N-glycosidase (PNGase). These samples were subsequently subjected to Western blotting for prostasin. B: 30 μM aprotinin was applied to the apical surface of control and CF HAEC for 24 h, and prostasin Western blotting was performed on the apical biotinylated proteins and whole cell lysate.
Prostasin is synthesized as a zymogen and as such requires proteolytic processing to be active (2). Therefore, we questioned whether the 37-kDa band in HAEC represents a proteolytically processed form of prostasin. We reasoned that serine protease inhibition would prevent the formation of the 37-kDa band if this molecular species represents prostasin that has undergone proteolytic processing. As shown in Fig. 2B, 24 h of apical aprotinin exposure increased the abundance of the 40-kDa band and correspondingly decreased the intensity of the 37-kDa band. This suggests that the 37-kDa molecular species represents a form of prostasin that has been processed by a serine protease. The molecular masses of the two prostasin bands are consistent with a recently published report of prostasin zymogen processing by matriptase (CAP3) in mouse epidermis (37). Therefore, both prostasin proteolytic processing and secretion occur selectively on the apical cell surface. This suggests that polarized processing of prostasin is not due to its GPI anchor but, rather, a consequence of polarized protease and/or phospholipase activity.
Prostasin expression is increased in CF HAEC
The results comparing the posttranslational modification of prostasin in CF and non-CF HAEC (Fig. 2) suggested that epithelia cultured from CF patients express more processed prostasin on the apical membrane than control HAEC. Furthermore, Tarran et al. (42) recently reported that prostasin gene expression was increased nearly fivefold in CF compared with non-CF HAEC in three tissue donors. To determine whether CF epithelia express more prostasin, we performed semiquantitative Western blotting on the apical secretions, apically biotinylated proteins, and whole cell lysate of nine CF tissue donors and morphologically matched non-CF donors (Fig. 3). More prostasin was present in the CF HAEC apical secretions (5,747 ± 213 vs. 4,677 ± 372 mean normalized densitometry units, P = 0.027, n = 9 tissue donors) and apical biotinylated proteins (4,399 ± 387 vs. 2,760 ± 472 mean normalized densitometry units, P = 0.025, n = 9 tissue donors). These results confirm that primary cultured CF airway epithelium expresses increased amounts of prostasin on the cell surface, where the protease is believed to activate ENaC. Furthermore, as shown in Fig. 3D, the ratio of processed (37 kDa) to total (37 kDa + 40 kDa) prostasin was significantly higher in CF HAEC when the two bands were clearly discernable by densitometry (0.78 ± 0.02 vs. 0.54 ± 0.08, P = 0.004, n ≥ 3 tissue donors). These results demonstrate that there is an increased amount of processed prostasin on the CF cell surface.
Fig. 3.
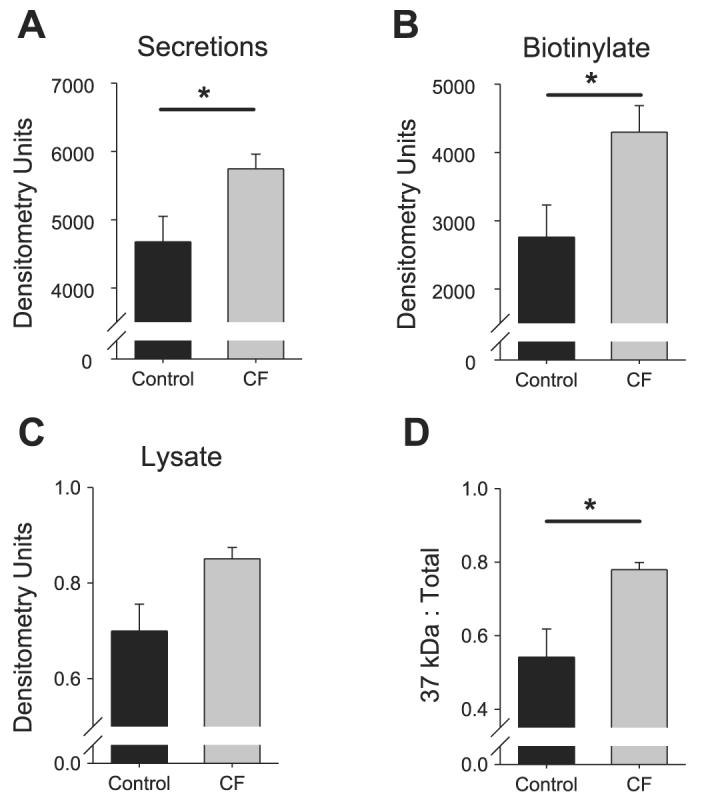
Increased prostasin expression in CF HAEC. Densitometry was performed on the Western blots of the apical secretions (A), recovered apical biotinylated proteins (B), and whole cell lysate (C) of control and CF HAEC. Data are means ± SE expressed as normalized mean densitometry units; n ≥ 4 cultures each from ≥9 tissue donors. When the 37- and 40-kDa bands were clearly discernable by densitometry, the ratio of 37 kDa prostasin to total prostasin was compared between control and CF HAEC (D). Data are means ± SE expressed as ratios; n = 4 tissue donors. *P < 0.05, significant difference between control and CF HAEC.
HAEC coordinate prostasin expression with ASL volume
Next, we determined whether HAEC regulate prostasin expression in response to changes in the ASL volume. To expand the ASL volume, we applied 100 μl of PBS to the apical surface of differentiated HAEC for 24 h. The apical secretions that had accumulated over the 24-h interval were collected, and the apical surface was biotinylated. As shown in the representative Western blot in Fig. 4A, prostasin expression increased in HAEC under ASL volume expansion conditions, suggesting that cellular mechanisms coordinate prostasin expression with ASL volume. To confirm these findings, we performed semi-quantitative Western blotting for prostasin on HAEC cultured from additional tissue donors under air-liquid and ASL volume expansion conditions. Although the amount of secreted prostasin between HAEC under control and ASL volume expansion conditions was not statistically increased (12,493 ± 1,223 vs. 9,029 ± 1,770 normalized densitometry units, P = 0.13), there was a large increase in the cell surface prostasin expression with ASL expansion (5,537 ± 356 vs. 3,547 ± 570 normalized densitometry units, P = 0.007, n = 12 tissue donors). Similarly, prostasin expression in the whole cell lysate also increased with ASL expansion (0.9 ± 0.03 vs. 0.62 ± 0.06 densitometry units normalized to β-actin, P < 0.001 by Mann-Whitney U-test, n = 12 tissue donors). ASL volume expansion had a similar effect on prostasin expression in CF HAEC (data not shown). Therefore, airway surface epithelium regulates prostasin expression in response to changes in the ASL volume.
Fig. 4.

Airway surface liquid (ASL) volume expansion increases prostasin expression. A: prostasin Western blot of the apical secretions, apical biotinylated proteins, and whole cell lysate of HAEC with or without ASL volume expansion (100 μl of PBS) for 24 h. B: mean normalized densitometry of the apical secretions, apical biotinylated proteins, and whole cell lysate of HAEC with or without ASL volume expansion. Data are mean ± SE; n ≥ 8 tissue donors (>2 cultures per donor). *P < 0.05, significant difference between control and ASL expansion conditions.
Next, we assessed the time dependence of the increase in prostasin expression following ASL expansion. Our previous results indicate that ASL expansion rapidly increases Na+ absorption with a half time (t1/2) of ∼30 min and a maximal response after 24 h (34). Real-time PCR and Western blotting for prostasin were performed on HAEC after 0, 2, 6, 12, and 24 h of ASL expansion (Fig. 5, A and B). Prostasin expression increased significantly after 12 h of ASL expansion. Because prostasin expression increased on a protein and message level over the same time interval, the increase in prostasin expression associated with ASL expansion appears to be transcriptionally mediated. These results suggest that ASL expansion may increase Na+ absorption by at least two mechanisms: 1) an initial rapid increase due to the dilution of soluble protease inhibitors in the ASL (34, 42) and 2) a slower increase due to upregulation of prostasin expression.
Fig. 5.
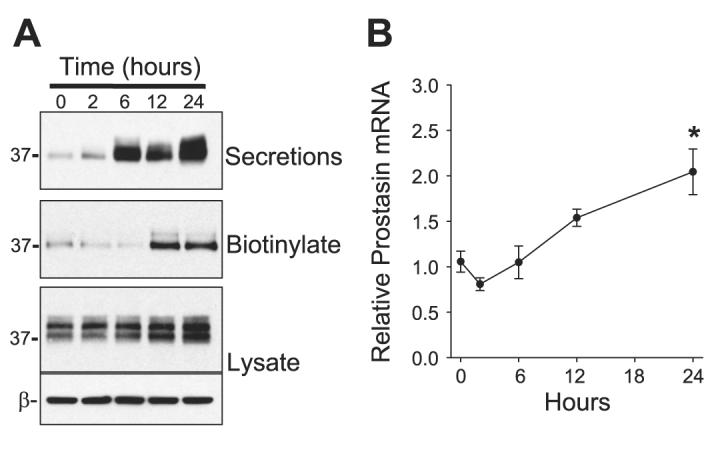
Kinetics of prostasin expression following ASL volume expansion. The ASL volume of differentiated HAEC filters was expanded (100 μl PBS) for 0, 2, 6, 12, and 24 h, followed by determination of prostasin expression by Western blotting (A) and real-time PCR (B). Data in B are means ± SE of prostasin mRNA expression values relative to air-liquid control cultures; n = 4 cultures. *P < 0.05, significantly different from time 0 as determined by ANOVA with Bonferroni post hoc analysis.
Endogenous PN-1 regulates ENaC activity through prostasin interactions
Previously, PN-1, or SERPINE2, has been shown to act as a suicide inhibitor of prostasin through formation of an inactive SDS-stable complex with prostasin (8, 9). In Xenopus oocytes, coexpression of PN-1 with αβγENaC leads to an inhibition of the amiloride-sensitive current INa (47). Furthermore, PN-1 knockdown, using siRNA, augments Na+ absorption in the mouse cortical collecting duct cell line M-1 (47). On the basis of these studies, we hypothesized that PN-1 may act as an endogenous regulator of prostasin and ENaC activity in the ASL of airway epithelium. To confirm the expression of PN-1 in HAEC, we cloned PN-1 from HAEC using real-time PCR, as described by Chen et al. (9), and the sequence was confirmed to be PN-1 (data not shown). As shown in Fig. 6A, Western blotting of HAEC lysate with anti-PN-1 monoclonal antibody revealed an ∼45-kDa band with electrophoretic mobility similar to that of recombinant human PN-1 (rhPN-1), confirming PN-1 expression in HAEC on a protein level. To determine whether the increased prostasin expression in CF HAEC could be explained by a deficiency in PN-1 expression, we compared PN-1 expression between CF and control HAEC (Fig. 6B). There was no difference in PN-1 expression between control and CF HAEC (1.442 ± 0.044 vs. 1.342 ± 0.038 PN-1 densitometry units normalized to β-actin, P = 0.097, n = 3 tissue donors). Thus PN-1 is expressed similarly in CF and non-CF HAEC, suggesting that the increased prostasin present in CF is not due to deficiency of PN-1.
Fig. 6.
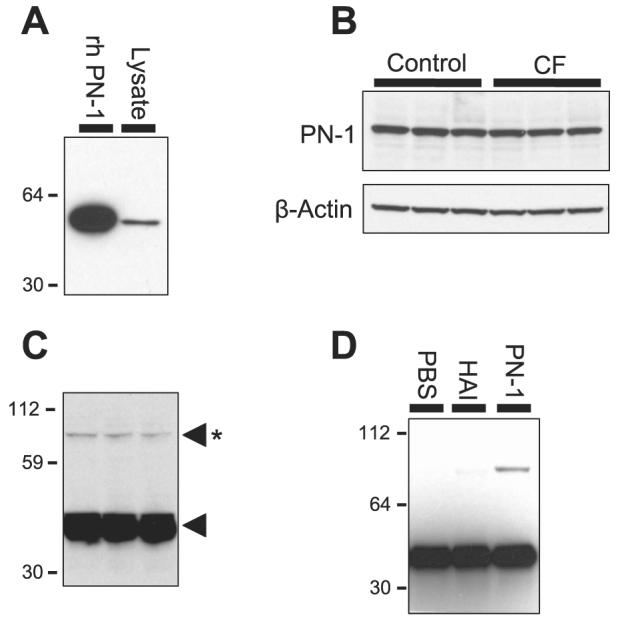
Protease nexin-1 (PN-1) expression in control and CF HAEC. A: PN-1 Western blot of HAEC cell lysate. Recombinant human PN-1 (rhPN-1) is shown for size comparison. B: PN-1 Western blot of control and CF HAEC lysate. C: prostasin Western blot of HAEC apical secretions with longer exposure. Asterisk denotes the 82-kDa SDS-stable prostasin molecular complex, and the lone arrowhead denotes prostasin. D: prostasin Western blot of HAEC apical secretions 24 h after ASL washout by apical exposure to PBS, bikunin (HAI), or rhPN-1. The 82-kDa PN-1-prostasin complex was only seen after exposure to PN-1.
Next, we determined whether PN-1 forms a complex with prostasin in HAEC. As shown in Fig. 6C, when prostasin Western blots of HAEC secretions were exposed for a prolonged period, an ∼82-kDa SDS-stable complex was observed. This suggests that PN-1 forms a complex in HAEC, because the prostasin complex with recombinant PN-1 migrates as an 82-kDa band (8, 9). To confirm that the 82-kDa band on the prostasin Western blots represents a complex with PN-1, we replaced the endogenous apical secretions of HAEC with 100 μl of PBS for 24 h. After apical washout, the 82-kDa band in the apical secretions was no longer present (Fig. 6D), suggesting that this molecular species represents a prostasin complex with a factor in the ASL. The 82-kDa band returned when 1 μM PN-1 was included in the apical solution, suggesting that this band represents a prostasin-PN-1 complex. The addition of other serine protease inhibitors, such as bikunin, known to inhibit both prostasin and Na+ current (3), did not lead to the formation of the 82-kDa prostasin complex. These findings suggest that PN-1 is an endogenous regulator of prostasin in the airway.
Because prostasin inhibition would be expected to inhibit ENaC activity, we determined whether PN-1 has an inhibitory effect on the Na+ conductance of HAEC. Increasing concentrations of rhPN-1 were applied to the apical surface of HAEC for 4 h before measurement of the amiloride-sensitive short-circuit current (INa). As demonstrated in Fig. 7A, PN-1 induced a dose-dependent decrease in INa with an IC50 of 5.5 × 10−7 M. The effects of PN-1 on INa were similar in CF HAEC (IC50 = 2.6 × 10−7 M, n = 4 cultures), suggesting that CF epithelia respond normally to protease inhibition by PN-1. As a protein control, we compared the effects of PN-1 with those of ovalbumin, which is a serpin with no inhibitory activity, on INa. Whereas 1 μM PN-1 decreased INa to 40.4 ± 1.6% of control, 1 μM ovalbumin did not affect Na+ conductance (100.9 ± 0.5% of control, P < 0.001, n = 3 cultures). These data indicate that PN-1 inhibits Na+ transport in airway epithelium.
Fig. 7.

PN-1 inhibits Na+ current and prostasin processing. A: dose-dependent inhibition of the amiloride-sensitive short-circuit current INa by PN-1 in HAEC. The apical surface of HAEC was washed with PBS and then exposed to increasing concentrations of rhPN-1 in 20 μl of PBS for 4 h before measurement of INa. Data are means ± SE of the amiloride-sensitive current normalized to control filters; n ≥ 3 cultures. B: comparison of pretrypsin INa (baseline) and the increase in INa following apical trypsin addition (Δtrypsin) in the presence of increasing PN-1 concentration ([PN-1]). Data are means ± SE of INa at baseline and the change from baseline INa following trypsin treatment; n = 4 cultures. C: prostasin expression in the apical secretions of HAEC following 4 h of treatment with increasing concentrations of rhPN-1. Note the dose-dependent increase in the presence of the 82-kDa SDS-stable prostasin complex (asterisk). D: prostasin expression in the apical biotinylated proteins of HAEC following 4 h of treatment with 1 μM PN-1.
Proteases activate ENaC by limited proteolysis of the extra-cellular loop of the α- and γ-subunits, which increases the open probability of the channel (4, 6, 7, 17-19). Therefore, if the inhibitory effect of PN-1 on Na+ current is due to prevention of protease-mediated ENaC activation, one would expect to find an increase in the number of unprocessed inactive channels and a corresponding decrease in the number of processed active channels. To assess this, we measured the INa in HAEC exposed to increasing concentrations of PN-1 before and after apical exposure to 1 μM trypsin (Fig. 7B). The increase in INa following trypsin exposure (Δtrypsin) provides a measurement of the number of unprocessed channels present on the apical surface (1, 5, 22, 34). In parallel to the decrease in INa associated with increasing PN-1 concentration, there was a reciprocal increase in the Δtrypsin (P = 0.01 by ANOVA for both the INa and Δtrypsin trends, n = 4 cultures). After trypsin treatment, there was no difference in the INa among the HAEC exposed to the increasing PN-1 concentrations (P = 0.59 by ANOVA, n = 4 cultures), suggesting that the PN-1 exposure decreased Na+ absorption by interfering with channel gating and not by altering channel number. These results indicate that PN-1 inhibits proteolytic processing of ENaC on the cell surface, leading to a reduction in Na+ conductance.
To determine whether PN-1 inhibits INa though prostasin inhibition, we assessed the effect of PN-1 on prostasin processing. If the inhibitory effects of PN-1 are solely due to formation of an inactive prostasin complex, then the amount of the PN-1-prostasin complex should increase in proportion to INa inhibition. To assess this, we applied increasing concentrations of PN-1 to the apical surface of differentiated HAEC and subjected the apical secretions to prostasin Western blotting. As shown in Fig. 7C, there was a dose-dependent increase in the abundance of the 82-kDa complex over the same PN-1 concentration that caused a large decrease in INa. However, the potent inhibition of ENaC by PN-1 was greater than what would be expected based on the ratio of inactive 82-kDa prostasin complex to total prostasin. This suggests that the decrease in Na+ conductance following PN-1 treatment may not be fully explained on the basis of the formation of an inactive complex of PN-1 with prostasin.
Because prostasin is synthesized as an inactive proenzyme, proteolytic processing is required to activate the enzyme. Therefore, we examined whether PN-1 prevents prostasin processing as an additional means to inhibit prostasin activity. As demonstrated in Fig. 7D, treatment with 1 μM PN-1 caused an increase in appearance of the 40-kDa band and a decrease in the 37-kDa band in the apically biotinylated proteins of HAEC. These results suggest that PN-1 inhibits prostasin by preventing the conversion of zymogen to active enzyme in addition to formation of an inactive prostasin complex. Furthermore, because prostasin has not been shown to be capable of autoactivation (2), these results suggest that PN-1 inhibits an upstream prostasin-activating protease.
To further characterize the mechanism by which PN-1 inhibits INa and prostasin processing, we performed a series of experiments to determine whether PN-1 inhibits an additional protease involved in ENaC channel activation and prostasin processing. Because the type II membrane serine protease matriptase is expressed in airway epithelium (21), has broad serpin reactivity (40), and is believed to be the human ortholog of CAP3 (46), we reasoned that matriptase might be an additional target for PN-1. Furthermore, matriptase is a key physiological activator of prostasin, and this newly described matriptase-prostasin cascade has been demonstrated to be fundamental to normal mouse epidermal differentiation (10, 24-26, 37). Initial tests demonstrated that the enzymatic activity of recombinant human matriptase was abolished by a 10-fold molar excess of aprotinin, AEBSF, or PN-1 (Fig. 8A). These results suggest that PN-1 is capable of inhibiting matriptase, which is a protease that activates both ENaC and prostasin.
Fig. 8.

PN-1 inhibits matriptase. A: inhibition of matriptase activity by PN-1, aprotinin, and 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF). Matriptase (10 nM) was incubated with 100 nM PN-1, aprotinin, AEBSF, or ovalbumin for 30 min, and the residual enzymatic activity was determined by adding the fluorogenic substrate Boc-QAR-AMC and measuring the emitted fluorescence. Data are relative fluorescence units (RFU) generated during a 15-min interval. B: the stoichiometry of inhibition for the interaction between PN-1 and matriptase. Increasing molar ratios of PN-1 were incubated with 10 nM matriptase, and the residual enzymatic activity was normalized to that of uninhibited controls (fractional activity, VI/VO). Data are mean values from ≥3 individual experiments. C: complex formation between PN-1 and matriptase. Recombinant matriptase was incubated with a fivefold molar excess of PN-1, and the product was separated by SDS-PAGE and immunoblotted for PN-1 and recombinant human matriptase (rMat).
To further characterize the interaction between PN-1 and matriptase, we determined the molar ratio at which PN-1 inhibits matriptase and assessed whether PN-1 forms a classic covalent complex with matriptase. The stoichiometry of inhibition (SI) was determined by incubating increasing molar ratios of PN-1 with matriptase and measuring the residual enzymatic activity (Fig. 8B). By extrapolation, the enzymatic activity of matriptase was abolished at an ∼5.5:1 molar ratio. These results indicate that PN-1 acts as a matriptase inhibitor, albeit at a high molar ratio in these in vitro conditions. We next determined whether PN-1 forms a covalent complex with matriptase by incubating a fivefold molar excess of PN-1 with matriptase. As shown in Fig. 8C, the product of the PN-1 and matriptase reaction contained the catalytic domain of matriptase (26 kDa), PN-1 (45 kDa), and the 71-kDa SDS-stable matriptase complex. In addition, an ∼40-kDa band was resolved, presumably due to removal of the PN-1 COOH terminus at the reactive site. These data demonstrate that PN-1 is capable of inhibiting matriptase through formation of a covalent serpin complex and suggest that the proteolytic activation of ENaC on the airway surface involves a proteolytic cascade involving prostasin, matriptase, and PN-1.
DISCUSSION
An accumulating body of research supports the hypothesis that the activity of Na+ channels is regulated by limited proteolysis of the extracellular loops of the α- and γENaC subunits (4, 7, 14, 16-19, 22, 28). Previous work from our laboratory (34) and Tarran et al. (42) has demonstrated that the degree of proteolytic activation of ENaC is modulated by the balance between channel-activating protease activity and soluble protease inhibitor concentrations present on the apical surface of airway epithelium. In the current studies, we have demonstrated that the expression of prostasin, a channel-activating protease, is regulated by changes in the ASL volume. Accordingly, 1) prostasin transcription and apical expression increase in response to ASL volume expansion, and 2) prostasin processing occurs on the apical cell surface and is mediated by a proteolytic cascade involving PN-1 and upstream prostasin-activating proteases, including matriptase. Most importantly, increased expression of processed prostasin is present on the CF cell surface, suggesting that abnormal regulation of prostasin contributes to the excessive Na+ and ASL absorption characteristic of CF airways.
Under air-liquid conditions, normal airway epithelia possess a reserve pool of inactive Na+ channels that are activated when exposed to CAPs such as trypsin (6), neutrophil elastase (5), or endogenous proteases when the ASL volume is expanded (34, 42). The ability of HAEC to increase Na+ absorption in response to changes in the ASL volume has been attributed to the dilution of endogenous soluble protease inhibitors and a corresponding CAP-mediated ENaC activation (34, 42). In CF HAEC, the reserve pool of unprocessed or partially processed Na+ channels appears to be constitutively activated, leading to an impaired ability to regulate Na+ and ASL absorption. The mechanism responsible for the protease-protease inhibitor imbalance in CF epithelia remains uncertain, since the concentrating effects of excessive ASL absorption would be predicted to increase the protease inhibitor concentration and mitigate protease-mediated ENaC activation. Na+ currents in CF epithelia are inhibited by both PN-1 (Fig. 7) and Kunitz-type protease inhibitors (3, 34, 42), suggesting that the CAPs in CF HAEC are responsive to protease inhibition. The current studies suggest that the increased level of proteolytic ENaC activation in the CF airways is, at least in part, the result of increased expression of prostasin.
The mechanism by which prostasin activates Na+ channels has proven to be complex. ENaC processing by prostasin leads to cleavage of the γ-subunit and a corresponding dramatic increase in the open probability of the channel (4). This increase in channel activity appears to be due to the release of an inhibitory 43-amino acid tract (γ-43) that lies between the furin cleavage site and the putative prostasin cleavage site (4, 17). In support of the theory that prostasin activates ENaC through proteolytic cleavage, prostasin fails to activate ENaC when the prostasin cleavage site on γENaC is mutated or the protease activity is inhibited by aprotinin (4, 13). Interestingly, Andreasen et al. (2) reported that when the catalytic triad of prostasin is mutated, the protease maintains its ability to activate the channel, suggesting that perhaps prostasin's proteolytic activity is not required for channel activation. Furthermore, the Na+ current of oocytes expressing ENaC is increased by prostasin coexpression despite the biochemical absence of proteolytically processed prostasin and under neutral pH conditions where the catalytic activity of the protease is dramatically decreased (2, 48). Subsequently, Bruns et al. (4) reported that γENaC processing occurs in oocytes expressing the catalytic site mutant prostasin, suggesting that either the catalytic mutant prostasin retains a portion of its protease activity or prostasin may facilitate the action of another channel-activating protease. Therefore, although it remains unclear whether the catalytic activity of prostasin is required, γENaC processing near the first membrane-spanning region appears to be critical for channel activation by proteases (1, 4, 16).
The kinetics of increased prostasin expression and INa with ASL volume expansion in this and our previous report (34) suggest two mechanisms whereby ASL volume regulates ENaC activity. The increase in prostasin expression (Fig. 5) occurs at a slower rate than the increase in Na+ absorption associated with ASL expansion [t1/2 of ∼30 min (34)]. We hypothesize that the rapid increase in INa during ASL expansion occurs as a result of protease inhibitor dilution and subsequent CAP-mediated ENaC activation. After prolonged periods of ASL expansion, however, prostasin expression increases as an additional mechanism to maintain heightened levels of Na+ absorption. Further studies are needed to elucidate the mechanism by which HAEC “sense” the ASL volume and to determine the cellular mechanisms that regulate prostasin transcription and protein processing.
PN-1 appears to be a critical regulator of a proteolytic cascade that leads to Na+ channel activation. PN-1 knockdown led to a 1.6-fold increase in INa in a mouse cortical collecting duct cell line (47), and extracellular exposure to PN-1 inhibited INa by ∼60% in HAEC (Fig. 7A). PN-1 appears to decrease INa by multiple mechanisms: 1) direct prostasin inactivation, 2) prevention of prostasin zymogen processing, and 3) direct inhibition of matriptase. Thus the recently described matriptase-prostasin cascade, counterbalanced by PN-1, appears to regulate Na+ channel activity.
PN-1 has recently been shown to be a susceptibility gene for the development of chronic obstructive pulmonary disease in several large population studies (11, 49). Although the functional significance of these single-nucleotide polymorphisms on PN-1 function or expression remain to be determined, it is possible that the airflow obstruction reported in these studies may be due to Na+ hyperabsorption as a result of altered PN-1 activity or expression associated with these polymorphisms. Whether similar polymorphisms could account for a portion of the heterogeneity in CF lung disease among patients with the ΔF508 mutation remains to be determined.
In summary, our data suggest that the airway epithelium regulates prostasin expression as a means of achieving ASL volume homeostasis. These regulatory mechanisms involve prostasin transcription, processing, and inactivation by PN-1. As a result of these complex regulatory networks, prostasin expression increases when the ASL volume is expanded. Thus Na+ and ASL absorption in the airway is regulated by a proteolytic cascade involving prostasin and matriptase, as well as the serpin PN-1. CF epithelia fail to properly regulate prostasin, leading to an increase in the cell surface expression of processed prostasin. These results suggest that abnormal prostasin regulation in CF epithelia leads to excessive proteolytic activation of ENaC and that this plays a significant role in the Na+ hyperabsorption characteristic of CF airway disease. Further defining the regulation of proteolytic cascades on the airway cell surface may reveal novel targets for CF therapeutics beyond the protease inhibitors currently under development.
ACKNOWLEDGMENTS
We thank Dr. Kenneth McCurry and the Lung Transplant Program at the University of Pittsburgh Medical Center for facilitating tissue acquisition and Joseph Latoche and Elisa Heidrich-Ohare for technical assistance. Real-time quantitative PCR was conducted by the TaqMan core facility of the Genomics and Proteomics Core Laboratories of the University of Pittsburgh.
GRANTS
This work was supported by the Cystic Fibrosis Foundation Shwachman Award (M. M. Myerburg) and the Research Development Program to the University of Pittsburgh (R. A. Frizzell and J. M. Pilewski), Institutional National Research Services Award T32 HL007653 (M. M. Myerburg), National Institutes of Health Grants R01 DK065161 (T. R. Kleyman), R01 DK054814 (R. A. Frizzell), and P30 DK072506 (R. A. Frizzell and J. M. Pilewski), and the American Lung Association (J. M. Pilewski).
REFERENCES
- 1.Adebamiro A, Cheng Y, Rao US, Danahay H, Bridges RJ. A segment of γ ENaC mediates elastase activation of Na+ transport. J Gen Physiol. 2007;130:611–629. doi: 10.1085/jgp.200709781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andreasen D, Vuagniaux G, Fowler-Jaeger N, Hummler E, Rossier BC. Activation of epithelial sodium channels by mouse channel activating proteases (mCAP) expressed in Xenopus oocytes requires catalytic activity of mCAP3 and mCAP2 but not mCAP1. J Am Soc Nephrol. 2006;17:968–976. doi: 10.1681/ASN.2005060637. [DOI] [PubMed] [Google Scholar]
- 3.Bridges RJ, Newton BB, Pilewski JM, Devor DC, Poll CT, Hall RL. Na+ transport in normal and CF human bronchial epithelial cells is inhibited by BAY 39-9437. Am J Physiol Lung Cell Mol Physiol. 2001;281:L16–L23. doi: 10.1152/ajplung.2001.281.1.L16. [DOI] [PubMed] [Google Scholar]
- 4.Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, Hughey RP, Kleyman TR. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the gamma subunit. J Biol Chem. 2007;282:6153–6160. doi: 10.1074/jbc.M610636200. [DOI] [PubMed] [Google Scholar]
- 5.Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol. 2005;288:L813–L819. doi: 10.1152/ajplung.00435.2004. [DOI] [PubMed] [Google Scholar]
- 6.Caldwell RA, Boucher RC, Stutts MJ. Serine protease activation of near-silent epithelial Na+ channels. Am J Physiol Cell Physiol. 2004;286:C190–C194. doi: 10.1152/ajpcell.00342.2003. [DOI] [PubMed] [Google Scholar]
- 7.Carattino MD, Sheng S, Bruns JB, Pilewski JM, Hughey RP, Kleyman TR. The epithelial Na+ channel is inhibited by a peptide derived from proteolytic processing of its alpha subunit. J Biol Chem. 2006;281:18901–18907. doi: 10.1074/jbc.M604109200. [DOI] [PubMed] [Google Scholar]
- 8.Chen LM, Skinner ML, Kauffman SW, Chao J, Chao L, Thaler CD, Chai KX. Prostasin is a glycosylphosphatidylinositol-anchored active serine protease. J Biol Chem. 2001;276:21434–21442. doi: 10.1074/jbc.M011423200. [DOI] [PubMed] [Google Scholar]
- 9.Chen LM, Zhang X, Chai KX. Regulation of prostasin expression and function in the prostate. Prostate. 2004;59:1–12. doi: 10.1002/pros.10346. [DOI] [PubMed] [Google Scholar]
- 10.Chen M, Chen LM, Lin CY, Chai KX. The epidermal growth factor receptor (EGFR) is proteolytically modified by the matriptase-prostasin serine protease cascade in cultured epithelial cells. Biochim Biophys Acta. 2007 doi: 10.1016/j.bbamcr.2007.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demeo DL, Mariani TJ, Lange C, Srisuma S, Litonjua AA, Celedon JC, Lake SL, Reilly JJ, Chapman HA, Mecham BH, Haley KJ, Sylvia JS, Sparrow D, Spira AE, Beane J, Pinto-Plata V, Speizer FE, Shapiro SD, Weiss ST, Silverman EK. The SERPINE2 gene is associated with chronic obstructive pulmonary disease. Am J Hum Genet. 2006;78:253–264. doi: 10.1086/499828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devor DC, Bridges RJ, Pilewski JM. Pharmacological modulation of ion transport across wild-type and ΔF508 CFTR-expressing human bronchial epithelia. Am J Physiol Cell Physiol. 2000;279:C461–C479. doi: 10.1152/ajpcell.2000.279.2.C461. [DOI] [PubMed] [Google Scholar]
- 13.Donaldson SH, Hirsh A, Li DC, Holloway G, Chao J, Boucher RC, Gabriel SE. Regulation of the epithelial sodium channel by serine proteases in human airways. J Biol Chem. 2002;277:8338–8345. doi: 10.1074/jbc.M105044200. [DOI] [PubMed] [Google Scholar]
- 14.Ergonul Z, Frindt G, Palmer LG. Regulation of maturation and processing of ENaC subunits in the rat kidney. Am J Physiol Renal Physiol. 2006;291:F683–F693. doi: 10.1152/ajprenal.00422.2005. [DOI] [PubMed] [Google Scholar]
- 15.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 16.Harris M, Firsov D, Vuagniaux G, Stutts MJ, Rossier BC. A novel neutrophil elastase inhibitor prevents elastase activation and surface cleavage of the epithelial sodium channel expressed in Xenopus laevis oocytes. J Biol Chem. 2007;282:58–64. doi: 10.1074/jbc.M605125200. [DOI] [PubMed] [Google Scholar]
- 17.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem. 2004;279:18111–18114. doi: 10.1074/jbc.C400080200. [DOI] [PubMed] [Google Scholar]
- 18.Hughey RP, Bruns JB, Kinlough CL, Kleyman TR. Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J Biol Chem. 2004;279:48491–48494. doi: 10.1074/jbc.C400460200. [DOI] [PubMed] [Google Scholar]
- 19.Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR. Maturation of the epithelial Na+ channel involves proteolytic processing of the alpha- and gamma-subunits. J Biol Chem. 2003;278:37073–37082. doi: 10.1074/jbc.M307003200. [DOI] [PubMed] [Google Scholar]
- 20.Jiang C, Finkbeiner WE, Widdicombe JH, McCray PB, Jr, Miller SS. Altered fluid transport across airway epithelium in cystic fibrosis. Science. 1993;262:424–427. doi: 10.1126/science.8211164. [DOI] [PubMed] [Google Scholar]
- 21.Kim MG, Chen C, Lyu MS, Cho EG, Park D, Kozak C, Schwartz RH. Cloning and chromosomal mapping of a gene isolated from thymic stromal cells encoding a new mouse type II membrane serine protease, epithin, containing four LDL receptor modules and two CUB domains. Immunogenetics. 1999;49:420–428. doi: 10.1007/s002510050515. [DOI] [PubMed] [Google Scholar]
- 22.Knight KK, Olson DR, Zhou R, Snyder PM. Liddle's syndrome mutations increase Na+ transport through dual effects on epithelial Na+ channel surface expression and proteolytic cleavage. Proc Natl Acad Sci USA. 2006;103:2805–2808. doi: 10.1073/pnas.0511184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knowles M, Gatzy J, Boucher R. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N Engl J Med. 1981;305:1489–1495. doi: 10.1056/NEJM198112173052502. [DOI] [PubMed] [Google Scholar]
- 24.Leyvraz C, Charles RP, Rubera I, Guitard M, Rotman S, Breiden B, Sandhoff K, Hummler E. The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J Cell Biol. 2005;170:487–496. doi: 10.1083/jcb.200501038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.List K, Currie B, Scharschmidt TC, Szabo R, Shireman J, Molinolo A, Cravatt BF, Segre J, Bugge TH. Autosomal ichthyosis with hypotrichosis syndrome displays low matriptase proteolytic activity and is phenocopied in ST14 hypomorphic mice. J Biol Chem. 2007;282:36714–36723. doi: 10.1074/jbc.M705521200. [DOI] [PubMed] [Google Scholar]
- 26.List K, Hobson JP, Molinolo A, Bugge TH. Co-localization of the channel activating protease prostasin/(CAP1/PRSS8) with its candidate activator, matriptase. J Cell Physiol. 2007;213:237–245. doi: 10.1002/jcp.21115. [DOI] [PubMed] [Google Scholar]
- 27.Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- 28.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone-mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest. 1999;104:R19–R23. doi: 10.1172/JCI7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsui H, Davis CW, Tarran R, Boucher RC. Osmotic water permeabilities of cultured, well-differentiated normal and cystic fibrosis airway epithelia. J Clin Invest. 2000;105:1419–1427. doi: 10.1172/JCI4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- 31.Matsui H, Randell SH, Peretti SW, Davis CW, Boucher RC. Coordinated clearance of periciliary liquid and mucus from airway surfaces. J Clin Invest. 1998;102:1125–1131. doi: 10.1172/JCI2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayor S, Riezman H. Sorting GPI-anchored proteins. Nat Rev. 2004;5:110–120. doi: 10.1038/nrm1309. [DOI] [PubMed] [Google Scholar]
- 33.McAllister F, Henry A, Kreindler JL, Dubin PJ, Ulrich L, Steele C, Finder JD, Pilewski JM, Carreno BM, Goldman SJ, Pirhonen J, Kolls JK. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol. 2005;175:404–412. doi: 10.4049/jimmunol.175.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Myerburg MM, Butterworth MB, McKenna EE, Peters KW, Frizzell RA, Kleyman TR, Pilewski JM. Airway surface liquid volume regulates ENaC by altering the serine protease-protease inhibitor balance: a mechanism for sodium hypersabsorption in cystic fibrosis. J Biol Chem. 2006;281:27942–27949. doi: 10.1074/jbc.M606449200. [DOI] [PubMed] [Google Scholar]
- 35.Myerburg MM, Latoche JD, McKenna EE, Stabile LP, Siegfried JS, Feghali-Bostwick CA, Pilewski JM. Hepatocyte growth factor and other fibroblast secretions modulate the phenotype of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1352–L1360. doi: 10.1152/ajplung.00328.2006. [DOI] [PubMed] [Google Scholar]
- 36.Narikiyo T, Kitamura K, Adachi M, Miyoshi T, Iwashita K, Shiraishi N, Nonoguchi H, Chen LM, Chai KX, Chao J, Tomita K. Regulation of prostasin by aldosterone in the kidney. J Clin Invest. 2002;109:401–408. doi: 10.1172/JCI13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Netzel-Arnett S, Currie BM, Szabo R, Lin CY, Chen LM, Chai KX, Antalis TM, Bugge TH, List K. Evidence for a matriptase-prostasin proteolytic cascade regulating terminal epidermal differentiation. J Biol Chem. 2006;281:32941–32945. doi: 10.1074/jbc.C600208200. [DOI] [PubMed] [Google Scholar]
- 38.Planes C, Leyvraz C, Uchida T, Angelova MA, Vuagniaux G, Hummler E, Matthay M, Clerici C, Rossier B. In vitro and in vivo regulation of transepithelial lung alveolar sodium transport by serine proteases. Am J Physiol Lung Cell Mol Physiol. 2005;288:L1099–L1109. doi: 10.1152/ajplung.00332.2004. [DOI] [PubMed] [Google Scholar]
- 39.Shipway A, Danahay H, Williams JA, Tully DC, Backes BJ, Harris JL. Biochemical characterization of prostasin, a channel activating protease. Biochem Biophys Res Commun. 2004;324:953–963. doi: 10.1016/j.bbrc.2004.09.123. [DOI] [PubMed] [Google Scholar]
- 40.Szabo R, Netzel-Arnett S, Hobson JP, Antalis TM, Bugge TH. Matriptase-3 is a novel phylogenetically preserved membrane-anchored serine protease with broad serpin reactivity. Biochem J. 2005;390:231–242. doi: 10.1042/BJ20050299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tarran R, Grubb BR, Gatzy JT, Davis CW, Boucher RC. The relative roles of passive surface forces and active ion transport in the modulation of airway surface liquid volume and composition. J Gen Physiol. 2001;118:223–236. doi: 10.1085/jgp.118.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tarran R, Trout L, Donaldson SH, Boucher RC. Soluble mediators, not cilia, determine airway surface liquid volume in normal and cystic fibrosis superficial airway epithelia. J Gen Physiol. 2006;127:591–604. doi: 10.1085/jgp.200509468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tong Z, Illek B, Bhagwandin VJ, Verghese GM, Caughey GH. Prostasin, a membrane-anchored serine peptidase, regulates sodium currents in JME/CF15 cells, a cystic fibrosis airway epithelial cell line. Am J Physiol Lung Cell Mol Physiol. 2004;287:L928–L935. doi: 10.1152/ajplung.00160.2004. [DOI] [PubMed] [Google Scholar]
- 44.Vallet V, Chraibi A, Gaeggeler HP, Horisberger JD, Rossier BC. An epithelial serine protease activates the amiloride-sensitive sodium channel. Nature. 1997;389:607–610. doi: 10.1038/39329. [DOI] [PubMed] [Google Scholar]
- 45.Verghese GM, Gutknecht MF, Caughey GH. Prostasin regulates epithelial monolayer function: cell-specific Gpld1-mediated secretion and functional role for GPI anchor. Am J Physiol Cell Physiol. 2006;291:C1258–C1270. doi: 10.1152/ajpcell.00637.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vuagniaux G, Vallet V, Jaeger NF, Hummler E, Rossier BC. Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus oocytes. J Gen Physiol. 2002;120:191–201. doi: 10.1085/jgp.20028598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wakida N, Kitamura K, Tuyen DG, Maekawa A, Miyoshi T, Adachi M, Shiraishi N, Ko T, Ha V, Nonoguchi H, Tomita K. Inhibition of prostasin-induced ENaC activities by PN-1 and regulation of PN-1 expression by TGF-beta1 and aldosterone. Kidney Int. 2006;70:1432–1438. doi: 10.1038/sj.ki.5001787. [DOI] [PubMed] [Google Scholar]
- 48.Yu JX, Chao L, Chao J. Prostasin is a novel human serine proteinase from seminal fluid. Purification, tissue distribution, and localization in prostate gland. J Biol Chem. 1994;269:18843–18848. [PubMed] [Google Scholar]
- 49.Zhu G, Warren L, Aponte J, Gulsvik A, Bakke P, Anderson WH, Lomas DA, Silverman EK, Pillai SG. The SERPINE2 gene is associated with chronic obstructive pulmonary disease in two large populations. Am J Respir Crit Care Med. 2007;176:167–173. doi: 10.1164/rccm.200611-1723OC. [DOI] [PubMed] [Google Scholar]