

Abstract
Producing complex chemicals using synthetic metabolic pathways in microbial hosts can have many advantages over chemical synthesis but is often complicated by deleterious interactions between pathway intermediates and the host cell metabolism. With the maturation of functional genomic analysis, it is now technically feasible to identify modes of toxicity associated with the accumulation of foreign molecules in the engineered bacterium. Previously, Escherichia coli was engineered to produce large quantities of isoprenoids by creating a mevalonate-based isopentenyl pyrophosphate biosynthetic pathway (V. J. J. Martin et al., Nat. Biotechnol. 21:796-802, 2003). The engineered E. coli strain produced high levels of isoprenoids, but further optimization led to an imbalance in carbon flux and the accumulation of the pathway intermediate 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA), which proved to be cytotoxic to E. coli. Using both DNA microarray analysis and targeted metabolite profiling, we have studied E. coli strains inhibited by the intracellular accumulation of HMG-CoA. Our results indicate that HMG-CoA inhibits fatty acid biosynthesis in the microbial host, leading to generalized membrane stress. The cytotoxic effects of HMG-CoA accumulation can be counteracted by the addition of palmitic acid (16:0) and, to a lesser extent, oleic acid (cis-Δ9-18:1) in the growth medium. This work demonstrates the utility of using transcriptomic and metabolomic methods to optimize synthetic biological systems.
Production of chemicals via synthetic enzymatic pathways in heterologous hosts has proven useful for many important classes of molecules, including isoprenoids (61), polyketides (49, 50), nonribosomal peptides (61), bioplastics (1), and chemical building blocks (45). Due to the inherent modularity of biological information, synthetic biology holds great potential for expanding this list of microbially produced compounds even further. Yet embedding a novel biochemical pathway in the metabolic network of a host cell can disrupt the subtle regulatory mechanisms that the cell has evolved over the millennia. Indeed, the final yield of a compound is often limited by deleterious effects on the engineered cell's metabolism that are difficult to predict due to our limited understanding of the complex interactions that occur within the cell. The unregulated consumption of cellular resources (18, 37), metabolic burden of heterologous protein production (19, 23, 24), and accumulation of pathway intermediates/products that are inhibitory (5, 62, 63) or toxic (6) to the host are all significant issues that may limit overall yield. A systematic, bottom-up survey to identify potential interactions that limit titer can be time-consuming and requires extensive iteration. Here we report on a top-down, systems biology approach to characterize the toxicity associated with the accumulation of a synthetic pathway intermediate, using DNA microarray analysis coupled with targeted metabolite detection.
Previously, Escherichia coli was engineered to produce amorpha-4,11-diene, the sesquiterpene precursor to the antimalarial compound artemisinin, using a multigene, heterologous pathway (44). Artemisinin, a sesquiterpene lactone endoperoxide extracted from Artemisia annua L (family Asteraceae; commonly known as sweet wormwood), is highly effective against multidrug-resistant Plasmodium spp. and is the key component of the artemisinin-based combination therapies now endorsed as the frontline therapy to treat the disease (2). However, the drug is in short supply and unaffordable to most malaria sufferers. Although total synthesis of artemisinin is difficult and costly, the semisynthesis of artemisinin or any derivative from microbially sourced artemisinic acid, its immediate precursor, represents a cost-effective, environmentally friendly, high-quality, and reliable source of artemisinin for use in artemisinin-based combination therapies. During the design optimization of the amorpha-4,11-diene pathway in E. coli, it was discovered that high expression of the first three genes in this synthetic pathway (atoB, ERG13, and tHMG1) resulted in a severe growth defect. The enzymes encoded by these three genes convert acetyl-coenzyme A (acetyl-CoA) to mevalonate via two intermediate compounds, acetoacetyl-CoA and 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) (Fig. 1A). While acetoacetyl-CoA is a compound endogenous to E. coli, HMG-CoA is not, and the growth inhibition was shown to correlate with the intracellular accumulation of this exogenous small molecule (51). However, the reason for its toxicity, and thus a solution to the problem, was not known.
FIG. 1.

(A) The heterologous pathway produces mevalonate from acetyl-CoA in three biochemical steps. The data presented in this article show that the growth inhibition associated with the pathway intermediate HMG-CoA is due to inhibition of malonyl-CoA:ACP transacylase (FabD) in E. coli's type II FAB pathway. The inhibition of the FAB pathway invokes a generalized stress response in the mevalonate-producing strain. As can be seen, the heterologous intermediate is similar in structure to malonyl-CoA, the native substrate for FabD. Abbreviations: A-CoA, acetyl-CoA; AA-CoA, acetoacetyl-CoA; HMG-CoA, hydroxymethylglutaryl-CoA; G6P, glucose-6-phosphate; FDP, fructose-1,6-bisphosphate; G3P, glyceraldehyde-3-phosphate; DHAP, dihydroxyacetone phosphate; PEP, phosphoenolpyruvate. (B) Genes of the heterologous mevalonate-producing pathway in E. coli as well as the inactive-pathway control, which due to a point mutation in ERG13, cannot produce HMG-CoA or mevalonate. Abbreviations: atoB, acetoacetyl-CoA thiolase gene; Erg13, HMG-CoA synthase gene; tHMGR, truncated HMG-CoA reductase gene; Erg13(C159A), inactive HMG-CoA synthase gene.
The toxicity associated with the accumulation of HMG-CoA provided an ideal model system with which to study the utility of top-down systems biology analysis in metabolic engineering. With the maturation of DNA microarray analysis, it is now possible to study the complex interplay between a synthetic biochemical pathway and the endogenous metabolic network of the heterologous host. Since a DNA microarray study gathers data on the entire transcriptome, it is not limited by any initial hypothesis and can identify potential deleterious interactions between an engineered pathway and the host metabolism that would have been difficult to predict a priori. Thus, transcriptional profiling, especially combined with other targeted methods such as metabolite analysis, is a powerful tool for diagnosing problems in the design of an engineered microorganism. Using this approach to study the toxicity caused by accumulation of HMG-CoA in bacteria, we report here that the growth inhibition observed in the mevalonate-producing E. coli strain is due to inhibition of the endogenous type II fatty acid biosynthesis (FAB) pathway by high levels of HMG-CoA (Fig. 1A). This work highlights the unpredictable interactions that can occur in engineering a bacterial cell and illustrates the utility of approaching problems in metabolic engineering by using the tools of systems biology.
MATERIALS AND METHODS
Strains, plasmids, and media.
The strains and plasmids used in this study are described below and in Table 1 (44, 51).
TABLE 1.
Strains used in this study
| Strain or plasmid | Genotype or description | Source or reference |
|---|---|---|
| Strains | ||
| DH10B | F−mcrA Δ(mrr-hsdRMS-mcrBC) F80lacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara leu)7697 galU galK λ−rpsL (Strr) nupG | Invitrogen |
| DP10 | DH10B Δ(araFGH) φ(ΔaraEp Pcp8-araE) | 51 |
| Plasmids | ||
| pBAD18 | Cloning vector containing Apr cassette, modified pBR322 origin with a truncated rop gene, araC, and PBAD | 22 |
| pBAD33 | Cloning vector containing Cmr cassette, pACYC184 origin, araC, and PBAD | 22 |
| pBAD33MevT | pBAD33 containing atoB, ERG13, and tHMGR under the control of PBAD; Cmr | 44, 51 |
| pMevT(C159A) | pBAD33MevT derivative containing ERG13(C159A) | 51 |
In E. coli DH10B, the arabinose-inducible araBAD promoter (PBAD) system suffers from all-or-none induction (42), in which subsaturating concentrations of arabinose give rise to subpopulations of cells that are fully induced and uninduced. DP10 is an E. coli DH10B derivative containing chromosomal araE, encoding the low-affinity, high-capacity arabinose transport protein, under the control of a constitutive promoter, PCP8, and a deletion of the genes encoding the secondary arabinose transporter, i.e., araFGH. The modifications allow regulatable control of PBAD in a homogeneous population of cells (41).
The low-copy-number plasmid pBAD33MevT contains the previously constructed MevT operon under regulated control of the arabinose-inducible PBAD (44). The operon consists of the genes atoB from E. coli, encoding acetoacetyl-CoA thiolase, and ERG13 and tHMG1 from Saccharomyces cerevisiae, encoding HMG-CoA synthase and truncated HMG-CoA reductase 1, respectively, whose expression allows the in vivo conversion of acetyl-CoA to mevalonate.
In order to investigate the source of toxicity in cells expressing the MevT operon, an inactive-pathway operon was constructed by replacing the catalytic cysteine of S. cerevisiae HMGS (at amino acid position 159), encoded by pBAD33MevT, with an alanine (51). Expression of the modified operon produced active acetoacetyl-CoA thiolase, active HMG-CoA reductase, and full-length, catalytically inactive HMG-CoA synthase. The resulting plasmid was pMevT(C159A) (51).
Medium components and chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and Fisher Scientific (Pittsburgh, PA). For propagation of engineered E. coli strains, Luria broth with Miller's modification (Sigma) was used with appropriate antibiotics for plasmid selection, and 0.06% glucose was used for the repression of the PBAD promoter system. Studies to characterize the cell physiology, metabolite levels, and mRNA transcript levels of the engineered strains were performed using defined C medium (30) supplemented with 3.4% glycerol, all individual amino acids per the work of Neidhardt et al. (46), 4.5 μg/ml thiamine-HCl, micronutrients, and 50 μg/ml (each) chloramphenicol and carbenicillin.
Fatty acids for medium supplementation were purchased from Sigma and employed in defined medium at a concentration of 100 μg/ml in the presence of 400 μg/ml Brij detergent (Sigma).
Growth, metabolite, and mRNA transcript analysis of engineered cells.
Cell growth, metabolite concentrations, and mRNA transcript levels of E. coli DP10 expressing genes encoding the active and inactive MevT operons were assayed. Starter cultures of E. coli DP10 harboring pBAD33MevT and pBAD18 (empty vector) or pMevT(C159A) and pBAD18 were inoculated from single colonies and incubated overnight at 37°C in defined C medium supplemented with 0.06% glucose (to repress PBAD) and antibiotics. Overnight starter cultures were diluted to an optical density at 600 nm (OD600) of 0.05 in fresh medium, incubated at 37°C with continuous shaking, and induced with the addition of 1.33 mM (0.02%) arabinose at an OD600 of ∼0.2 to 0.3. During experiments involving metabolite or transcript level analysis, the engineered strains were incubated in baffled shake flasks. Samples were taken at multiple time points during the course of the experiments to assay for cell growth, mevalonate production, transcripts, and intracellular metabolites. Samples for microarray analysis were taken preinduction and during logarithmic growth to capture the early response dynamics of the host to the HMG-CoA toxicity. The OD600 of shake flask samples was measured using a UV spectrophotometer (Beckman), and values for dry cell weight (DCW) were calculated using an equivalence factor of 1 g DCW/liter to an OD600 of 2.5 (4). Experiments were repeated in duplicate to confirm trends in growth and metabolite concentrations.
To screen for fatty acids that alleviate HMG-CoA-associated growth inhibition, cultures were grown in 96-well microtiter plates at 37°C, using defined C medium as described above. The optical density was measured using a microtiter plate reader (SpectraMax; Molecular Devices).
Transcript analysis sample preparation.
Biomass designated for transcript analysis was snap-frozen in liquid nitrogen immediately and stored at −80°C until analysis. RNAs were extracted from the cell samples by use of an RNeasy Midi kit (Qiagen). Using 40-μg aliquots of extracted RNA from each sample point, prelabeled cDNA was synthesized using random-primed reverse transcription in a 40-μl volume containing 12.5 μg primers (Invitrogen, Carlsbad, CA), 1× reverse transcriptase buffer (Invitrogen, Carlsbad, CA), 0.01 mM dithiothreitol (Invitrogen, Carlsbad, CA), 1 unit/μl Superase-In (Ambion, Austin, TX), a 0.5 mM concentration (each) of dATP, dCTP, and dGTP (Invitrogen, Carlsbad, CA), 0.1 mM dTTP (Invitrogen, Carlsbad, CA), 0.4 mM amino-allyl-dUTP (Ambion, Austin, TX), and 10 units/μl Superscript II (Invitrogen, Carlsbad, CA), following the enzyme manufacturer's instructions. The cDNA was base hydrolyzed in 100 mM NaOH-10 mM EDTA at 65°C for 10 minutes and then neutralized in HEPES, pH 7.0, at a final concentration of 500 mM. The Tris remaining in the cDNA suspension was removed by three buffer exchange spins using Micron YM-30 columns (Millipore) and eluted in a final volume of 15 μl of water. The cDNA was then labeled using either Alexa 555 or Alexa 647 (Invitrogen, Carlsbad, CA) following the manufacturer's protocol.
Microarray hybridization.
Glass microarrays printed with full-length double-stranded DNA prepared by PCR or 70-mer oligonucleotides (Operon) designed to probe every open reading frame of E. coli MG1655 were hybridized in a Tecan hybridization station with ∼6 to 10 μg of labeled cDNA per channel of detection. The hybridization program included a prehybridization step (5× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate]-0.2% sodium dodecyl sulfate [SDS]-1% bovine serum albumin, 42°C, 60 min), a 15-hour hybridization step (Ambion Hyb solution 3, 40°C, medium agitation), two low-stringency washes (1× SSC-0.2% SDS, 42°C, 2 min [each]), two high-stringency washes (0.1× SSC-0.2% SDS, 25°C, 2 min [each]), and two final washes (0.1× SSC, 25°C, 2 min [each]). Following hybridization, the slides were scanned with an Axon 4500 scanner.
Transcriptional profile data analysis.
The raw scans were globally normalized using Genepix software and then exported to SNOMAD (9) for loess normalization to correct for any hybridization artifacts and to generate local Z scores (see reference 52 for a review of local Z score use in microarray analysis). The local Z scores generated by SNOMAD as well as by serial analysis for microarray (SAM) (60) software were used as guides to determine biologically significant gene expression changes among the replicate hybridization data sets. This list of significant genes was then mined using hierarchal clustering (13) (Cluster 3.0) to determine a base set of clusters in each data set. Once a base set of clusters was chosen, k-means clustering was also used to search the data set for temporal patterns in gene expression (Cluster 3.0).
GC-MS quantification of mevalonate.
The mevalonate (mevalonic acid) concentration in cultures of engineered E. coli was determined by gas chromatography-mass spectrometry (GC-MS) analysis as described previously (51). Briefly, culture aliquots were acidified with HCl in glass GC vials to convert mevalonate to mevalonic acid lactone. The acidified cultures were extracted with an equal volume of ethyl acetate containing (−)-trans-caryophyllene (internal standard), and the organic layer was diluted into fresh ethyl acetate prior to GC-MS analysis. Ethyl acetate extracts were analyzed using GC-MS by scanning for only ions m/z 71 and 58, corresponding to mevalonic acid lactone, and ions m/z 189 and 204, corresponding to (−)-trans-caryophyllene. The retention time, mass spectrum, and concentration of extracted mevalonic acid lactone were confirmed using commercial dl-mevalonic acid lactone (Sigma).
Intracellular metabolite extraction and analysis.
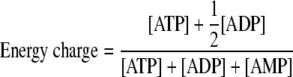
The concentrations of intracellular acyl-CoAs and adenylate pool were determined by liquid chromatography-MS (LC-MS) analysis of trichloroacetic acid (TCA) culture extracts as described previously (51). To simultaneously and rapidly quench cellular metabolism, isolate E. coli cells from growth medium, and extract metabolites, cells were centrifuged through a layer of silicone oil into a denser solution of TCA. TCA extracts, neutralized with tri-n-octylamine, were analyzed using an Agilent 1100 series LC-MS employing electrospray ionization and operating in positive mode. The following selected ions corresponding to the protonated molecular ion of each metabolite were monitored: for ATP, m/z 508; for ADP, m/z 428; for AMP, m/z 348; for CoA, m/z 768; for acetyl-CoA, m/z 810; for propionyl-CoA, m/z 824; for crotonyl-CoA, m/z 836; for acetoacetyl-CoA, m/z 852; for malonyl-CoA, m/z 854; for succinyl-CoA, m/z 868; for methylmalonyl-CoA, m/z 868; and for HMG-CoA, m/z 912. Retention times, mass spectra, and concentrations of extracted metabolites were confirmed using commercial standards (Sigma). The adenylate energy charge of each strain was calculated from the adenylate pool measurement as defined by Atkinson (3), as follows:
 |
Analysis of cellular fatty acid composition.
The fatty acid composition and the fatty acid fraction of engineered cells were determined by fatty acid methyl ester (FAME) analysis of lyophilized cell pellets (55). Sample aliquots of the bacterial cultures were centrifuged to pellet the cells, and the biomass was snap-frozen in liquid nitrogen. The frozen pellets were then lyophilized for 24 h at −80°C under vacuum, using a freeze drier. FAME analysis of lyophilized cell pellets was performed by Microbial ID (Newark, DE).
RESULTS
Transcriptomic and metabolic analyses were performed to characterize the HMG-CoA-induced toxicity in the growth-inhibited, mevalonate-producing strain (E. coli DP10 harboring pBAD33MevT). Since the metabolic burden of producing heterologous protein is a well-documented phenomenon (32, 39, 41, 48), we previously designed a control in which the ERG13 gene had a point mutation in its active site and whose expression resulted in a full-length, catalytically inactive protein (54). Thus, this inactive-pathway control [E. coli DP10 harboring pMevT(C159A) and pBAD18] (Fig. 1B) grew under the full burden of heterologous protein production yet suffered no HMG-CoA-associated growth defect since this intermediate could not be synthesized by the mutated ERG13 protein. Because of this, the inactive-pathway strain, E. coli DP10 harboring pMevT(C159A) and pBAD18, provided an ideal control for two-color microarray analysis of the mevalonate-producing, active-pathway strain, E. coli DP10 harboring pBAD33MevT and pBAD18 (Fig. 1B).
Malonyl-CoA accumulates during HMG-CoA-mediated growth inhibition.
The growth and acyl-CoA profiles of the active-pathway (mevalonate-producing) strain and the inactive-pathway control strain were analyzed by LC-MS to monitor metabolite changes that correlate with HMG-CoA growth inhibition (Fig. 2). In comparison to the inactive-pathway control, growth of E. coli expressing MevT from pBAD33MevT was inhibited (Fig. 2A), and the growth inhibition correlated directly with the accumulation of HMG-CoA (Fig. 2D), as previously reported (51). This pathway intermediate accumulated to ∼125 nmol/g DCW in the active-pathway strain during log-phase growth and did not accumulate in the inactive-pathway control strain (Fig. 2D).
FIG. 2.

Growth and metabolite time course profiles for inactive-pathway control strain [E. coli DP10 harboring pMevT(C159A) and pBAD18] (□) and the growth-inhibited, mevalonate-producing strain (E. coli DP10 harboring pBAD33MevT and pBAD18) (▴). Arabinose induction was done at 0.1 g DCW/liter, and the first sample for metabolite extraction was taken at 0.5 h postinduction. Shown are cell growth, in g DCW/liter (A), mevalonate concentration (B), intracellular malonyl-CoA (C), intracellular HMG-CoA (D), intracellular free CoA (E), intracellular acetyl-CoA (F), intracellular acetoacetyl-CoA (G), and energy charge (H).
The most interesting trend noted in the acyl-CoA concentration profiles was the significant diversion of the total acyl-CoA pool to malonyl-CoA during HMG-CoA-associated growth inhibition (Fig. 2C). Malonyl-CoA is a key substrate for the initiation of fatty acid elongation and is typically maintained at relatively low levels in E. coli by the coordinated regulation of long-chain acyl-ACP demand and FAB (<100 nmol/g DCW in the inactive-pathway control strain). Normally, when demand for long-chain acyl-ACP decreases, unused malonyl-CoA is recycled to acetyl-CoA by KAS1 (fabB) and malonyl-CoA-ACP transacylase (encoded by fabD) (28). Yet the growth-inhibited strain converted ∼34% of its total acyl-CoA pool to malonyl-CoA, which represented a >4-fold increase in the intracellular concentration of this fatty acid precursor compared to that in the inactive-pathway control.
Acetyl-CoA, an important central metabolite and precursor to mevalonate, was observed to be twofold lower in the active-pathway strain (Fig. 2F). This decrease in acetyl-CoA concentration following arabinose induction of the MevT genes can be explained partially by diversion of carbon into the engineered pathway. The anomalous accumulation of malonyl-CoA most likely played a role in the lower concentration of acetyl-CoA observed in the active-pathway strain as well, suggesting that the normal regulation of fatty acid metabolite levels was impaired by high concentrations of intracellular HMG-CoA. The accumulation of malonyl-CoA in E. coli DP10 containing pBAD33MevT may also account for the slightly lower level of acetoacetyl-CoA (Fig. 2G) than that in E. coli DP10 harboring pMevT(C159A). While it has been reported that low free CoA levels can inhibit protein production and growth (34), the free CoA concentration in the mevalonate-producing strain was similar to that in the inactive-pathway control strain (Fig. 2E).
The remaining acyl-CoAs that were tracked (propionyl-CoA and succinyl-CoA) did not vary significantly among strains. The adenylate energy charges of all cultures were >0.80 during early logarithmic-phase growth, indicating rapid quenching of cellular metabolism and negligible degradation of adenylates or acyl-CoAs during extraction from the cell (8).
FAB is altered by HMG-CoA accumulation.
DNA microarray analysis was performed on the growth-inhibited, mevalonate-producing strain and the inactive-pathway control strain to study the early transcriptional response to HMG-CoA toxicity. RNA was isolated from biomass sampled from each culture just prior to induction with arabinose and at 1 and 3 hours postinduction; the RNA was used to synthesize labeled cDNA. In order to fully elucidate the transcriptional response to HMG-CoA accumulation, the following three separate expression profiles (with technical replicates) were generated by two-color hybridization of the cDNA to DNA microarrays: profile A, the 1- and 3-hour-postinduction transcript profile for the inactive-pathway control [E. coli DP10 containing pMevT(C159A) and pBAD18] relative to a preinduction profile; profile B, the mevalonate-producing strain (E. coli DP10 containing pBAD33MevT and pBAD18) 1- and 3-hour-postinduction transcript profile relative to a preinduction profile; and profile C, the mevalonate-producing strain's 1- and 3-hour-postinduction transcript profile relative to the inactive-pathway strain's 1- and 3-hour-postinduction transcript profile.
There was a consistent up-regulation observed in the expression of the β-ketoacyl-ACP synthase I (encoded by fabB), both over the growth time course of the active-pathway strain (postinduction compared to preinduction) (data not shown) and in the cross-strain comparison (active-pathway strain versus inactive-pathway control strain) (Fig. 3). There was also an up-regulation in transcription of malonyl-CoA-ACP transacylase (encoded by fabD) observed in the ∼3-h postinduction sample, with a smaller up-regulation of fabH. Both fabD and fabH share a common promoter, though there can be extensive posttranscriptional processing of these transcripts (10). FabD is the only enzyme in E. coli that interacts with malonyl-CoA directly, and this occurs during the transfer of the malonyl moiety from CoA to the acyl carrier protein (ACP) (10). Additionally, there were several genes involved in the initial steps of FAB whose expression was up-regulated in mevalonate-producing E. coli, including those encoding biotin biosynthetic enzymes and acetyl-CoA carboxylase (Fig. 3).
FIG. 3.

Transcript profiles of the initial steps of type II FAB in Escherichia coli. Malonyl-CoA is synthesized from acetyl-CoA by the action of acetyl-CoA carboxylase, a heterotetramer composed of subunits encoded by accABCD. The malonate moiety is transferred from CoA to ACP by the action of malonyl-CoA:ACP transacylase (FabD). See the work of Magnuson et al. for a full review of the E. coli FAB pathway (43). Also shown (inset) are the expression values and Z scores (in parentheses) for FAB genes that exhibited a biologically significant up-regulation in the mevalonate-producing strain (E. coli DP10 containing pBAD33MevT and pBAD18) relative to the inactive-pathway control strain [E. coli DP10 containing pMevT(C159A) and pBAD18] in the microarray analysis. Also shown are the expression profiles for the CFA synthase gene (cfa) as well as two genes in the biotin synthesis pathway, the gene encoding 7,8-diaminopelargonic acid synthase (bioA) and one encoding biotin synthase (bioB).
The alteration in expression of FAB genes and the fourfold increase in malonyl-CoA levels observed in the mevalonate-producing strain indicated that HMG-CoA accumulation altered fatty acid anabolism. Thus, FAME analysis was performed to determine if there were detectable changes in the fatty acid profile that correlated with HMG-CoA accumulation. Indeed, an enrichment of unsaturated fatty acids (UFA) and a decrease in the percentage of saturated fatty acids (SFA) were observed in the membrane lipids of the active-pathway strain compared to the inactive-pathway strain (Fig. 4B and C). Additionally, there was a marked decrease in the amount of fatty acid as a percentage of DCW in the growth-inhibited culture (Fig. 4A).
FIG. 4.

Cellular fatty acid compositions of the mevalonate-producing strain (E. coli DP10 containing pBAD33MevT and pBAD18) and the inactive-pathway strain [E. coli DP10 containing pMevT(C159A) and pBAD18] obtained by FAME analysis. Open symbols in each graph represent the inactive-pathway strain, and solid symbols represent the mevalonate-producing strain. (A) Total fatty acids as a percentage of DCW. (B) UFA as a percentage of total fatty acids in each strain. ▴ and ▵, 16-carbon UFA (includes 16-carbon CFA); ▪ and □, 18-carbon UFA (includes 18-carbon CFA); and • and ○, CFA (both 16-carbon and 18-carbon CFA). (C) SFA as a percentage of total fatty acids. ▴ and ▵, 16-carbon SFA; ▪ and □, 14-carbon SFA; and • and ○, 12-carbon SFA.
A dramatic up-regulation in the expression of the cyclopropane fatty acid (CFA) synthase gene (cfa) was also observed in the active-pathway strain (Fig. 3). While there has been extensive effort to elucidate the role of CFA in bacteria, there are few clues as to what physiological role they serve (21). Expression of cfa is known to increase upon entry into the stationary phase, and it has been reported that the synthesis of CFA provides E. coli with a method of altering membrane fluidity and/or integrity when normal fatty acid or phospholipid biosynthesis is impeded (21). Again, the fatty acid profiles of membrane lipids correlated with the microarray data quite well, as there was a significant accumulation of CFA observed in the HMG-CoA-stressed cells (Fig. 4B). The high percentage of CFA observed during the log-phase growth of the active-pathway E. coli strain indicated a high degree of stress and a more severe perturbation of FAB than was seen in the inactive-pathway control strain.
The accumulation of HMG-CoA induces a cascade of stress responses.
E. coli has evolved several stress response regulons that allow the organism to adapt rapidly to environmental changes. The transcriptional modulation of these regulons (box plots in Fig. 5) provided insight into the toxicity associated with HMG-CoA accumulation in the mevalonate-producing strain. These box plots show the regulation of groups of related genes as a whole based upon EcoCyc (40) and gene ontology classification.
FIG. 5.

(A) Box plots of transcript expression Z scores for the genes in the σ32 heat shock regulon, oxidative stress-regulated genes (OxyR and SoxS oxidative stress regulons), and osmotically induced operons at 1 hour and 3 hours postinduction. The box plots include the Z scores for all genes detected in the respective functional category based upon EcoCyc (40) annotation and gene ontology classification. The local Z score generated from the SNOMAD analysis represents a change value weighted to account for the confidence in the data, and any gene with a Z score with an absolute value of >1.96 was considered differentially expressed (9). The box plots show the values of the greatest down-regulation, lower quartile (Q1), median, upper quartile (Q3), and largest up-regulation observed for each profile for the regulon or gene group indicated. There were three expression profiles generated in this experiment, as follows: profile A, the 1- and 3-hour-postinduction transcript profile for the inactive-pathway control [E. coli DP10 containing pMevT(C159A) and pBAD18] relative to a preinduction profile; profile B, the mevalonate-producing strain (E. coli DP10 containing pBAD33MevT and pBAD18) 1- and 3-hour-postinduction transcript profile relative to a preinduction profile; and profile C, the mevalonate-producing strain's 1- and 3-hour-postinduction transcript profile relative to the inactive-pathway strain's 1- and 3-hour-postinduction transcript profile. A strong activation of the oxidative and osmotic stress regulons was observed in the active-pathway strain but not in the inactive-pathway control strain. The heat shock regulon was activated early in the active-pathway strain, but overall expression of the regulon was lowered at the 3-hour time point. The heat shock regulon remained highly activated in the inactive-pathway control strain. (B) Time course of expression of genes encoding the 30S and 50S ribosomal proteins as well as the ribosome modulation factor (rmf) observed in the cross-strain microarray analysis (mevalonate-producing strain versus the inactive-pathway control strain). The log2(expression ratio) is defined such that positive values (green) represent up-regulation and negative values (red) represent down-regulation in the mevalonate-producing strain compared to the inactive-pathway control. There was a significant down-regulation of ribosomal protein genes in E. coli DP10 harboring pBAD33MevT (active-pathway strain) compared to their expression in E. coli DP10 harboring pMevT(C159A) (the inactive-pathway control strain).
As shown in the box plots of Z scores (Fig. 5A; Table 2), there was a significant up-regulation of genes involved in osmoregulation in the active-pathway strain, including those encoding trehalose biosynthetic enzymes (otsAB operon), an osmoprotectant/proton symporter (proP), betaine biosynthetic proteins (bet operon), and osmotically inducible genes whose products have not been annotated fully (osmC, osmY, and osmE). No similar osmotic stress response was observed in the inactive-pathway control strain, and indeed, when the two strain's mRNA profiles were compared directly by cross-strain DNA microarray hybridization, the expression levels of the osmoregulatory genes were significantly higher in the growth-inhibited, mevalonate-producing strain than in the inactive-pathway control strain. Of those genes that had biologically significant changes in expression (Table 2), up-regulation of the osmotic response in the active-pathway strain was as high as fivefold for some genes at 3 hours postinduction.
TABLE 2.
Transcript expression ratios of genes with significant differential expression in the heat shock, osmotic stress, and oxidative stress responsesa
| Gene | Fold change (local Z score)
|
Functional activity | |||||
|---|---|---|---|---|---|---|---|
| Profile A
|
Profile B
|
Profile C
|
|||||
| 1 h | 3 h | 1 h | 3 h | 1 h | 3 h | ||
| Heat shock response genes | |||||||
| clpA | 1.9 (1.5) | 2.9 (4.3) | 2.3 (2.9) | 1.5 (1.2) | ATP-dependent molecular chaperone | ||
| clpB | 9.4 (5.8) | 22.3 (8.8) | 5.1 (5.5) | 2.7 (3.1) | 4.3 (4.0) | 1.3 (1.7) | Molecular chaperone |
| clpP | 1.5 (1.7) | 1.91 (2.5) | 2.7 (2.2) | 1.5 (0.5) | 2.5 (2.3) | 0.7 (1.3) | Serine protease |
| dnaJ | 2.1 (1.5) | 2.5 (3.5) | 2.5 (2.7) | 2.1 (1.5) | 3.3 (2.9) | 1.5 (1.1) | DnaJ/DnaK/GrpE chaperone system |
| dnaK | 6.3 (3.7) | 6.4 (7.7) | 6.0 (3.5) | 2.3 (2.0) | 3.4 (2.5) | 0.8 (1.4) | DnaJ/DnaK/GrpE chaperone system |
| grpE | 5.3 (4.2) | 5.0 (6.0) | 4.9 (3.6) | 1.3 (0.9) | 2.0 (2.1) | 1.8 (1.4) | DnaJ/DnaK/GrpE chaperone system |
| hsIU | 3.1 (2.4) | 2.6 (5.3) | 1.9 (3.3) | 1.7 (1.1) | 3.4 (2.9) | 0.9 (0.1) | Component of HsIVU protease |
| hsIV | 2.1 (1.9) | 2.6 (3.9) | 2.5 (3.1) | 1.4 (1.9) | 3.3 (3.0) | 0.9 (1.6) | Component of HsIVU protease |
| htpG | 3.4 (2.2) | 3.4 (5.0) | 2.1 (3.6) | 2.0 (1.4) | Molecular chaperone | ||
| htpX | 1.8 (1.4) | 2.5 (3.5) | 1.8 (2.1) | 1.4 (1.3) | Heat shock protein | ||
| htrA | 2.1 (3.6) | 0.8 (1.7) | Periplasmic serine protease | ||||
| ibpA | 5.7 (3.3) | 42.1 (7.8) | 18.1 (5.2) | 9.2 (4.2) | 3.7 (4.6) | 3.4 (3.3) | Inclusion body protein |
| ibpB | 5.1 (2.4) | 37.5 (9.4) | 14.2 (6.6) | 5.3 (3.3) | 6.8 (5.9) | 2.8 (4.0) | Inclusion body protein |
| ion | 2.1 (1.7) | 2.1 (3.2) | ATP-dependent protease | ||||
| mopA | 3.4 (1.9) | 3.1 (4.9) | 4.5 (5.2) | 2.1 (1.2) | 2.5 (3.8) | 0.6 (1.6) | Member of GroES molecular chaperone family |
| mopB | 4.0 (2.2) | 4.9 (6.4) | 3.1 (4.2) | 2.7 (2.1) | 2.0 (3.6) | 1.1 (0.9) | Member of GroES molecular chaperone family |
| Oxidative stress response genes | |||||||
| acrA | 1.1 (0.6) | 3.5 (2.5) | Periplasmic component of multidrug efflux pump | ||||
| ahpC | 1.4 (1.4) | 3.1 (3.4) | 1.1 (0.5) | 1.9 (2.2) | Subunit of alkylhydroperoxide reductase | ||
| dps | 1.4 (1.1) | 2.1 (2.3) | 2.9 (3.1) | 4.1 (4.0) | 2.2 (3.1) | 3.2 (12.1) | Iron binding protein/DNA protection |
| fumC | 1.1 (0.6) | 3.3 (2.4) | Fumarase | ||||
| gor | 1.9 (0.3) | 2.8 (2.4) | Unknown function | ||||
| grxA | 0.8 (0.2) | 3.1 (2.6) | Glutaredoxin 1 | ||||
| katE | 1.2 (2.6) | 3.0 (3.6) | 3.8 (3.2) | Heme d synthase/hydroperoxidase | |||
| katG | 1.1 (0.9) | 2.3 (2.7) | 2.6 (2.8) | 3.8 (3.1) | Hydroperoxidase I | ||
| poxB | 1.8 (2.6) | 2.2 (3.8) | Acetoin synthesis/pyruvate oxidase | ||||
| sodA | 1.3 (0.3) | 1.8 (2.5) | 0.7 (1.0) | 1.9 (2.7) | Superoxide dismutase (Mn) | ||
| sodB | 1.3 (1.2) | 2.1 (2.4) | 0.8 (0.9) | 1.9 (2.0) | Superoxide dismutase (Fe) | ||
| soxS | 1.0 (0.4) | 1.8 (2.4) | Transcriptional dual regulator | ||||
| sufA | 2.1 (3.1) | 3.6 (4.0) | 1.6 (2.1) | 2.4 (4.7) | Fe-S cluster repair | ||
| sufB | 1.4 (2.4) | 3.2 (3.3) | 1.6 (2.2) | 2.4 (6.5) | Fe-S cluster repair | ||
| sufC | 2.1 (2.3) | 2.4 (3.2) | 1.7 (2.2) | 2.2 (4.0) | Fe-S cluster repair | ||
| sufD | 1.8 (2.9) | 2.9 (3.7) | 1.9 (2.2) | 2.1 (3.7) | Fe-S cluster repair | ||
| trxC | 1.4 (0.9) | 1.9 (2.1) | 1.2 (1.3) | 2.4 (3.9) | 1.5 (0.3) | 4.2 (2.5) | Thioredoxin 2 |
| zwf | 1.1 (0.1) | 2.6 (3.1) | 0.4 (−0.9) | 5.5 (3.4) | Glucose 6-phosphate-1-dehydrogenase | ||
| Osmotic stress response genes | |||||||
| betA | 1.5 (4.6) | 2.8 (4.1) | 1.3 (2.7) | 3.2 (3.7) | Choline dehydrogenase | ||
| betB | 1.7 (2.1) | 2.5 (3.5) | 1.6 (2.7) | 2.4 (2.8) | Betaine aldehyde dehydrogenase | ||
| betT | 0.8 (2.0) | 1.5 (2.7) | Choline transporter | ||||
| kdpA | 1.8 (2.1) | 1.5 (−0.8) | 1.2 (1.8) | 1.9 (2.8) | Subunit of potassium ion transporter | ||
| osmB | 0.9 (2.8) | 3.2 (3.0) | Osmotically inducible lipoprotein | ||||
| osmC | 1.7 (2.3) | 3.2 (2.7) | 3.3 (4.2) | 5.5 (6.0) | 3.2 (4.1) | 4.6 (3.5) | Osmotically inducible peroxidase |
| osmE | 2.4 (3.8) | 3.0 (4.5) | 1.3 (2.2) | 2.1 (3.3) | Osmotically inducible protein | ||
| osmY | 1.3 (1.7) | 3.2 (2.6) | 2.9 (3.8) | 6.6 (6.3) | 2.3 (4.6) | 3.6 (5.8) | Osmotically inducible protein |
| otsA | 1.8 (2.4) | 3.9 (5.2) | 1.2 (2.5) | 5.3 (4.5) | Trehalose-6-phosphate synthase | ||
| otsB | 1.9 (2.4) | 2.9 (4.6) | 1.3 (4.2) | 5.3 (4.9) | Trehalose-6-phosphate phosphatase | ||
| proP | 1.6 (1.6) | 2.1 (3.3) | 1.4 (2.3) | 1.9 (3.3) | Proline/betaine MFS transporter | ||
| proV | −2.3 (−2.2) | −1.7 (−2.6) | Subunit of betaine transporter | ||||
Profile A, 1- and 3-hour-postinduction transcript profile for the inactive-pathway control [E. coli DP10 containing pMevT(C159A) and pBAD18] relative to a preinduction profile; profile B, the mevalonate-producing strain (E. coli DP10 containing pBAD33MevT and pBAD18) 1- and 3-hour-postinduction transcript profile relative to a preinduction profile; and profile C, the mevalonate-producing strain's 1- and 3-hour-postinduction transcript profile relative to the inactive-pathway strain's 1- and 3-hour-postinduction transcript profile. The expression of genes not listed in a comparison did not change significantly in the respective comparison.
The active-pathway strain also exhibited a significant increase in expression of oxidative stress-associated genes 3 hours after induction (box plot of SoxS and OxyR-regulated genes in Fig. 5A). The up-regulated genes in the HMG-CoA-stressed cells, over the individual time course or in the strain-to-strain comparison, included most of the OxyR regulon, including dps (encoding a DNA binding protein), the suf operon (involved in iron-sulfur cluster repair), grxA (encoding glutaredoxin 1), trxC (encoding thioredoxin 2), gor (encoding glutathione reductase), katG (encoding peroxidase), and ahpC (encoding alkylhydroperoxide reductase) (Table 2). The up-regulation of this stress response regulon is indicative of increased hydrogen peroxide production in the mevalonate-producing strain, especially at the later time point. Again, this trend was not observed in the inactive-pathway control strain; therefore, the H2O2 response appears to be specific to the accumulation of HMG-CoA.
The expression of the mevalonate pathway also elicited a moderate heat shock response consistent with heterologous protein production. As shown in the time course expression profile box plots of σ32-regulated genes in cells harboring the active pathway (Fig. 5A), expression of many members of the heat shock regulon was induced at 1 h postinduction, but then expression of most of these genes decreased at 3 h postinduction. The heat shock regulon genes most highly expressed in the active-pathway strain were those encoding the chaperones ClpB, DnaK, GroEL, and GroES as well as the inclusion body proteins IbpA and IbpB. In contrast, the inactive-pathway control continued to express genes in the heat shock regulon at high levels 3 hours after induction, and the same trend was observed when the two strain's transcriptional profiles were compared directly.
Coincident with expression of the heat shock regulon, there was a strong, two- to fivefold down-regulation of the genes encoding the ribosomal proteins observed in the active-pathway strain at 1 and 3 hours postinduction (time course clustering) (Fig. 5B). A strong heat shock response in E. coli is known to result in a down-regulation of the ribosomal protein genes (12, 53), yet the inactive-pathway strain, which had a much stronger up-regulation of the heat shock genes (Fig. 5A, profile A), did not exhibit a similar down-regulation of the ribosomal genes. Interestingly, the decreased transcription of the ribosomal protein genes in the active-pathway strain correlated with the fourfold up-regulation of the gene encoding the ribosome modulation factor, rmf. This gene, whose product dimerizes the 70S ribosome and reduces translational capacity, is generally expressed during the transition to stationary phase or under conditions of stress (33).
Thus, the two strains both exhibited an activation of the heat shock regulon, but each had a quite different response dynamic. There was a strong expression of this regulon in the active-pathway strain 1 hour after induction, but that response decayed by the third hour. The inactive-pathway control exhibited a steadily increasing induction of the heat shock genes. Heterologous protein production often increases the transcription of heat shock genes encoding molecular chaperones and proteases (12). Thus, the strong down-regulation of the ribosomal proteins, likely part of a coordinated response in the HMG-CoA-stressed cells, limited heterologous protein synthesis and, in turn, led to the general down-regulation of the heat shock regulon observed for the time course of cells harboring pBAD33MevT. Conversely, since the inactive-pathway strain was not stressed by HMG-CoA accumulation, it was able to maintain a higher rate of protein synthesis, which induced increased transcription of heat shock regulon genes, as observed at the later time point. This could explain the observation previously made that HMG-CoA reductase activity is higher in the inactive-pathway strain (51).
Medium supplementation tests.
Based on the differential fatty acid auxotrophies reported in the literature for E. coli strains with various mutations of FAB genes, we designed a panel of medium supplementation tests to screen for fatty acids that could relieve the HMG-CoA-associated growth defect. E. coli strain DP10 was transformed with either pBAD33MevT (active pathway), pMevT(C159A) (inactive-pathway control), or pBAD33 (empty vector control) and grown in a defined medium (C medium) in a 96-well plate growth assay. The growth medium was supplemented with 100 μg/ml of oleic acid (cis-Δ9-18:1), palmitoleic acid (cis-Δ9-16:1), palmitic acid (16:0), a combination (16:0 plus cis-Δ9-16:1), or no supplement at all. Both control strains grew well in all medium formulations tested (Fig. 6).
FIG. 6.

Effect of fatty acid supplementation on growth of mevalonate-producing and control strains of E. coli DP10. (A) Growth of the active-pathway strain [E. coli DP10(pBAD33MevT)] in a defined medium supplemented with 100 μg/ml of oleic acid (cis-Δ9-18:1) (⋄), palmitoleic acid (cis-Δ9-16:1) (□), palmitic acid (16:0) (▵), a combination of fatty acids (16:0 plus cis-Δ9-16:1) (+), or no fatty acid supplement (○) in a 96-well incubator/plate reader monitored continuously at OD600. The growth of the inactive-pathway strain [E. coli DP10 carrying pMevT(C159A)] in nonsupplemented medium (▪) is shown as well. Growth profiles of the inactive-pathway control strain and an empty vector control [E. coli DP10(pBAD33)] were equivalent in medium supplemented with the various fatty acids. (B) Shake flask growth of E. coli DP10(pBAD33MevT) with (•) and without (○) a palmitic acid (16:0) supplement compared to that of the inactive-pathway control [E. coli DP10 carrying pMevT(C159A)] (▪).
The growth inhibition associated with the expression of the active-pathway plasmid was alleviated by 16:0 SFA and 18:1 UFA supplementation and exaggerated by 16:1 UFA supplementation (Fig. 6A). The cultures supplemented with the combination of palmitic and palmitoleic acid grew slightly slower than the cultures with no supplement. Since the growth dynamics in a 96-well plate may not be completely predictive of the physiology of a fully aerobic culture, we grew the active-pathway and inactive-pathway strains in baffled shake flasks in defined medium (C medium), with and without the palmitic acid supplement, and induced expression of the mevalonate pathway genes. Again, the growth inhibition associated with expression of MevT from pBAD33MevT was relieved by the presence of the 16:0 fatty acid (Fig. 6B).
DISCUSSION
Engineering the heterologous mevalonate pathway into E. coli exposed the host's metabolic network to a biochemical intermediate that the organism had not evolved to counteract. Given the complexity of the metabolic network and the extensive number of interactions that occur in the E. coli cytosol, it is not surprising that a deleterious interaction would arise. While it can be relatively simple to determine that an engineered synthetic biochemical pathway is not functioning in the heterologous host, it is often a far more challenging task to determine exactly what is causing the problem. Using microarray and metabolite analysis of just such a design problem, we have demonstrated that the growth inhibition associated with HMG-CoA accumulation in E. coli DP10 harboring pBAD33MevT is due to inhibition of one or more enzymes involved in the elongation or priming steps of FAB.
It has previously been reported that there is significant conversion of the acyl-CoA pool to malonyl-CoA in E. coli when the initial steps of FAB are inhibited by chemicals, such as the antibiotics thiolactomycin (28, 29) and cerrulenin (17, 29), or genetic methods, such as disruption of protein function (58) and mutation (36). E. coli coordinates FAB with lipid requirements (and hence growth), and the accumulation of long-chain acyl-ACPs is a key signal of slowing growth, which results in the coordinated down-regulation of fabB, fabA, and accAB (27, 28, 35, 36) as well as the inhibition of enzymatic activity (27). During this down-regulation of FAB, excess malonyl-CoA is recycled back to the acetyl-CoA pool through the actions of KAS1 (fabB) and malonyl-CoA-ACP transacylase (fabD) (28). Thus, the impressive accumulation of malonyl-CoA observed in the mevalonate-producing cells is an indication that this complex regulation has been disrupted.
Supplementation of the growth medium with certain fatty acids abolished the HMG-CoA-induced growth inhibition in the active-pathway strain by removing the need for fatty acid anabolism and confirmed the hypothesis that FAB was inhibited, either directly or indirectly, by HMG-CoA accumulation. The results from the entire panel of fatty acid supplements offer insight as to where HMG-CoA may inhibit the FAB pathway. E. coli requires both SFA and UFA to maintain proper membrane fluidity. Harder et al. observed that temperature-sensitive fabB mutants were auxotrophic for trans-UFA or a combination of SFA and cis-UFA at the nonpermissive temperature (25), while fabD mutants grew at the nonpermissive temperature only when the medium was supplemented with 16:0 SFA or 18:1 UFA but not 16:1 UFA (26). Additionally, when this fabD mutant was grown in nonsupplemented medium at a temperature that only partially inhibited growth, the membrane lipids were enriched for 16:1 SFAs. Since the supplementation of UFA or a combination of SFA and UFA failed to relieve the HMG-CoA-associated toxicity, FabB does not appear to be the target of HMG-CoA inhibition in the FAB pathway. Instead, our results suggest that FabD was the target, since inhibited growth of the active-pathway strain in nonsupplemented medium enriched UFA and the addition of palmitic acid (16:0) (and, to a lesser extent, oleic acid [cis-Δ9-18:1]) markedly improved the growth of this strain.
FabD is the only enzyme in E. coli that interacts with malonyl-CoA directly, and this occurs during the transfer of the malonyl moiety from CoA to ACP (10). This reaction is critical to FAB since it provides the two-carbon units needed to extend fatty acids. The reaction mechanism is mediated by an active-site serine that attacks the thioester carbonyl and releases free CoA, leaving a malonyl-FabD complex. Next, the malonyl-FabD ester carbonyl is attacked by the phosphopantetheinyl thiol of ACP, which ultimately yields malonyl-ACP (47). HMG-CoA is structurally very similar to malonyl-CoA and may interfere with substrate binding to the active site. Supporting this hypothesis is the evidence that another acyl-CoA, acetyl-CoA, has been shown to be a weak inhibitor of malonyl-CoA-ACP transacylase activity (38, 47).
The up-regulation of accBC, fabB, fabD, and fabH observed in the growth-inhibited, mevalonate-producing strain is consistent with the hypothesis that HMG-CoA inhibits FAB. This inhibition reduced the availability of long-chain acyl-ACPs, which was sensed by the heterologous host as a decoupling of the rates of FAB and growth. The resulting up-regulation of FAB genes was the host cell's attempt to maintain growth by up-regulating the flux of carbon into fatty acid anabolism. A similar transcriptional response to chemical inhibition of FAB has been documented for Mycobacterium tuberculosis, where exposure to thiolactomycin elicited an up-regulation of the genes encoding β-ketoacyl-ACP synthase I (fabB), malonyl-CoA-ACP transacylase (fabD), and alkyl hydroperoxide reductase C (ahpC) (7), just as observed in the mevalonate-producing strain.
The up-regulation of FAB genes allowed the heterologous host to overcome the most deleterious effect of HMG-CoA, a complete cessation of growth, which is similar in principle to the resistance to thiolactomycin that E. coli gains when fabB is expressed on a multicopy plasmid (11). Yet while the up-regulation of the FAB system was sufficient to allow slow growth in the presence of high levels of cytosolic HMG-CoA, there were consequences to the cell. Jackowski and Rock observed that inhibition of the initial steps of FAB increased the UFA/SFA ratio in the membrane (34) because the inhibition reduced the availability of fatty acids, which, in turn, induced expression of the β-ketoacyl-ACP synthase I (fabB). This enzyme has multiple roles in E. coli's FAB pathway, including both SFA synthesis and the diversion of saturated long-chain acyl-ACPs into the desaturation pathway that produces UFA. Indeed, the increase in FAB gene expression observed in the HMG-CoA-inhibited cells resulted in a coordinate enrichment of UFA in the cell membrane and an overall decrease in fatty acids as a percentage of DCW.
The altered UFA/SFA ratio in the membrane that resulted from HMG-CoA inhibition of fatty acid anabolism in the mevalonate-producing strain could explain the cascade of osmotic, oxidative, and heat shock stress responses observed in that strain's transcriptional profile. Recent studies of the osmosensor ProP in E. coli have discovered that membrane composition is a key signal governing the long-term response to alterations in medium osmolality (59). The oxidative stress response in the active-pathway strain was also an indication of membrane-associated stress, since H2O2 is mostly a by-product of the respiratory electron transport chain in E. coli (20), and both degradation of membrane integrity and osmotic stress have been reported to induce transcription of H2O2 defense genes (14, 57). Finally, the coordinated heat shock response to H2O2 is well established (15, 16), and the sum of all these stress responses appeared ultimately to result in a down-regulation of the translational machinery in the mevalonate-producing strain. Thus, the HMG-CoA-induced changes in membrane composition resulted in a coordinated modulation of many stress response regulons, which may be the ultimate cause for the slow growth observed in HMG-CoA-stressed cells.
In summary, the model for the growth inhibition observed in E. coli expressing the MevT operon is one where the cell's type II fatty acid anabolism was impeded, most likely by inhibition of FabD activity, which in turn limited the availability of long-chain acyl-ACPs. This reduced availability increased the transcription of genes in the initial steps of fatty acid synthesis, which overcame the HMG-CoA inhibition enough to allow slow growth. As a consequence of low levels of acyl-ACP and increased expression of FabB activity, however, the membrane lipids were enriched in UFA. This in turn altered the membrane structural properties and induced the transcription of genes associated with osmotic, oxidative, and heat shock stress. The sum of these responses ultimately limited the growth rate in the mevalonate-producing culture. It should be noted that a similar deleterious interaction does not appear to occur during accumulation of HMG-CoA in S. cerevisiae (56), which has a nondisassociated, type I FAB pathway, and interestingly, the type II pathway antibiotic thiolactomycin is not active against the S. cerevisiae type I FAB pathway. This report highlights the unexpected interactions that can occur when a novel, heterologous biochemical pathway is engineered into a host organism as well as the utility of a systems biology approach to the design problems that inevitably arise in metabolic engineering.
Acknowledgments
This research was funded by the Bill and Melinda Gates Foundation (through a grant from The Institute of OneWorld Health).
We acknowledge Jack Newman for his assistance.
Footnotes
Published ahead of print on 14 March 2008.
REFERENCES
- 1.Aldor, I. S., and J. D. Keasling. 2003. Process design for microbial plastic factories: metabolic engineering of polyhydroxyalkanoates. Curr. Opin. Biotechnol. 14:475-483. [DOI] [PubMed] [Google Scholar]
- 2.Anonymous. 2006. Guidelines for the treatment of malaria. World Health Organization, Geneva, Switzerland.
- 3.Atkinson, D. E. 1968. The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 7:4030-4034. [DOI] [PubMed] [Google Scholar]
- 4.Bailey, J. E., and D. F. Ollis. 1986. Biochemical engineering fundamentals, 2nd ed. McGraw Hill, New York, NY.
- 5.Barbirato, F., J. P. Grivet, P. Soucaille, and A. Bories. 1996. 3-Hydroxypropionaldehyde, an inhibitory metabolite of glycerol fermentation to 1,3-propanediol by enterobacterial species. Appl. Environ. Microbiol. 62:1448-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berry, A., T. C. Dodge, M. Pepsin, and W. Weyler. 2002. Application of metabolic engineering to improve both the production and use of biotech indigo. J. Ind. Microbiol. Biotechnol. 28:127-133. [DOI] [PubMed] [Google Scholar]
- 7.Betts, J. C., A. McLaren, M. G. Lennon, F. M. Kelly, P. T. Lukey, S. J. Blakemore, and K. Duncan. 2003. Signature gene expression profiles discriminate between isoniazid-, thiolactomycin-, and triclosan-treated Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 47:2903-2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman, A. G., L. Fall, and D. E. Atkinson. 1971. Adenylate energy charge in Escherichia coli during growth and starvation. J. Bacteriol. 108:1072-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colantuoni, C., G. Henry, S. Zeger, and J. Pevsner. 2002. SNOMAD (standardization and normalization of microarray data): web-accessible gene expression data analysis. Bioinformatics 18:1540-1541. [DOI] [PubMed] [Google Scholar]
- 10.Cronan, J. E., and C. O. Rock. September 1996, posting date. Biosynthesis of membrane lipids. In R. Curtiss III et al. (ed.), EcoSal—Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, DC. http://www.ecosal.org.
- 11.de Mendoza, D., A. Klages Ulrich, and J. E. Cronan, Jr. 1983. Thermal regulation of membrane fluidity in Escherichia coli. Effects of overproduction of beta-ketoacyl-acyl carrier protein synthase I. J. Biol. Chem. 258:2098-2101. [PubMed] [Google Scholar]
- 12.Dong, H., L. Nilsson, and C. G. Kurland. 1995. Gratuitous overexpression of genes in Escherichia coli leads to growth inhibition and ribosome destruction. J. Bacteriol. 177:1497-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eisen, M. B., P. T. Spellman, P. O. Brown, and D. Botstein. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95:14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farr, S. B., and T. Kogoma. 1991. Oxidative stress responses in Escherichia coli and Salmonella typhimurium. Microbiol. Rev. 55:561-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fredriksson, A., M. Ballesteros, S. Dukan, and T. Nystrom. 2005. Defense against protein carbonylation by DnaK/DnaJ and proteases of the heat shock regulon. J. Bacteriol. 187:4207-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fredriksson, A., M. Ballesteros, S. Dukan, and T. Nystrom. 2006. Induction of the heat shock regulon in response to increased mistranslation requires oxidative modification of the malformed proteins. Mol. Microbiol. 59:350-359. [DOI] [PubMed] [Google Scholar]
- 17.Furukawa, H., J. T. Tsay, S. Jackowski, Y. Takamura, and C. O. Rock. 1993. Thiolactomycin resistance in Escherichia coli is associated with the multidrug resistance efflux pump encoded by emrAB. J. Bacteriol. 175:3723-3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glick, B. R. 1995. Metabolic load and heterologous gene expression. Biotechnol. Adv. 13:247-261. [DOI] [PubMed] [Google Scholar]
- 19.Goff, S. A., and A. L. Goldberg. 1985. Production of abnormal proteins in E. coli stimulates transcription of lon and other heat shock genes. Cell 41:587-595. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Flecha, B., and B. Demple. 1995. Metabolic sources of hydrogen peroxide in aerobically growing Escherichia coli. J. Biol. Chem. 270:13681-13687. [DOI] [PubMed] [Google Scholar]
- 21.Grogan, D. W., and J. E. Cronan, Jr. 1997. Cyclopropane ring formation in membrane lipids of bacteria. Microbiol. Mol. Biol. Rev. 61:429-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guzman, L. M., et al. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121-4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harcum, S. W., and W. E. Bentley. 1999. Heat-shock and stringent responses have overlapping protease activity in Escherichia coli. Implications for heterologous protein yield. Appl. Biochem. Biotechnol. 80:23-37. [DOI] [PubMed] [Google Scholar]
- 24.Harcum, S. W., and W. E. Bentley. 1993. Response dynamics of 26-, 34-, 39-, 54-, and 80-kDa proteases in induced cultures of recombinant Escherichia coli. Biotechnol. Bioeng. 42:675-685. [DOI] [PubMed] [Google Scholar]
- 25.Harder, M. E., I. R. Beacham, J. E. Cronan, Jr., K. Beacham, J. L. Honegger, and D. F. Silbert. 1972. Temperature-sensitive mutants of Escherichia coli requiring saturated and unsaturated fatty acids for growth: isolation and properties. Proc. Natl. Acad. Sci. USA 69:3105-3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harder, M. E., R. C. Ladenson, S. D. Schimmel, and D. F. Silbert. 1974. Mutants of Escherichia coli with temperature-sensitive malonyl coenzyme A-acyl carrier protein transacylase. J. Biol. Chem. 249:7468-7475. [PubMed] [Google Scholar]
- 27.Heath, R. J., and C. O. Rock. 1996. Regulation of fatty acid elongation and initiation by acyl-acyl carrier protein in Escherichia coli. J. Biol. Chem. 271:1833-1836. [DOI] [PubMed] [Google Scholar]
- 28.Heath, R. J., and C. O. Rock. 1995. Regulation of malonyl-CoA metabolism by acyl-acyl carrier protein and beta-ketoacyl-acyl carrier protein synthases in Escherichia coli. J. Biol. Chem. 270:15531-15538. [DOI] [PubMed] [Google Scholar]
- 29.Heath, R. J., S. W. White, and C. O. Rock. 2002. Inhibitors of fatty acid synthesis as antimicrobial chemotherapeutics. Appl. Microbiol. Biotechnol. 58:695-703. [DOI] [PubMed] [Google Scholar]
- 30.Helmstetter, C., S. Cooper, O. Pierucci, and E. Revelas. 1968. On the bacterial life sequence. Cold Spring Harbor Symp. Quant. Biol. 33:809-822. [DOI] [PubMed] [Google Scholar]
- 31.Hoffmann, F., and U. Rinas. 2000. Kinetics of heat-shock response and inclusion body formation during temperature-induced production of basic fibroblast growth factor in high-cell-density cultures of recombinant Escherichia coli. Biotechnol. Prog. 16:1000-1007. [DOI] [PubMed] [Google Scholar]
- 32.Hoffmann, F., and U. Rinas. 2004. Stress induced by recombinant protein production in Escherichia coli. Adv. Biochem. Eng. Biotechnol. 89:73-92. [DOI] [PubMed] [Google Scholar]
- 33.Izutsu, K., A. Wada, and C. Wada. 2001. Expression of ribosome modulation factor (RMF) in Escherichia coli requires ppGpp. Genes Cells 6:665-676. [DOI] [PubMed] [Google Scholar]
- 34.Jackowski, S., and C. O. Rock. 1986. Consequences of reduced intracellular coenzyme A content in Escherichia coli. J. Bacteriol. 166:866-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.James, E. S., and J. E. Cronan. 2004. Expression of two Escherichia coli acetyl-CoA carboxylase subunits is autoregulated. J. Biol. Chem. 279:2520-2527. [DOI] [PubMed] [Google Scholar]
- 36.Jiang, P., and J. E. Cronan, Jr. 1994. Inhibition of fatty acid synthesis in Escherichia coli in the absence of phospholipid synthesis and release of inhibition by thioesterase action. J. Bacteriol. 176:2814-2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones, K. L., S. W. Kim, and J. D. Keasling. 2000. Low-copy plasmids can perform as well as or better than high-copy plasmids for metabolic engineering of bacteria. Metab. Eng. 2:328-338. [DOI] [PubMed] [Google Scholar]
- 38.Joshi, V. C., and S. J. Wakil. 1971. Studies on the mechanism of fatty acid synthesis. XXVI. Purification and properties of malonyl-coenzyme A-acyl carrier protein transacylase of Escherichia coli. Arch. Biochem. Biophys. 143:493-505. [DOI] [PubMed] [Google Scholar]
- 39.Kanemori, M., H. Mori, and T. Yura. 1994. Induction of heat shock proteins by abnormal proteins results from stabilization and not increased synthesis of sigma 32 in Escherichia coli. J. Bacteriol. 176:5648-5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karp, P. D., M. Riley, M. Saier, I. T. Paulsen, J. Collado-Vides, S. M. Paley, A. Pellegrini-Toole, C. Bonavides, and S. Gama-Castro. 2002. The EcoCyc database. Nucleic Acids Res. 30:56-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khlebnikov, A., K. A. Datsenko, T. Skaug, B. L. Wanner, and J. D. Keasling. 2001. Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology 147:3241-3247. [DOI] [PubMed] [Google Scholar]
- 42.Khlebnikov, A., O. Risa, T. Skaug, T. A. Carrier, and J. D. Keasling. 2000. Regulatable arabinose-inducible gene expression system with consistent control in all cells of a culture. J. Bacteriol. 182:7029-7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magnuson, K., S. Jackowski, C. O. Rock, and J. E. Cronan, Jr. 1993. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol. Rev. 57:522-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin, V. J. J., D. J. Pitera, S. T. Withers, J. D. Newman, and J. D. Keasling. 2003. Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. 21:796-802. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura, C. E., and G. M. Whited. 2003. Metabolic engineering for the microbial production of 1,3-propanediol. Curr. Opin. Biotechnol. 14:454-459. [DOI] [PubMed] [Google Scholar]
- 46.Neidhardt, F. C., P. L. Bloch, and D. F. Smith. 1974. Culture medium for enterobacteria. J. Bacteriol. 119:736-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oefner, C., H. Schulz, A. D'Arcy, and G. E. Dale. 2006. Mapping the active site of Escherichia coli malonyl-CoA-acyl carrier protein transacylase (FabD) by protein crystallography. Acta Crystallogr. D 62:613-618. [DOI] [PubMed] [Google Scholar]
- 48.Parsell, D. A., and R. T. Sauer. 1989. Induction of a heat shock-like response by unfolded protein in Escherichia coli: dependence on protein level not protein degradation. Genes Dev. 3:1226-1232. [DOI] [PubMed] [Google Scholar]
- 49.Peiru, S., H. G. Menzella, E. Rodriguez, J. Carney, and H. Gramajo. 2005. Production of the potent antibacterial polyketide erythromycin C in Escherichia coli. Appl. Environ. Microbiol. 71:2539-2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pfeifer, B. A., S. J. Admiraal, H. Gramajo, D. E. Cane, and C. Khosla. 2001. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science 291:1790-1792. [DOI] [PubMed] [Google Scholar]
- 51.Pitera, D. J., C. J. Paddon, J. D. Newman, and J. D. Keasling. 2007. Balancing a heterologous mevalonate pathway for improved isoprenoid production in Escherichia coli. Metab. Eng. 9:193-207. [DOI] [PubMed] [Google Scholar]
- 52.Quackenbush, J. 2002. Microarray data normalization and transformation. Nat. Genet. 32:496-501. [DOI] [PubMed] [Google Scholar]
- 53.Rinas, U. 1996. Synthesis rates of cellular proteins involved in translation and protein folding are strongly altered in response to overproduction of basic fibroblast growth factor by recombinant Escherichia coli. Biotechnol. Prog. 12:196-200. [DOI] [PubMed] [Google Scholar]
- 54.Rokosz, L. L., D. A. Boulton, E. A. Butkiewicz, G. Sanyal, M. A. Cueto, P. A. Lachance, and J. D. Hermes. 1994. Human cytoplasmic 3-hydroxy-3-methylglutaryl coenzyme A synthase: expression, purification, and characterization of recombinant wild-type and Cys129 mutant enzymes. Arch. Biochem. Biophys. 312:1-13. [DOI] [PubMed] [Google Scholar]
- 55.Sasser, M. 1990. Identification of bacteria by gas chromatography of cellular fatty acids. Technical note 101. Microbial ID, Newark, DE.
- 56.Seker, T., K. Moller, and J. Nielson. 2005. Analysis of acyl-CoA ester intermediates of the mevalonate pathway in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 67:119-124. [DOI] [PubMed] [Google Scholar]
- 57.Smirnova, G. V., N. G. Muzyka, and O. N. Oktyabrsky. 2000. The role of antioxidant enzymes in response of Escherichia coli to osmotic upshift. FEMS Microbiol. Lett. 186:209-213. [DOI] [PubMed] [Google Scholar]
- 58.Subrahmanyam, S., and J. E. Cronan, Jr. 1998. Overproduction of a functional fatty acid biosynthetic enzyme blocks fatty acid synthesis in Escherichia coli. J. Bacteriol. 180:4596-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsatskis, Y., J. Khambati, M. Dobson, M. Bogdanov, W. Dowhan, and J. M. Wood. 2005. The osmotic activation of transporter ProP is tuned by both its C-terminal coiled-coil and osmotically induced changes in phospholipid composition. J. Biol. Chem. 280:41387-41394. [DOI] [PubMed] [Google Scholar]
- 60.Tusher, V. G., R. Tibshirani, and G. Chu. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98:5116-5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watts, K. T., B. N. Mijts, and C. Schmidt-Dannert. 2005. Current and emerging approaches for natural product biosynthesis in microbial cells. Adv. Synth. Catal. 347:927-940. [Google Scholar]
- 62.Zhu, M. M., P. D. Lawman, and D. C. Cameron. 2002. Improving 1,3-propanediol production from glycerol in a metabolically engineered Escherichia coli by reducing accumulation of sn-glycerol-3-phosphate. Biotechnol. Prog. 18:694-699. [DOI] [PubMed] [Google Scholar]
- 63.Zhu, M. M., F. A. Skraly, and D. C. Cameron. 2001. Accumulation of methylglyoxal in anaerobically grown Escherichia coli and its detoxification by expression of the Pseudomonas putida glyoxalase I gene. Metab. Eng. 3:218-225. [DOI] [PubMed] [Google Scholar]