

Short abstract
An analysis of the seven genes of the tryptophan pathway in the Sargasso Sea metagenome shows that the majority of contigs and scaffolds contain whole or split operons that are similar to previously analyzed trp gene organizations.
Abstract
Background
The enormous database of microbial DNA generated from the Sargasso Sea metagenome provides a unique opportunity to locate genes participating in different biosynthetic pathways and to attempt to understand the relationship and evolution of those genes. In this article, an analysis of the Sargasso Sea metagenome is made with respect to the seven genes of the tryptophan pathway.
Results
At least 5% of all the genes that are related to amino acid biosynthesis are tryptophan (trp) genes. Many contigs and scaffolds contain whole or split operons that are similar to previously analyzed trp gene organizations. Only two scaffolds discovered in this analysis possess a different operon organization of tryptophan pathway genes than those previously known. Many marine organisms lack an operon-type organization of these genes or have mini-operons containing only two trp genes. In addition, the trpB genes from this search reveal that the dichotomous division between trpB_1 and trpB_2 also occurs in organisms from the Sargasso Sea. One cluster was found to contain trpB sequences that were closely related to each other but distinct from most known trpB sequences.
Conclusion
The data show that trp genes are widely dispersed within this metagenome. The novel organization of these genes and an unusual group of trpB_1 sequences that were found among some of these Sargasso Sea bacteria indicate that there is much to be discovered about both the reason for certain gene orders and the regulation of tryptophan biosynthesis in marine bacteria.
Background
The tryptophan pathway and the organization of the trp genes involved in its synthesis have been a model system for many years and these genes continue to receive attention [1,2]. With the availability of extensive DNA sequences, it has been found that trp genes are not identically organized in all organisms. The classical structure of the trp operon contains genes for all seven catalytic domains in the following order: promoter, trpE, trpG, trpD, trpC, trpF, trpB and trpA. In some organisms each catalytic domain is encoded by a different gene. As shown in Figure 1, there are seven catalytic domains that carry out the reactions that convert chorismate and L-glutamine to L-tryptophan.
Figure 1.

The biochemical pathway of tryptophan biosynthesis. The genetic nomenclature for the seven genes that encode the enzymes is that for Bacillus subtilis. PR-Anth, N-(5'-phosphoribosyl)-anthranilate; CdRP, 1-(o-carboxy-phenylamino)-1-deoxyribulose-5-phosphate; InGP, indole 3-glycerol phosphate. trpE encodes the large aminase subunit of anthranilate synthase; trpG encodes for small glutamine binding subunit of anthranilate synthase and catalyzes the glutaminase reaction; trpD encodes anthranilate-phosphoribosyl transferase; trpF encodes phosphoribosyl-anthranilate isomerase; trpC encodes indoleglycerol phosphate synthase; trpA, the a subunit of tryptophan synthase which converts InGP to indole; trpB encodes the b subunit of tryptophan synthase and converts indole and serine to tryptophan and glyceraldehydes-3-phosphate.
To date, several deviations from the classical structure have been reported. Gene fusion may result in a single polypeptide carrying two or more catalytic domains. The most extreme exception is found in the eukaryote Euglena in which a single gene encodes a polypeptide with five catalytic domains [3]. In split operons, the trp genes are organized into two or more sub-operons [4]. Other events include gene reshuffling, gene insertions and gene deletions. An analysis of more than 100 genomes showed that the evolution of trp operon is both the result of vertical genealogy and lateral gene transfer. It has been found that, if events of lateral gene transfer and paralogy can be sorted out, the vertical transfer of the trp genes becomes apparent [4,5].
As a result of the publication of the Sargasso Sea metagenome by Venter et al. [6], it may be possible to deduce the evolutionary relationships between the trp genes of different marine organisms from the Sargasso Sea. This metagenome is composed of more than one million non-redundant sequences, or reads, that have been estimated to derive from 1,800 different genomes, including 148 phylotypes. These sequences were assembled and scanned for the presence of open reading frames, which were then annotated and analyzed [6]. Overall, more than 1.2 million putative genes were identified, including 37,118 genes for amino acid biosynthesis. Tryptophan pathway genes should be widely represented among these sequences. A vast amount of information about the trp genes from various bacterial species exists in the literature and the Sargasso Sea metagenome data should contribute much to our knowledge of the evolution and organizational diversity of these important genes [7], in particular those from a marine environment. Marine bacteria live in an exacting environment that makes selective demands on its inhabitants-in quite a different way to the terrestrial environment.
We have made an extensive search for tryptophan pathway genes within the metagenome data. Our major goal was to determine whether the classical structure of the trp operon predominates in marine microorganisms and whether novel structures are present. This information should help us look at questions about the origin of the trp genes and the genetic and selective processes that have acted on them including their lateral transfer between different bacterial species
Results
Computer search for tryptophan pathway genes
Contigs and scaffolds from the Sargasso Sea metagenome were screened for trp genes. The search was run seven times, each using the amino acid sequence of a different Bacillus subtilis trp gene. Among contigs and scaffolds, we found 2,926 that had trp genes. Of these, 879 contained 2 or more trp genes and 2,047 contained only a single trp gene. After removing repeats resulting from sequences carrying several trp genes, we found 1,928 trp genes that were associated with at least one other trp gene, which makes it very likely that these are trp genes. A total of 4,009 trp-like genes were found but some of these might be pseudogenes. That is, a minimum of 5% of all the genes for amino acid biosynthesis (37,118 genes [6]) are trp-like genes
The gene order E-G-D-C-F-B-A was taken as the prototype for complete operons. For "split-operons", the prototypes used were E-G-D-C and F-B-A. Table 1 shows the distribution of the contigs for different trp genes. The assembly of important scaffolds and contigs (see Table 2) was verified by re-assembling their reads using the SEQUENCHER program version 4.1.2 by Gene Codes Corporation (Ann Arbor, MI, USA). The resulting assembly was found to be consistent with that previously generated by the Celera Assembler [6] The amount of coverage gives an estimate of the frequency of a contig within the population of organisms sampled and was determined for each contig. The results of this search are presented in Table 2. Full and split operons with a classical structure are widely represented.
Table 1.
Distribution of trp gene appearances on scaffolds and contigs in the Sargasso metagenome
| Gene | Total number of copies* | With other trp genes† | Alone‡ |
|---|---|---|---|
| trpE | 663 | 277 | 386 |
| trpG | 826 | 396 | 430 |
| trpD | 426 | 278 | 148 |
| trpC | 382 | 153 | 229 |
| trpF | 378 | 235 | 143 |
| trpB | 892 | 408 | 484 |
| trpA | 442 | 215 | 227 |
| 4,009 | 879 | 2,047 |
* Total number of copies, number of occurrences of the gene in the Sargasso Sea metagenome. † With other trp genes, number of occurrences on scaffolds and contigs containing more than one trp gene. ‡ Alone, number of occurrences on scaffolds and contigs with no other trp genes
Table 2.
Coverage and gene order of different contigs and scaffolds
| Contig/Scaffold | Actual length* | Coverage† | Gene order‡ |
|---|---|---|---|
| AACY01037482 | 5934 | 10.81 | D→C→F→B→A |
| AACY01011678 | 5668 | 10.66 | Full operon |
| CH026811 | 14769 | 8.78 | Full operon |
| AACY01096779 | 10932 | 8.69 | E→G→D→C |
| AACY01096698 | 2822 | 8.51 | E→G→D→C |
| AACY01104100 | 6690 | 8.21 | E→G→D→C→B→A |
| AACY01008961 | 7081 | 7.36 | E→G→D→C |
| AACY01117014 | 7301 | 5.94 | E→G→D→C |
| AACY01092457 | 4603 | 4.45 | E→G→D→C |
| AACY01074747 | 3876 | 4.26 | E→G→PLPDE_IV |
| AACY01046473 | 3887 | 3.96 | E→G→D→C |
| AACY01056517 | 4373 | 3.85 | E→G→D→C |
| CH025535 | 76373 | 3.72 | E→G→D→C→F→B→X→A |
| AACY01039569 | 5041 | 3.45 | E→G→D→C |
| AACY01065695 | 3747 | 3.37 | E→G→D→C |
| AACY01088195 | 7958 | 3.27 | E→G→D→C |
| CH020599 | 17648 | 3.18 | G→D→C→F |
| AACY01010663 | 3644 | 3.17 | E→G→D→C |
| CH006047 | 9399 | 3.03 | Full operon |
| AACY01056487 | 4038 | 2.91 | E→G→D→C |
| CH025058 | 36,150 | 2.69 | B→A→E→G→D→C |
| CH025585 | 10777 | 2.59 | Full operon |
| CH006071 | 68188 | 2.53 | Full operon |
| AACY01110889 | 4437 | 2.43 | F→(EG) |
| AACY01063516 | 4094 | 2.35 | E→G→D→C |
| AACY01027084 | 3981 | 2.21 | D→C→F→B→A |
| AACY01064621 | 5161 | 2.02 | E→G→D→C |
| AACY01052709 | 2451 | 2.00 | E→G→D→C |
| AACY01079380 | 1515 | 1.89 | G→C |
| AACY01015506 | 2202 | 1.35 | E→G→D→C |
| CH200199 | 1879 | 1.00 | E→G→D→C |
| CH199785 | 1823 | 1.00 | E→G→D→C |
| CH174161 | 1722 | 1.00 | E→G→D→C |
*Actual length, number of known nucleotides; †Coverage, average number of reads covering each nucleotide; ‡Gene order, of different contigs and scaffolds.
Table 1 also gives the results for each separate gene. It shows that different genes are not represented with equal frequency: trpE, trpG and trpB are over-represented. A possible explanation for this is that trpE and trpG homologues take part in other biochemical pathways such as the pathway for para-amino benzoic acid [8] and have been incorrectly identified as trp genes.
A computer search of this type cannot determine the actual enzymatic activity of a particular coding region and this can lead to an over-representation of certain genes. An analysis of the trpG and pabA genes, which are almost certainly derived from a common source, showed that these cannot be distinguished from one another unless they are associated with an adjacent trp gene (for trpG) or a pab gene (for pabA). In the cases where there is no ambiguity as to their identity, it was found that these two genes from the same organism were often more closely related than when they were compared to their counterparts in other organisms (data not shown). An analysis of the trpE and pabB genes, which also have a common origin, gave similar results. Gene duplication could also cause an apparent over-representation and this is discussed below in reference to the occurrence of the two kinds of trpB genes. Genes that encode enzymes that act in more than one pathway and catalyze similar reactions can either appear in searches done on two different pathways or not appear in either search. An example of this phenomenon is the trpF gene, which is discussed below.
In order to determine the extent of coverage by this search method, an analysis of the trpE, trpD and trpA genes was made using the genes from the ten different organisms listed in Table 3 as probes. The results of these searches for trpD and trpA are shown in Table 3.
Table 3.
Search for trpD and trpA genes using multiple probes
| Species and strain* | matches† | both‡ | probe only§ | Bacillus only¶ | % new¥ |
|---|---|---|---|---|---|
| trpD | |||||
| Sulfolobus solfataricus P2 | 454 | 444 | 10 | 24 | 2 |
| Thermoplasma acidophilum DSM 1728 | 409 | 404 | 5 | 64 | 1 |
| Nostoc sp. PCC 7120 | 436 | 430 | 6 | 38 | 1 |
| Thermoanaerobacter tengcongensis MB4 | 493 | 467 | 26 | 1 | 6 |
| Rhodopirellula baltica SH 1 | 448 | 442 | 6 | 26 | 1 |
| Bacteroides fragilis NCTC 9343 | 424 | 419 | 5 | 49 | 1 |
| Corynebacterium jeikeium K411 | 443 | 433 | 10 | 35 | 2 |
| Methanosphaera stadtmanae DSM 3091 | 441 | 433 | 8 | 35 | 2 |
| Neisseria meningitidis FAM18 | 474 | 458 | 16 | 10 | 3 |
| Clostridium kluyveri DSM 555 | 492 | 464 | 28 | 4 | 6 |
| All# | 514 | 468 | 46 | 0 | 10 |
| trpA | |||||
| Sulfolobus solfataricus P2 | 222 | 222 | 0 | 241 | 0 |
| Nostoc sp. PCC 7120 | 471 | 445 | 26 | 18 | 6 |
| Pseudomonas putida KT2440 | 498 | 457 | 41 | 6 | 9 |
| Rhodopirellula baltica SH1 | 478 | 456 | 22 | 7 | 5 |
| Corynebacterium jeikeium K4111 | 463 | 432 | 31 | 31 | 7 |
| Bacteroides fragilis NCTC 9343 | 437 | 431 | 6 | 32 | 1 |
| Clostridium kluyveri DSM 555 | 475 | 443 | 32 | 20 | 7 |
| Thermoplasma acidophilum DSM 1728 | 25 | 25 | 0 | 438 | 0 |
| Neisseria meningitidis 053442 | 479 | 452 | 27 | 11 | 6 |
| Leptospira biflexa serovar Patoc | 474 | 451 | 23 | 12 | 5 |
| All# | 517 | 463 | 54 | 0 | 12 |
* Species and strain, those used to probe the database † Matches, number of genes detected using the specific probe ‡ Both, genes detected by both the specific probe and that from Bacillus; § Probe only, those sequences detected by the specific probe but not by that from Bacillus ¶ Bacillus only, those sequences detected by the Bacillus probe but not by the specific probe ¥ % new, per cent of new sequences not detected by the Bacillus probe # All, the total number of sequences found by all probes; those that were common to Bacillus and one or more of the specific probes; the number of genes found with specific probes but not by that from Bacillus (new sequences); those found by the Bacillus probe but not by the others; the per cent of new sequences, that is the number of new sequences divided by the number of Bacillus sequences times 100. The data given in the table are raw data without the elimination of sequences that are somewhat doubtful because in this table we are trying to maximally expand the search parameters.
The analysis of trpE sequences is complicated by the concomitant detection of pabB sequences. New trpE sequences were uncovered and these usually represent about 10% of those detected using the Bacillus probe. Using probes of ten species to search for trpD led to the discovery of an average of about 3% for each probe. However as many of the new genes will appear in more than one search, only an additional 10% (46/468) of new trpD genes were found in toto. Table 3 also presents the data for trpA, another gene for which little ambiguity is anticipated. That search again led to the discovery of new genes (an average of 4.5% per search) but again the total of new trpA genes from the ten probes was only 12% (54/463). Therefore, the coverage provided by the Bacillus probes, while not complete, renders a fairly accurate picture of the trp genes in the Sargasso Sea metagenome database. We would expect that using more and more probes would be subject to the law of diminishing returns.
Operon structures
Table 4 summarizes the number of scaffolds and contigs that contain several trp genes. Some scaffolds have all seven trp genes grouped together. The descriptions of several scaffolds of particular interest are presented in Table 5. Eleven of the 24 scaffolds and contigs containing 4 trp genes were lacking flanking sequences, and therefore could not be considered as split operons. The other 13 had genes unrelated to the trp operon on both ends, or at least after the trpC gene (for split operons of the EGDC type), and therefore fit the definition of split operons. In the 61 scaffolds and contigs that have three genes together, only 16 contain trp genes flanked by those that are unrelated and can be unambiguously denoted as split-operons. The following previously described split-operons were found: E→G→D→C, F→B→A, F→B→X→A. Calculations of frequencies of gene pairs (Figure 2) hint that the first two split operons are the most abundant within the Sargasso Sea metagenome, while other organizations, including the classical full operon, are much less abundant. This conclusion may be supported by the very few C→F pairs that have been found.
Table 4.
Number of contigs and scaffolds containing multiple trp genes
| No. of trp genes | No. of contigs and scaffolds |
|---|---|
| 7 | 8 |
| 6 | 3 |
| 5 | 3 |
| 4 | 24 |
| 3 | 61 |
| 2 | 780 |
| 1 | 2,046 |
Table 5.
Description of selected scaffolds
| Scaffold | No of trp genes in the scaffold | Gene order | Comments |
|---|---|---|---|
| CH027495 | 6 | EGD(CF)B | Lack of trpA gene Gap of unsequenced DNA between trpB and those genes that are unrelated to trp genes may contain gene trpA. |
| CH027608 | 5 | DCFBA | Lack of trpE and trpG genes. However, the region between trpD and genes unrelated to trp is missing. |
| CH011919 | 5 | EGDCBA | Lack of a trpF gene There is a gap in the sequence between two neighboring contigs that contain E-G-D-C on the one hand and B-A on the other. Until the connecting pieces are found in both these cases, no decision can be made as to whether the missing genes are separate from the other trp genes. |
| CH005689 | 5 | EGDFB | Lacks both trpC and trpA. While the absence of trpC is not in doubt because trpD is adjacent to trpF, and on the same contig, trpA is probably missing due to the incompleteness of the sequence. |
| CH026313 | 4 | DCFB | Lack of trpE trpG and trpA genes. Not definite that this is a split operon because of gaps between trpD/trpB and their neighboring genes. Moreover the gap between trpD and trpC challenge the correctness of assembly |
| AACY01051805 AACY01049273 | 7 | EGDCFBA | Shewanella oneidensis, SAR-1 and SAR-2 |
| CH004526 CH004459 | Split operon: 4 and 3 | EGDC FBXA | One interesting feature of the trp genes of Burkholderia SAR-1 should be mentioned: in all previously known genomes of Burkholderia sp., the split-operons contain F→B→X→A where "X" is unrelated to known trp genes. The sequence from the Sargasso Sea metagenome of SAR-1 Burkholderia-like sequences contains an F→X→A split operon. The computer program used by Venter and colleagues failed to identify a trpB gene within the sequence. However when a search was made using the Burkholderia trpB sequence as a probe, a trpB gene was detected between trpF and X, as is true for all other Burkholderia species and there were no non-trp genes between trpF and trpB. |
Figure 2.

Distribution of neighboring genes involving at least one trp gene. (a) Each arrow connects neighboring genes, its size and color represents number of pairs found in the Sargasso metagenome (see legend, only pairs observed more than 30 times are shown). Pairs of genes composing the two split operons E→G→D→C and F→B→A are abundant while the pair C→F was rarely found. This may hint that the trp genes are usually organized as split operons rather than as full operons. (b) The representation of classical full and split trp operons.
As illustrated in Figure 3, most of the complete and incomplete trp gene clusters maintain the structure of the prototype trp operon. All genes within these clusters have the same direction of transcription and the same gene order. Two of the split operons, [GenBank: AACY01080023] and [GenBank: AACY01120345], seem to be from the genome of Burkholderia SAR-1, while two full operons described in Table 5 seem to come from Shewanella SAR 1 and 2. As the sequences of these do not differ from those found earlier for those organism and the probable source of these is a filter contamination as has been stated in several papers [9,10] they were not taken into account in our calculations.
Figure 3.

Alignment of trp sequences from different contigs and scaffolds. The following abbreviations are used: E, trpE; G, trpG (or sequences with a high similarity to pabA); C, trpC; D, trpD; F, trpF; B, trpB; A, trpA; Unk, an ORF with unknown function; truA, the tRNA pseudouridine synthase; moaC, a protein related to the molybdenum cofactor; SSL22, DNA or RNA helicases of superfamily II; lexA, the SOS-response transcriptional repressor.
Two contigs show a different type of organization than that generally found in bacteria. In one contig [GenBank: AACY01110889] trpF is followed by a gene that is a fusion between trpE and trpG. This contig is a part of a scaffold, [GenBank: CH022404], which shows no similarity to any known bacterium with regard to trpE and trpG. While the fusion of trpG and trpE has been found in bacteria such as Legionella pneumophila, Rhodopseudomonas palustris, Thermomonospora fusca, Anabaena sp. and Nostoc punctiforme, none of them contain the gene order F-(E-G). However, the gene order trpF-trpE-trpG has been found in some Archaea such as Halobacterium sp., Methanosarcina barkeri and Ferroplasma acidarmanus, but in these species trpE and trpG are separate genes. In a second contig [GenBank: AACY01079380] the gene order trpG-trpC has been observed. This gene order has already been described for Archaea such as Thermoplasma acidophilum, Thermoplasma volcanium, Ferroplasma acidarmanus and Sulfolobus solfataricus [4].
The order of adjacent trp genes within two scaffolds, [GenBank: CH025058] (gene order: B-A-E-G-D-C) and [GenBank: AACY01110889] (gene order: F-(EG)) are entirely novel and have not been observed to date. Both have a relatively high coverage in the database, which confirms the importance and abundance of these gene orders in marine populations. An analysis of other, non-trp genes within these scaffolds failed to reveal any significant similarity between them and known genomes.
A phylogenetic analysis of some of these complete and split operons was made against operons from known organisms. The results are presented in Figure 4. All the full operons are much more related to the full operons of known organisms than they are to the split operons of other known species. The figure also shows that most of the split operons are grouped with split operons from known organisms. The four exceptions to this rule are probably due to incomplete sequences and these are likely to be full operons. This analysis also supports our hypothesis that split operons are more prevalent than full operons (Figure 2) in the Sargasso Sea metagenome
Figure 4.
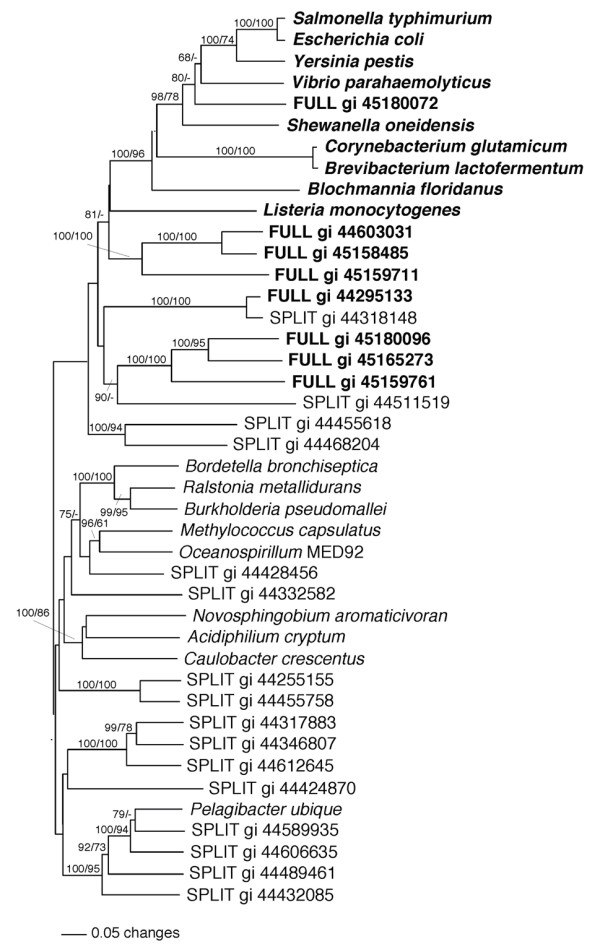
Phylogenetic analysis of scaffolds and contigs containing whole and complete operons. The concatenated amino acid sequences from genes trpE, trpG, trpD, and trpC were used to analyze the relationships among both known species and those from the Sargasso Sea metagenome. Full operons are written in bold whereas split operons are not.
Non-operon organization
As shown in Table 4, 70% of the contigs and scaffolds detected have a single trp gene. Those with two trp genes are also very prevalent (26%) even though some of these are probably partial segments of larger operons. As shown in Table 6, 133 scaffolds and contigs carry one or two trp genes enclosed between non-trp genes. While trpE and trpG may be overrepresented due to the existence of homologous genes as mentioned above, other trp genes are also observed in a "detached" manner. This indicates that the trp genes of marine organisms are frequently detached or occur as pairs.
Table 6.
Frequency of scaffolds and contigs containing unusual organizations of trp genes.
| Gene order (enclosed)* | No of occurences† | Gene order (partial) ‡ | No of occurences |
|---|---|---|---|
| X→E→X | 16 | E→X | 55 |
| X→G→X | 42 | X→G | 88 |
| X→D→X | 3 | G→X | 108 |
| X→C→X | 5 | X→D | 16 |
| X→F→X | 2 | D→X | 8 |
| X→B→X | 7 | X→C | 16 |
| X→A→X | 5 | F→X | 6 |
| X→B→A(→X) | 9 | X→B | 69 |
| (X→) E→G→X | 44 | B→X | 49 |
| X→A | 16 | ||
| Total | 133 | Total | 431 |
* Gene order (enclosed), organizations of one and two trp operons enclosed between non-trp genes; † Number of occurrences, number of contigs and scaffolds carrying the organization; ‡ Gene order (partial), pairs of trp and non-trp genes that are inconsistent with classical organization.
The existence of pairs of trp genes makes good sense biochemically. Anthranilate synthase is composed of an equal number of trpE and trpD encoded subunits. Tryptophan synthase contains two subunits each of the polypeptides from the trpA and trpB genes. The trpG when unfused to trpE or trpD leads to a polypeptide also found in equimolar amounts to those from trpE and trpD. Organizing these specific genes in pairs would seem to ensure that they are transcribed together and render the proper amounts of the translation products.
The occurrence of detached trp genes is apparently an adaptation to the particular environment in which marine organisms are found. Most of the bacteria previously analyzed probably encounter periods of feast and famine with regard to tryptophan. Therefore they need to respond to external conditions that vary. The existence of transport systems for concentrating externally found tryptophan and the organization of the trp biosynthetic genes into operons almost certainly reflect their environmental challenges. In contrast, marine organisms exist in a rather constant environment with respect to tryptophan. It is unlikely that tryptophan from external sources is available and this amino acid must be synthesized entirely within the bacterial cell. The main regulation of the pathway is expected to be at the level of feedback inhibition and it is probable that trp gene expression is constitutive rather than controlled by the mechanism of repression-derepression. The level of expression of a detached trp gene can be controlled simply by modifying the strength of the associated promoter. A trp repressor or repressors and attenuation become superfluous under such circumstances. This should extend to most or all of the other genes involved in amino acid biosynthesis. Therefore axenic cultures of some of these marine organisms are eagerly awaited.
Conserved non-trp flanking genes
Another way of examining the evolution of the trp genes and the relationships between various species is the analysis of genes not involved in tryptophan biosynthesis that either neighbor the trp genes or are inserted between them. Xie and colleagues have reported that trpF, trpB and trpA in split-pathway operons are flanked by conserved genes that are unrelated to tryptophan biosynthesis [4]. They have found genes that encode the β-subunit of acetyl-coenzymeA-carboxylase (accD), folylpolyglutamate synthase/dihydrofolate synthase (folC), fimbria V protein (lysM) and the tRNA pseudouridine synthase (truA). In most cases the genes accD and folC follow trpA. For the Thiobacillus-Pseudomonas-Azotobacter cluster and others, the trpF-trpB-trpA operon is flanked on the trpF side by lysM and truA. The presence of particular genes appearing near those of trp was examined using the Sargasso Sea metagenome data and the results of this analysis are shown in Table 7.
Table 7.
Genes flanking the trp operon
| Gene | Number of times in the metagenome | Percent found near trp genes |
|---|---|---|
| TruA | 30 | 93% are adjacent and before trpF |
| AccD | 53 | 86.8% are adjacent and after trpA 9.4% are adjacent and after trpB; trpA is elsewhere 3.8% are adjacent and after trpF; trpB and A are absent |
| FolC | 13 | 77% occur as trpA-accD-folC 23% occur in the order of trpB-accD-folC |
| PyrF | 60 | 77% are before trpF in split operons 23% are before trpB |
| LexA | 92 | 100% are adjacent and after trpC when trpF is elsewhere |
| MoaC | 25 | 100% neighbor and are after trpC |
| PLPDE_IV | 21 | 57% adjacent and after trpE 38% adjacent and after trpG |
The first three rows of Table 7 confirm previous publications. In addition, four other genes, not previously noted, were found with high frequencies near the trp genes of the Sargasso Sea metagenome: pyrF (orotidine-5'-phosphate decarboxylase), lexA (the SOS-response transcriptional repressor), moaC (a protein related to the molybdenum cofactor) and PLPDE_IV (the class of amino acid aminotransferases). It should be mentioned that PLPDE_IV is the only gene, besides aroG and aroH (see below), found near the trp genes that can be logically connected to tryptophan biosynthesis. This class of amino-transferases includes some D-amino acid transferases, pyridoxal-5-phosphate-dependent enzymes such as tryptophanase, and others. If in fact the cell is able to use D-tryptophan as a source of L-tryptophan via a D-amino acid transferase, then the inclusion of a gene encoding such an activity among the trp genes would make sense as this gene would undergo derepression in coordination with those involved in L-tryptophan biosynthesis.
It is clear that specific neighboring genes are very prevalent when a split trp operon occurs. It seems unlikely that the same event has occurred many times: strains with these particular flanking genes are most likely derived from a common ancestor.
Analysis of trpB genes
Surprisingly, it has been found that a significant number of organisms possess more than one trpB gene encoding the β-chain of tryptophan synthase. Usually, but not always, the 'extra' gene is unlinked to the trpA gene encoding the α chain of this enzyme. These extra trpB genes belong to a distinct subgroup encoding the β-chain which is termed trpB_2. This had been recognized in the COGs database as "alternative tryptophan synthase" - COG1350 [11] while the major group is denoted as trpB_1 and includes the well-studied polypeptides from such organisms as Escherichia coli, Salmonella typhimurium and Bacillus subtilis. The minor trpB_2 group includes mostly, but not exclusively, archaeal species. The evolution and properties of trpB_2, have been analyzed and discussed in a number of recent articles [12-15].
The 3-dimensional structure of tryptophan synthase from Salmonella typhimurium has been elucidated by X-ray crystallography to a resolution of 2.5 angstroms [16]. The enzyme is a αββα complex which forms an internal hydrophobic tunnel into which indole, produced by the a subunit, enters and then reaches the active site of the b subunit. The α monomers and β dimers contact one another via a highly specific mechanism of recognition. In addition, the genes encoding these two subunits are almost always closely linked and their expression is frequently translationally coupled [17,18].
The data collected from the Sargasso Sea metagenome were examined to determine whether the trpB sequences from the Sargasso Sea differ from those of known organisms and whether both trpB_1 and trpB_2 exist in this sample. When a phylogenetic analysis of trpB genes found in the present survey was conducted, it was found that the majority of these (Figure 5) fall into the trpB_1 group while a few trpB_2 genes also occur. Among the trpB_1 genes, one cluster is quite distinct and probably split off from major type at a relatively early stage. Genes in this cluster have a high similarity to the marine bacterium Pelagibacter ubique (Candidatus) HTCC1062 (SAR11) and the sequence identity of these to P. ubique at the amino acid level was between 64% and 87% while the genes neighboring some of these trpBs showed an even higher identity to their counterparts from SAR11. One of the most remarkable features of P. ubique is its extremely small genome that lacks any pseudogenes or recent gene duplications. It has only one copy of trpB, and therefore it can be concluded that this gene must be functional in tryptophan biosynthesis and not a pseudogene. P. ubique contains two split operons: trpE-trpG-trpD-trpC and trpF-trpB-trpA. The gene order of the neighboring, non-related trp genes of the second split operon is: (gene not mentioned above) himD-pyrF-trpF-trpB-trpA-accD-folC. The himD gene encodes a sequence-specific DNA-binding transcriptional activator. Comparison of the gene order between contigs containing SAR11-like trpB from the Sargasso Sea metagenome showed that most of the contigs have a gene order that is similar to SAR11. Three of 37 contigs lack trpF and 2 contigs contain only a trpB gene flanked by genes unrelated to trp and which are similar in sequence and order to that of SAR11. This indicates that most or all of these trpB genes are part of the SAR11 group. Since the trpB of SAR11 is more closely related to trpB_1 than to trpB_2 [19], it seems that the genes from this particular cluster should probably be considered to be of the trpB_1 type.
Figure 5.

Representation of Sargasso metagenome trpB sequences and those from known bacteria with respect to genetic distance. 40 representatives from trpB sequences analyzed here were chosen for this analysis. As can be seen, the constructed tree shows two distinct groups; however a third group appears which consists of only environmental sequences and the Ple (Pelagibacter ubique (Candidatus)) sequence. The abbreviation of trpB genes from known bacteria are listed in Table 8. For the environmental trpB sequences abbreviation the NCBI accession numbers were taken. Bootstraps for the main groups are shown.
Discussion
The tryptophan operon of bacteria has been studied for more than 50 years and its structure and regulation are known for many terrestrial organisms that can be grown in laboratory culture. With the explosive expansion of genomics during the last decade and the data thus generated, many trp sequences from both known and unknown marine species have become available. This provides an excellent opportunity for expanding our knowledge about the ways in which different organisms, particularly marine bacteria, have organized these genes. In the present research, trp pathway genes within the Sargasso Sea database were retrieved by BLAST analysis using known trp protein sequences. It was found that trp genes account for about 5% of all genes that were previously identified as genes for amino acid synthesis in the Sargasso Sea metagenome. In almost all cases in which the trp genes form an operon, the order and direction of transcription of the trp genes are similar to familiar prototypes. The reason for this conservation remains unknown. This might be explained in part by an advantage conferred when genes whose products form complexes are adjacent to one another and translational coupling occurs. Of the 85 contigs and scaffolds that contain three or four trp genes, only 29 could be unambiguously defined as containing split pathway operons. The following already known orders of split operons were found: E→G→D→C, F→B→A. In addition, we have found evidence for completely dispersed trp genes in the form of isolated and pairs of genes.
Since these marine organisms survive and grow in a very different environment from those organisms previously studied, they are likely to have been genetically separated from them and to have evolved to solve the particular regulatory problems that exist in their environment. It was expected that some marine bacteria would exhibit novel organizations of these genes and such organizations were in fact found. Among the trp genes organized into operon structures, most resemble examples already discovered. In addition, two previously unknown groupings were uncovered in the present search. However a notable quantity of genes that were either detached or in mini-operons containing only two trp genes was discovered. Novel organizations of the trp genes probably arise from adaptations to the marine environment and it is likely that some marine bacteria will have unusual regulatory features. Such features can only be elucidated when these organisms become amenable to axenic culture. Cloning and expressing these genes in the laboratory from those organisms that cannot yet be cultured may however provide a partial regulatory picture. In this regard, a search for genes related to the trpR gene of Escherichia coli (the gene that encodes the tryptophan repressor) in the Sargasso Sea metagenome was performed. This search failed to reveal any significant trpR homologs. This is not surprising, because regulatory circuits undoubtedly arise later than the genes for biosynthesis and are adaptations to specific environments.
Genes with unknown function have been previously found to be inserted within the trp operon [4]. Such genes were found between trpB and trpA in one contig from the Sargasso Sea metagenome, a location already observed for some species of Flavobacterium and Burkholderia. Another contig carried such a gene between trpF and trpB. While the reason for the presence of these non-trp genes is unclear and the possibility exists that they are simply morons [20], it is possible that they actually participate in tryptophan biosynthesis. That is, these genes may not be essential for tryptophan synthesis but rather aid it by increasing the catalysis of one of the enzymes or by being involved in complex formation. Even a very small advantage is expected to be of great importance for the survival of an organism in an oligotrophic environment such as that of the Sargasso Sea.
One should keep in mind that the arrangement of genes in operon confers both advantages and disadvantages. The most obvious advantage is that genes with similar function are transcribed together. The greatest disadvantage is that, unless some further level of regulation exists (differences in the amounts of mRNA or its stability, the strength of ribosomal binding sites, and so on), the amount of the polypeptides from these genes will be the same even though the resultant enzymes may have different catalytic rates [21]. The ones with slower rates will be the limiting factor. As a result, when the genes are transcribed together, an excess of some enzymes is likely to occur. However, the amount of mRNA and polypeptide synthesis is only one aspect of the control of the tryptophan pathway. Besides these, there are two other levels of control that affect the amount of tryptophan synthesis within the cell. The first of these is feed-back inhibition which influences the activity of the first two reactions [22], and thereby the amount of metabolites flowing through the pathway. The second is the formation of multi-enzyme complexes that greatly increases the catalytic efficiency of the various reactions. In complexes, the product of one reaction can be used directly by the next enzyme and the concentration of the substrate in the vicinity of the second enzyme is much higher than would occur were the two enzymes separate. Examples of such complexes are trpE-trpD (trpG) and trpA-trpB and the trpC-trpF gene fusion in Escherichia coli. In addition, one polypeptide can greatly enhance the activity of a second when a complex is formed (for example, in the trpA-trpB heterotetramer, αββα from Escherichia coli [23-25].
Different solutions to the problems of optimal synthesis of tryptophan and the regulation of trp gene expression would not be surprising since this amino acid is one of the most expensive in chemical terms. One solution might be to organize the trp genes in a different manner; another would be the creation of trp gene fusions. Both of these have been observed. Our analysis uncovered some known gene fusions, E-G [GenBank: AACY01100727] and C-F [GenBank: AACY01022048] and two novel fusion of a trp gene with a gene unrelated to the trp genes: E-PLPDE_IV [GenBank: AACY01077237] and F-TruA [GenBank: AACY01600616]. All of above indicate that there is quite a lot of genetic diversity among marine bacteria.
It was found that several specific genes are often neighbors of the trp genes of marine microorganisms. When present in contigs, lexA, pyrF and moaC were always placed after trpC. This may be a general phenomenon but our information is still too scanty to allow a definite conclusion to be drawn. Similarity in gene order is usually taken to indicate an evolutionary relationship between such segments. Of particular interest was the observation that in 3 cases aroH or aroG occur adjacent to trpA. For these examples, the distance between the end of trpA and the ensuing aro gene is 3, 18, or 20 base pairs, which makes it very likely that the two genes are expressed together. The synthesis and activity of the enzyme they encode, DAHP synthase, is involved in the synthesis of a precursor of chorismic acid and this aro gene is often regulated by the level of tryptophan. Therefore such an arrangement might make sense.
Since there is more than one kind of trpB gene, a comparison was made of amino acid sequences of trpB genes from the Sargasso Sea metagenome with those from known organisms. The majority of the metagenomic trpB sequences detected fall into the trpB_1 group while some others were related to the trpB_2 group. One cluster containing a number of trpB_1 sequences is quite distant from the usual type and has a high similarity to that of Pelagibacter ubique (Candidatus) HTCC1062 (SAR11). This cluster probably diverged rather early from the major trpB_1 line.
Conclusion
The present analysis has revealed that tryptophan genes are rather frequent within the Sargasso Sea metagenome. All trp genes that were found have enough similarity to COGs to be recognized. This seems to indicate, but does not prove, that all have come from a common ancestor. However, additional genes for tryptophan biosynthesis may exist which we were unable to detect with the probes employed. In this regard, it has been reported [26] that some organisms indeed lack a recognizable trpF in their genomes but are capable of growing without external tryptophan. A gene whose sequence is not homologous to known trpFs but whose product catalyzes this reaction has in fact been found in Streptomyces coelicolor A3 and Mycobacterium tuberculosis HR37Rv [26]. This trpF gene is an example of reticulate evolution because it can catalyze reactions in both the histidine and tryptophan pathways [27,28]. A BLAST search with the amino acid sequence of the trpF gene from Streptomyces coelicolor A3 gene (SCO2050) against the Sargasso Sea metagenome data showed more than 500 hits that can be identified as hisA proteins. Thus, only a functional analysis of these environmental sequences can prove whether they can take part in both pathways or not. The fact that a group of marine trpB_1 sequences are similar to one another but quite distant from the major trpB_1 group supports the idea that there may be trp genes that are not recognized as such by those sequences presently known.
While trp operons, both complete and split, exist in marine bacteria, many trp genes are no longer found in that framework. In contrast to most terrestrial bacteria, the operon structure is not used for the trp genes in some of marine origin. There are mini-operons of 2 genes in many cases (Table 5) and also an even more frequent occurrence of single trp genes. It is of course an open question whether what we observe is the result of the breakup of an original operon structure or that the trp operons at present have arisen from these unlinked genes. Since the marine environment is very exacting and selective, it is certain that organisms lacking an operon structure for the trp genes have found an evolutionary advantage in the organization of the trp genes that they possess. It should be mentioned that in Escherichia coli and Salmonella, about 50% of the genes encoding polypeptides involved in amino acid synthesis are separate although their trp genes are not. On the basis of our results in which novel trp gene orders were found, it appears likely that further studies of the trp genes and their regulation and organization will provide many future surprises.
Materials and methods
Analysis of Sargasso Sea metagenome database
Amino acid sequences with homology to each trp catalytic domain were obtained from an NCBI BLAST search of the Sargasso Sea metagenome database [29]. The amino acid sequences from Bacillus subtilis of each pathway catalytic domain were used as query entries for protein BLAST. Bacillus proteins were chosen as a starting point for the search because the catalytic domains are encoded by separate genes. In Bacillus six genes, except trpG, are organized into one operon and have been intensively studied at the level of DNA, RNA and protein levels [30-32]. For the trpB_2 search, the sequence of Chlorobium tepidum CT0192 (Q8KF11) was used. The list of trp genes has been generated in several steps. First, BLAST searches of trp genes against the Sargasso Sea metagenome has been performed, using an e-value threshold of 1e-5. For cross validation, both peptide and DNA sequence databases were searched and the results were compared. While 95% of the ORFs were identified in both searches, some were discovered only once. In such cases a manual check of the results has been performed. In addition, genes that are homologous to trp genes (PabA, PabB, PhzA and PhzB) were used to remove misclassified trpE and trpG genes. As a result, a list of contigs containing trp genes was created. Redundant contigs were removed based on BLAST searches with a 95% identity threshold. In the last step, contigs that belong to the same scaffolds were identified and treated. The results of the above semi-automatic process were validated by large-scale manual examinations.
In order to assemble a contig, Venter and colleagues used the Celera Assembler [6]. To validate the Sargasso Sea scaffolds the following procedure was performed. First, all singleton reads composing each scaffold were retrieved by conducting a BLASTn search for the scaffolds against the Sargasso Sea reads. Next, the SEQUENCHER program (Gene Codes Corporation) was used for re-assembling the reads and the results were compared to each original scaffold for validation. No significant differences between the assemblies of the Celera Assembler and SEQUENCHER were found.
Coverage was calculated by recruiting reads from Sargasso Sea using BLAST, considering only reads with 90% and higher identity to the scaffold and at least 80% of the read taking part in the alignment. These parameters are rather stringent, but give a good indication with respect to the distribution of each scaffold.
Phylogenetic analysis
Amino acid sequences of many trpB genes were used to analyze the phylogenetic relationships between different environmental samples. Only genes encoding more than 251 amino acids were analyzed. The alignment was done using the ClustalW program [33]. Neighbor joining (NJ) and maximum parsimony (MP) analyses were conducted on protein data sets using version 4.0b10 of PAUP [34]. Default parameters were used in all analyses. Bootstrap resampling of NJ (1000 replicates) and MP (1000 replicates) trees were performed in all analyses to evaluate the reliability of the inferred topologies. The resultant trees were viewed through the TreeView (Win32) program [35]. To understand the relationship between the sub-families each was analyzed both by comparing one group against the others and to representative trpB gene sequences that exist in the NCBI database.
Abbreviations
MP, Maximum Parsimony analysis; NJ, Neighbor Joining analysis; ORF, Open Reading Frame; SSM, Sargasso Sea Metagenome; trp, Tryptophan. Additionally Table 8 lists the species names used and their abbreviations
Table 8.
List of species names and their abbreviations
| Species name | Abbreviation used | NCBI number |
|---|---|---|
| Aeropyrum pernix | Ape-1 | Q9Y8T5 |
| Aeropyrum pernix | Ape-2 | Q9Y9H2 |
| Aquifex aeolicus | Aae | O67409 |
| Arabidopsis thaliana | Ath-1 | P14671 |
| Arabidopsis thaliana | Ath-2 | BAB10143 |
| Archaeoglobus fulgidus | Afu-1 | O28672 |
| Archaeoglobus fulgidus | Afu-2 | O29028 |
| Bordetella pertussis | Bpe | NP_882102 |
| Campylobacter jejuni | Cje | CAL34499 |
| Pelagibacter ubique (Candidatus) HTCC1062 | Ple | YP_265913 |
| Chlamydia psittaci | Cps | Q822W9 |
| Chlorobium tepidum | Cte | Q8KF11 |
| Corynebacterium diphtheriae | Cdip-1 | NP_940652 |
| Corynebacterium diphtheriae | Cdip-2 | NP_940660 |
| Escherichia coli | Eco | P0A879 |
| Geobacter sulfurreducens | Gsu | AAT73768 |
| Haemophilus influenzae | Hin | P43760 |
| Lactococcus lactis | Lla | Q01998 |
| Legionella pneumophila | Lpn | CAH15507 |
| Mesorhizobium loti | Mlo | NP_105798 |
| Methanobacterium thermoautotrophicum | Mth-1 | O27696 |
| Methanobacterium thermoautotrophicum | Mth-2 | O27520 |
| Methanococcus jannaschii | Mja | Q60179 |
| Methanosarcina barkeri | Mba | AAZ72487 |
| Mycobacterium bovis | Mbo | NP_855291 |
| Mycobacterium tuberculosis | Mtu | P66984 |
| Neisseria gonorrhoeae | Ngo | Q84GJ9 |
| Neisseria meningitides | Nme | AAF41116 |
| Pyrobaculum aerophilum | Paero | Q8ZV44 |
| Pyrococcus abyssi | Pab-2 | Q9V150 |
| Pyrococcus furiosus | Pfu-2 | Q8U0J5 |
| Pyrococcus horikoshii | Pho | NP_143439 |
| Rhodopseudomonas palustris | Rpa-1 | YP_779393 |
| Salmonella typhimurium | Sty | NP_460685 |
| Staphylococcus aureus | Sau | BAB42464 |
| Streptococcus mutans | Smu | NP_720974 |
| Sulfolobus solfataricus | Sso-1 | P50383 |
| Sulfolobus solfataricus | Sso-2 | AAK41396 |
| Thermomonospora fusca | Tfu | YP_289226 |
| Thermoplasma acidophilum | Tac | Q9HKD2 |
| Thermoplasma volcanium | Tvo | NP_111450 |
| Thermotoga maritime | Tma | Q9WZ09 |
| Xylella fastidiosa | Xfa | C82688 |
| Zea mays | Zma-1 | P43284 |
| Zea mays | Zma-2 | P43283 |
Authors' contributions
JKa and JKu conceived the idea for this analysis and JKu contributed the main guidelines and concepts of the article. JKa performed manual check of the results and was responsible for data organization. IS performed bioinformatics involved in this study. OB performed the phylogenetic analysis. JKu and JKa prepared the initial manuscript. All authors participated in the analysis of the data. All authors have read and approved the final article.
Contributor Information
Juliana Kagan, Email: kjuliana@tx.technion.ac.il.
Itai Sharon, Email: itaish@cs.technion.ac.il.
Oded Beja, Email: beja@tx.technion.ac.il.
Jonathan C Kuhn, Email: jkuhn@tx.technion.ac.il.
Acknowledgements
We would like to extend our gratitude to Michael Shmoish for his help with the statistical analyses.
References
- Yanofsky C. Using studies of tryptophan metabolism to answer basic biology questions. J Biol Chem. 2003;278:10859–10878. doi: 10.1074/jbc.X200012200. [DOI] [PubMed] [Google Scholar]
- Li Y, Wexler M, Richardson DJ, Bond PL, Johnston AWB. Screening a wide host-range, waste-water metagenomic library in tryptophan auxotrophs of Rhizobium leguminosarum and of Escherichia coli reveals different classes of cloned trp genes. Environ Microbiol. 2005;7:1927–1936. doi: 10.1111/j.1462-2920.2005.00853.x. [DOI] [PubMed] [Google Scholar]
- Schwarz T, Uthoff K, Klinger C, Meyer HE, Bartholmes P, Kaufmann M. Multifunctional tryptophan-synthesizing enzyme the molecular weight of the Euglena Gracilis protein is unexpectedly low. Journal of Biological Chemistry. 1997;272:10616–10623. doi: 10.1074/jbc.272.16.10616. [DOI] [PubMed] [Google Scholar]
- Xie G, Keyhani NO, Bonner CA, Jensen RA. Ancient origin of the tryptophan operon and the dynamics of evolutionary change. Microbiol Mol Biol Rev. 2003;67:303–342. doi: 10.1128/MMBR.67.3.303-342.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Bonner CA, Song J, Keyhani NO, Jensen RA. Inter-genomic displacement via lateral gene transfer of bacterial trp operons in an overall context of vertical genealogy. BMC Biology. 2004;2:15. doi: 10.1186/1741-7007-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, Fouts DE, Levy S, Knap AH, Lomas MW, Nealson K, White O, Peterson J, Hoffman J, Parsons R, Baden-Tillson H, Pfannkoch C, Rogers YH, Smith HO. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Valera F. Environmental genomics, the big picture? FEMS Microbiol Lett. 2004;231:153–158. doi: 10.1016/S0378-1097(04)00006-0. [DOI] [PubMed] [Google Scholar]
- Crawford IP, Milkman R. In: Evolution at the molecular level. Selander RK, Clark AG, Whittam TS, editor. Sunderland, MA: Sinauer and Assoc Inc; 1991. Orthologous and paralogous divergence, reticulate evolution, and lateral gene transfer in bacterial trp genes. pp. 77–95. [Google Scholar]
- Falkowski PG, De Vargas C. Genomics and evolution. Shotgun sequencing in the sea: a blast from the past? Science. 2004;304:58–60. doi: 10.1126/science.1097146. [DOI] [PubMed] [Google Scholar]
- DeLong EF. Microbial community genomics in the ocean. Nat Rev Microbiol. 2005;3:459–469. doi: 10.1038/nrmicro1158. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Frost C, Bonner CA, Jensen RA. Significance of two distinct types of tryptophan synthase beta chain in bacteria, archaea and higher plants. Genome Biol. 2001;3:0004.0001–0004.0013. doi: 10.1186/gb-2001-3-1-research0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkl R. Modelling the evolution of the archeal tryptophan synthase. BMC Evolutionary Biology. 2007;7:59. doi: 10.1186/1471-2148-7-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopoldseder S, Hettwer S, Sterner R. Evolution of multi-enzyme complexes: the case of tryptophan synthase. Biochemistry. 2006;45:14111–14119. doi: 10.1021/bi061684b. [DOI] [PubMed] [Google Scholar]
- Hettwer S, Sterner R. A novel tryptophan synthase b-subunit from the hyperthermophile Thermotoga maritima. Quarternary structure, steady-state kinetics, and putative physiological role. J Biol Chem. 2002;277:8194–8201. doi: 10.1074/jbc.M111541200. [DOI] [PubMed] [Google Scholar]
- Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- Oppenheim DS, Yanofsky C. Translational coupling during expression of the tryptophan operon of Escherichia coli. Genetics. 1980;95:785–795. doi: 10.1093/genetics/95.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber-Ban E, Hur O, Bagwell C, Banik U, Yang LH, Miles EW, Dunn MF. Investigation of allosteric linkages in the regulation of tryptophan synthase: the roles of salt bridges and monovalent cations probed by site-directed mutation, optical spectroscopy, and kinetics. Biochemistry. 2001;40:3497–3511. doi: 10.1021/bi002690p. [DOI] [PubMed] [Google Scholar]
- Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, Bibbs L, Eads J, Richardson TH, Noordewier M, Rappe MS, Short JM, Carrington JC, Mathur EJ. Genome streamlining in a cosmopolitan oceanic bacterium. Science. 2005;309:1242–1245. doi: 10.1126/science.1114057. [DOI] [PubMed] [Google Scholar]
- Hendrix RW, Lawrence JG, Hatfull GF, Casjens S. The origins and ongoing evolution of viruses. Trends Microbiol. 2000;8:504–508. doi: 10.1016/S0966-842X(00)01863-1. [DOI] [PubMed] [Google Scholar]
- Glansdorff N. On the origin of operons and their possible role in evolution toward thermophily. J Mol Evol. 1999;49:432–438. doi: 10.1007/PL00006566. [DOI] [PubMed] [Google Scholar]
- Pabst MJ, Kuhn JC, Somerville RL. Feedback regulation in the anthranilate aggregate from wild type and mutant strains of Escherichia coli. J Biol Chem. 1973;248:901–914. [PubMed] [Google Scholar]
- Balbinder E. Intergeneric complementation between A and B components of bacterial tryptophan synthetases. Biochem and Biophys Res Commun. 1964;17:770–774. doi: 10.1016/0006-291X(64)90429-2. [DOI] [Google Scholar]
- Dicamelli RF, Balbinder E. The association of tryptophan synthetase subunits from Escherichia coli and Salmonella typhimurium in homologous and heterologous combinations. Genet Res. 1976;27:323–333. doi: 10.1017/s0016672300016517. [DOI] [PubMed] [Google Scholar]
- Dicamelli RF, Balbinder E, Lebowits J. Pressure effects on the association of the 2 subunits of tryptophan synthetase from Escherichia coli and Salmonella typhimurium. Arch Biochem and Biophys. 1973;155:315–324. doi: 10.1016/0003-9861(73)90120-3. [DOI] [PubMed] [Google Scholar]
- Barona-Gómez F, Hodgson DA. Occurrence of a putative ancient-like isomerase involved in histidine and tryptophan biosynthesis. EMBO reports. 2003;4:296–300. doi: 10.1038/sj.embor.embor771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford IP, Milkman R. In: Evolution at the molecular level. Selander RK, Clark AG, Whittam TS, editor. Sunderland, MA: Sinauer and Assoc Inc; 1991. Orthologous and paralogous divergence, reticulate evolution, and lateral gene transfer in bacterial trp genes. pp. 77–95. [Google Scholar]
- Kane JF, Holmes WM, Jensen RA. Metabolic interlock. The dual function of a folate pathway gene as an extra-operonic gene of tryptophan biosynthesis. J Biol Chem. 1972;247:1587–1596. [PubMed] [Google Scholar]
- BLAST x. NCBI database. http://www.ncbi.nlm.nih.gov/blast/ http://www.ncbi.nlm.nih.gov/blast/
- Losick R, Sonenshein AL. Molecular biology. Turning gene regulation on its head. Science. 2001;293:2018–2019. doi: 10.1126/science.1064982. [DOI] [PubMed] [Google Scholar]
- Yanofsky C. Advancing our knowledge in biochemistry, genetics, and microbiology through studies on tryptophan metabolism. Annu Rev Biochem. 2001;70:1–37. doi: 10.1146/annurev.biochem.70.1.1. [DOI] [PubMed] [Google Scholar]
- Tam le T, Eymann C, Antelmann H, Albrecht D, Hecker M. Global gene expression profiling of Bacillus subtilis in response to ammonium and tryptophan starvation as revealed by transcriptome and proteome analysis. J Mol Microbiol Biotechnol. 2007;12:121–130. doi: 10.1159/000096467. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acid Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. PAUP*. Phylogenetic analysis using parsimony (*and other methods) 4.0. Sunderland, Massachusetts, USA Sinauer Associates Incorporated; 2002. [Google Scholar]
- Page RDM. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]