

Summary
Interleukin-6 (IL-6) is a major survival factor for malignant plasma cells involved in multiple myeloma. Using an RNase protection assay, we looked for gene expression of 10 anti- and pro-apoptotic Bcl-2-family proteins in 12 IL-6-dependent human myeloma cell lines (HMCL). A high Mcl-1 gene expression was found in all HMCLs and the other genes were variably expressed. Out of the 10 Bcl-2-family members, only the Mcl-1 gene was regulated by IL-6. Upon starvation of IL-6, Mcl-1 gene expression decreased in association with myeloma cell apoptosis and was up-regulated after adding IL-6 again in association with myeloma cell survival. A constitutive Mcl-1 expression was induced with an Mcl-1-GFP retrovirus in two IL-6-dependent HMCLs. The Mcl-1 HMCLs have a marked reduced apoptosis upon IL-6 starvation compared to HMCLs transduced with control GFP retrovirus and may grow without adding IL-6. These data emphasize the major role of Mcl-1 anti-apoptotic protein in the IL-6-induced survival of human myeloma cells.
Keywords: Antibodies, immunology, Antigens, CD, immunology, metabolism, Apoptosis, physiology, Cell Survival, physiology, Cytokine Receptor gp130, Genetic Vectors, Humans, Interleukin-6, metabolism, Membrane Glycoproteins, immunology, metabolism, Multiple Myeloma, metabolism, pathology, Neoplasm Proteins, metabolism, Proto-Oncogene Proteins c-bcl-2, biosynthesis, genetics, metabolism, Retroviridae, Transduction, Genetic, Tumor Cells, Cultured
Keywords: myeloma, IL-6, apoptosis, Mcl-1
Introduction
Human myeloma cells survive and proliferate in close contact with bone marrow stromal cells (Klein et al., 1989; Uchiyama et al., 1993; Lokhorst et al., 1994). Interleukin-6 (IL-6) is one of the survival factors delivered by stromal cells. In particular, myeloma cell lines, whose growth is strictly dependent on addition of exogenous IL-6, can be obtained from patients with extramedullary proliferation (Shimizu et al., 1989; Zhang et al., 1994; Westendorf et al., 1996). These IL-6-dependent myeloma cell lines are a model of choice to study the mechanisms of IL-6-induced survival and proliferation. After a long period of culture in vitro, IL-6-dependent cell lines may grow autonomously, probably due to additional mutations, as illustrated for the U266 cell line (Spets et al., 1997). Myeloma cells have been shown to express several anti-apoptotic proteins, namely Bcl-2, Bcl-xL and more recently Mcl-1 (Hamilton et al., 1991; Pettersson et al., 1992; Harada et al., 1998; Puthier et al., 1999b; Jourdan et al., 2000). IL-6 activates the JAK/Stat pathway (De Vos et al., 2000), in particular Stat3, and Stat3-binding motifs are present in the promoter region of Bcl-xL (Catlett-Falcone et al., 1999) and Mcl-1 (Epling-Burnette et al., 2001a). Using dominant-negative Stat3 protein, Catlett-Falcone et al. have shown that Bcl-xL was regulated by Stat3 in the U266 cell line (Catlett-Falcone et al., 1999). In other studies, Puthier et al. have shown that Bcl-xL was also regulated by IL-6 in the MDSN cell line through activation of the JAK/Stat pathway, unlike the MAP kinase pathway (Puthier et al., 1999a). With a large panel of 10 human myeloma cell lines, we also found that Bcl-2, Bcl-xL and Mcl-1 proteins were expressed in most cell lines (Jourdan et al., 2000). However, we found that only Mcl-1 expression was regulated by IL-6 or IFN-alpha in association with their anti-apoptotic effect. Our data suggest that Mcl-1 was the major mediator of the survival activity of IL-6 or IFN-alpha. Mcl-1 was recently shown to have a role in myeloma survival for three autonomously growing cell lines (Derenne et al., 2002). In this study, oligonucleotide antisenses to Mcl-1 were able to induce the apoptosis of these three cell lines. Oligonucleotide antisenses to Bcl-xL or Bcl-2 had no effect. These data are emphasized by the recent demonstration that Mcl-1 was involved in the actinomycin D-induced apoptosis of myeloma cells (Zhang et al., 2002). This contribution of Mcl-1 in myeloma cell survival was shown with autonomously growing myeloma cell lines because they can be easily transfected with oligonucleotide antisenses or vectors. Thus a role for Mcl-1 in the survival of myeloma cells that is strictly dependent on the addition of exogenous IL-6, similar to primary myeloma cells, still remains to be demonstrated.
In the present study, we have shown by RNase protection assay that, among 10 genes coding for anti- or pro-apoptotic proteins, only the Mcl-1 gene was regulated by IL-6 in cytokine-dependent myeloma cell lines. In addition, we show that a constitutive Mcl-1 expression makes survival of myeloma cells possible in the absence of IL-6. These data demonstrate that Mcl-1 is one of the major anti-apoptotic mediator induced by IL-6 in myeloma cells.
Results
Expression of Bcl-2 family genes in myeloma cells
In order to identify which member of the Bcl-2-family proteins are involved in myeloma cell survival, we used a sensitive RNase protection assay (RPA). This assay makes it possible to detect genes coding for five anti-apoptotic proteins (Bcl-w, Bcl-xL, Bfl-1/A1, Bcl-2 and Mcl-1) and five pro-apoptotic proteins (Bcl-xS, Bid, Bik, Bak and Bax). The RPA was performed in 12 IL-6-dependent HMCLs (XG-1-XG-16), two autonomously growing HMCLs (U266 and RPMI8226) and two EBV-transformed lymphoblastoid cell lines (LCL-TR and LCL-BR). The Mcl-1 gene was expressed in 14/14 HMCLs and in the two LCLs (Figure 1). Bcl-xL and Bcl-w were also expressed in a majority of HMCLs. A1 was not expressed in HMCLs (0/14), contrary to LCLs. These data fit our recent observation of a loss of A1 gene expression during differentiation of B cells into plasma cells detected with the Affymetrix microarray (Tarte et al., 2002). Bcl-2 was weakly expressed in the majority of HMCLs, unlike XG-5. This is in full agreement with our previous data showing a high level of Bcl-2 protein in XG-5 cells (Jourdan et al., 2000). Concerning the pro-apoptotic genes, an expression of Bax, Bak and Bid genes was found in a majority of HMCLs. The expression of Bcl-xS and Bik genes was weaker and detected in few HMCLs.
Figure 1. Expression of genes coding for anti- and pro-apoptotic proteins in myeloma cell lines and lymphoblastoid cell lines lines.

RNA was obtained from 12 IL-6-dependent myeloma cell lines (XG-1–XG-16), two autonomously growing myeloma cell lines (U266 and RPMI8226) and two EBV transformed lymphoblastoid cell lines (TR and BR). Cells were harvested during the exponential growth phase and the RNA extracted and probed for Bcl-2 family gene expression using a RNase protection assay. Results are those of one experiment representative of four.
Regulation of Bcl-2 family genes by IL-6
We looked for a regulation of the 10 Bcl-2 family genes in two IL-6-dependent HMCLs. These two HMCLs rapidly apoptosed after removal of IL-6 (Jourdan et al., 2000). They were starved of IL-6 and IL-6 was added again for 6 hours. Figure 2 shows an RPA of one representative experiment and Figure 3 the scanned results of three different experiments performed with the two HMCLs. Results show that only the Mcl-1 gene was significantly up-regulated upon IL-6 stimulation (P = 0.03). In particular, we found no regulation of the Bcl-xL gene (P = 1.0), in agreement with our previous data obtained by Western blot (Jourdan et al., 2000). We also found no regulation of the genes coding for the eight other Bcl-2 family-member genes (Figures 2 and 3).
Figure 2. Regulation of Bcl-2 family gene expression by IL-6.
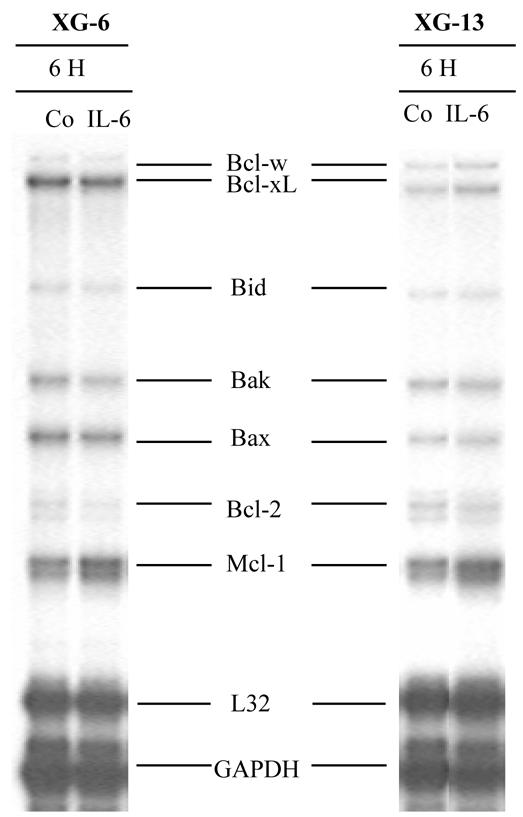
XG-6 and XG-13 myeloma cells were starved of IL-6 for 1 hour and cultured again with IL-6 for 6 hours. RNA was extracted and assayed for the Bcl-2 family gene expression using RPA. Results are those of one experiment representative of 3.
Figure 3. Reproducible up-regulation of Mcl-1 gene expression by IL-6.
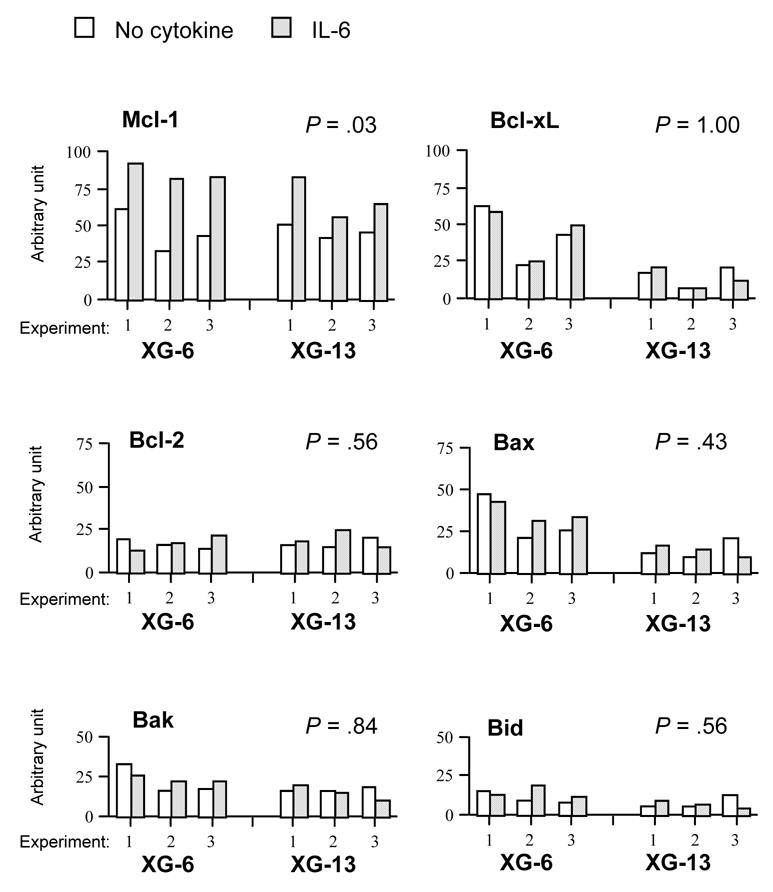
The blots of three independent RPA experiments were scanned and the values were normalized using the L32-band intensities as internal standards. Results are the values for the six main Bcl-2 family members expressed in XG-6 and XG-13 HMCLs starved of IL-6 and cultured for 6 hours with no cytokine (blank column) or with 2 ng/mL IL-6 (hatched column). Statistical analysis was performed with a Wilcoxon test for pairs, grouping data obtained with XG-6 and XG-13 cells.
Constitutive expression of Mcl-1 in myeloma cells transduced with an Mcl-1 retrovirus
The IL-6-dependent HMCLs are a choice model to study the biology of myeloma cells. In order to look for a biological role of Mcl-1 in their survival, we transduced two strictly IL-6-dependent HMCLs with a control green fluorescent protein (GFP) retrovirus or an Mcl-1-GFP retrovirus. We used retroviruses because these cell lines cannot be efficiently transfected with expression vectors or oligonucleotide antisenses. After selection with G418, both HMCLs highly expressed GFP as indicated by FACS analysis (Figure 4A). All cells, when cultured with exogenous IL-6, expressed Mcl-1 protein but XG-1Mcl-1 and XG-6Mcl-1 cells expressed a higher level of Mcl-1 whereas Bcl-xL levels were similar (Figure 4B). In order to show the reproducibility of obtaining Mcl-1 transfectants, five independent transductions were performed with XG-6 cells. After selection with G418, we obtained five XG-6 HMCLs transduced with the GFP control retrovirus and five XG-6 HMCLs transduced with the Mcl-1-GFP retrovirus. The 10 XG-6GFP and XG-6Mcl-1 HMCLs expressed GFP and the five XG-6Mcl-1 cell lines overexpressed Mcl-1 similar to the XG-6Mcl-1 HMCL shown in Figure 4A and 4B (results not shown).
Figure 4. Transduction of myeloma cells with Mcl-1-GFP retrovirus or control GFP retrovirus.
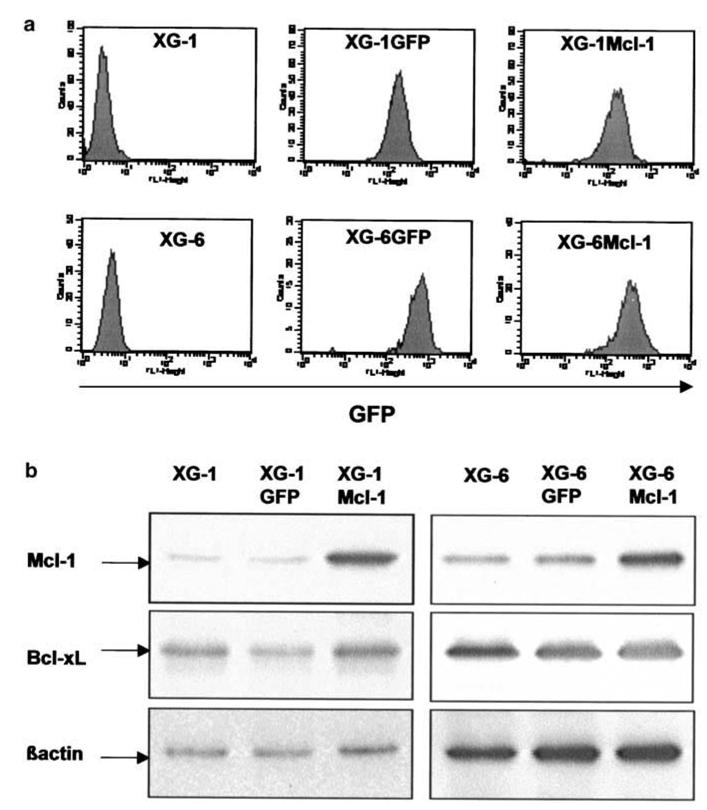
A: XG-1 and XG-6 myeloma cells were transduced with Mcl-1-GFP or GFP retroviruses and were selected with 700 μg/mL G418 and 2 ng/mL IL-6. All selected myeloma cells highly expressed GFP after 15 days of culture.
B: Parental XG cells, XG cells transduced with Mcl-1-GFP or GFP retroviruses were cultured with IL-6 and harvested for Western blot analysis. Blots were probed with anti-Mcl-1, anti-Bcl-xL or anti-β actin antibodies. Western blots are for one representative experiment out of three.
Constitutive expression of Mcl-1 reduced apoptosis in IL-6-deprived myeloma cell lines and induced IL-6-independent growth
We then conducted experiments to determine the effects of a constitutive Mcl-1 expression in the survival of XG-1 and XG-6 HMCLs. As shown in Figure 5, removal of IL-6 led to a rapid loss of Mcl-1 protein in the XG-6GFP and parental XG-6 cell lines. No down-regulation of Mcl-1 was found in XG-6Mcl-1 cells. In agreement with our previous data, no down-regulation of Bcl-xL was observed in the parental XG-6, XG-6GFP and XG-6Mcl-1 cells after IL-6 starvation. Similar results were found for XG-1GFP and XG-1Mcl-1 cells (results not shown). Removal of IL-6 resulted in the induction of maximum apoptosis in parental XG-6 and XG-1 cells or XG-6GFP and XG-1GFP cells. Detailed data from one experiment with XG-6 cells are shown in Figure 6A and the mean values +/− SD of 5 separate experiments with XG-6 and XG-1 cells in Figure 6B. Addition of IL-6 induced a strong survival of XG-6 and XG-1 or XG-6GFP and XG-1GFP myeloma cells. Apoptosis was 61 % lower in XG-6Mcl-1 cells (P = .0022) and 65 % lower in XG-1Mcl-1 cells (P = .0001) after 3 days of IL-6 removal (Figure 6B). Addition of IL-6 increased this survival. We then performed the same experiments with the various XG-6GFP or XG-6Mcl-1 cell lines obtained in five separate transduction experiments (Figure 7A). Results show that a strong apoptosis occurred in the five XG-6GFP HMCLs starved of IL-6 for 3 days. Transduction of Mcl1 made it possible to reduce this apoptosis by 47% (P = 0.0079).
Figure 5. IL-6-independent Mcl-1 expression in XG-6Mcl-1 myeloma cell line.
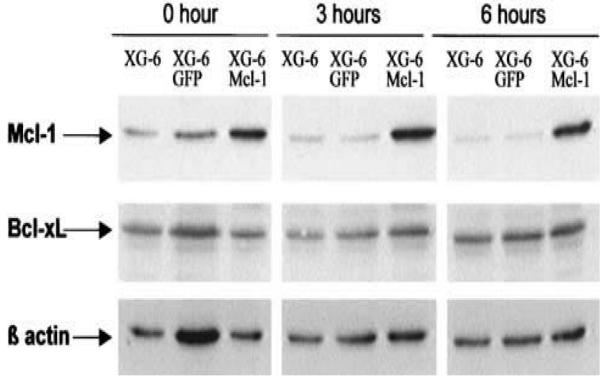
Parental XG-6 cells, XG-6GFP or XG6Mcl-1 cells were starved of IL-6 for 1 hour and stimulated without cytokine or with IL-6 for various times. Cells were harvested for Western blot analysis and blots were probed for Mcl-1, Bcl-xL and β actin. Western blots are for one representative experiment out of three.
Figure 6. Constitutive Mcl-1 expression prevents from apoptosis induced by IL-6 starvation starvation.
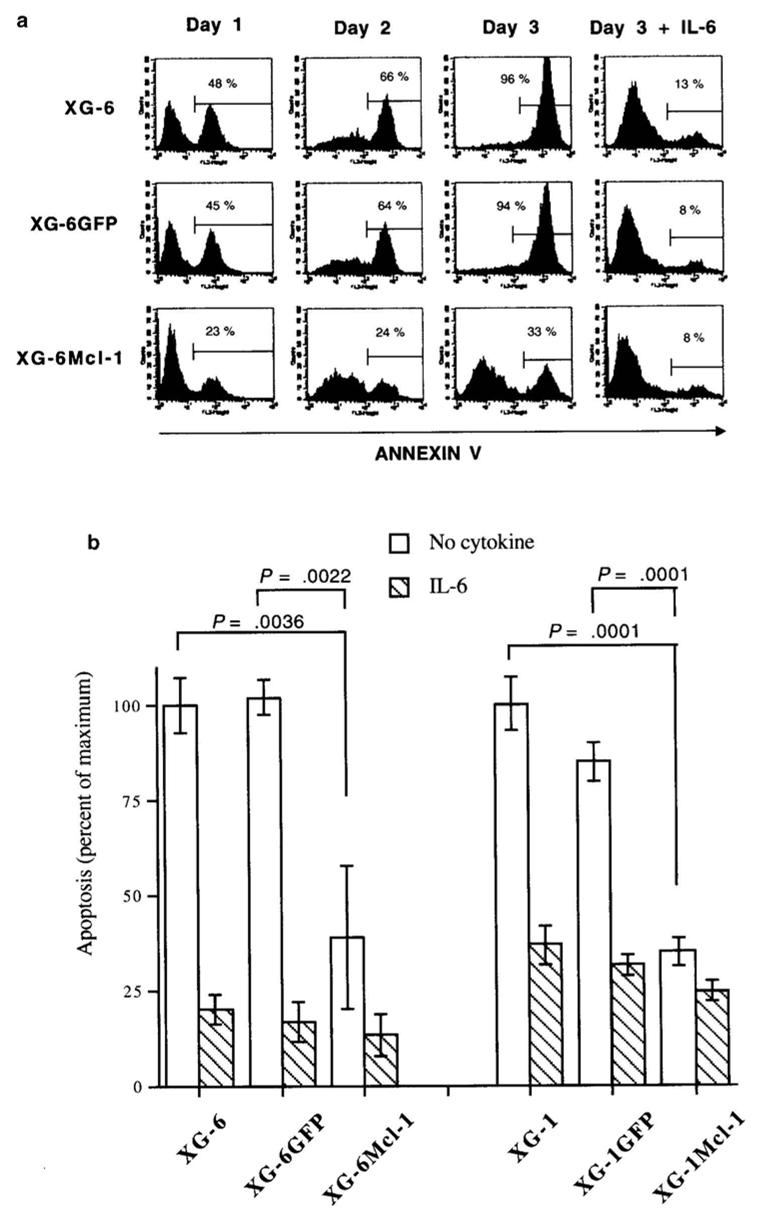
A: Parental XG-6 cells, XG-6GFP or XG6Mcl-1 cells were starved of IL-6 for 1 hour and cultured without cytokine or with 2 ng/mL IL-6. At various days of culture, cells were harvested and apoptotic cells were detected with annexin-V staining. Results are those of one experiment representative of five.
B: The experiment described in A was performed on five occasions with XG-1 and XG-6 cells. Results are the mean values ± SD of apoptotic cells (expressed as the percentage of the maximum observed with XG-6 or XG-1 cells cultured without IL-6) on day 3 of culture. The statistical significance was determined using a Student t test for pairs.
Figure 7. Transduction with Mcl-1 retrovirus obtains reproducible myeloma cell lines. surviving IL-6 starvation.

A: The 5 XG-6GFP and XG-6Mcl-1 clones were obtained in five separate transduction experiments with GFP or Mcl-1-GFP retroviruses starved of IL-6 for 1 hour and cultured for 3 days without cytokine. At day 3 of culture, the percentage of apoptotic cells were determined by staining with annexin V. Statistical analysis was performed with a Wilcoxon test.
B: XG-6 cells transduced with GFP or Mcl-1-GFP retroviruses were cultured at 105 cells/mL with or without 2 ng/mL of recombinant IL-6. Every 3–4 days, cells were counted and diluted at 105 cells/mL with fresh culture medium containing the initial cytokine concentration. Results are the cumulative cell numbers obtained from 18 days of culture. They are those of one experiment representative of two.
Finally, we looked for the possibility of growing the XG-6Mcl-1 cell line without IL-6 (Figure 7B). As expected, the XG-6GFP cell line did not grow without adding IL-6 and all cells were dead within 4 days. Addition of IL-6 prevented cell death and induced exponential growth, with a 1.2-day doubling time. Interestingly, the XG-6Mcl-1 was able to grow without adding IL-6. The growth rate was slower than that observed with the XG-6GFP cultured with IL-6 (1.7 day). Addition of IL-6 accelerated the growth rate that was close to that obtained with XG-6GFP.
The myeloma cell survival induced by up-regulated Mcl-1 expression was not abrogated by a neutralizing anti-gp130 antibody
As illustrated in Figure 8, virtually all XG-6GFP cells underwent apoptosis when starved of IL-6 for 3 days. Addition of IL-6 together with the control G4 nonneutralizing anti-gp130 monoclonal antibody (mAb) prevented this apoptosis. The survival effect of IL-6 was abrogated with the A1 neutralizing anti-gp130 mAb. Interestingly, the survival mediated by the constitutive Mcl-1 expression in XG-6Mcl-1 cells was not abrogated by the A1 neutralizing anti-gp130 mAb. IL-6 further reduced the apoptosis in XG-6Mcl-1, in agreement with above data and this effect was abrogated by the A1 anti-gp130 mAb.
Figure 8. The myeloma cell survival induced by constitutive Mcl-1 expression is not abrogated by a neutralizing anti-gp130 IL-6 transducer antibody.
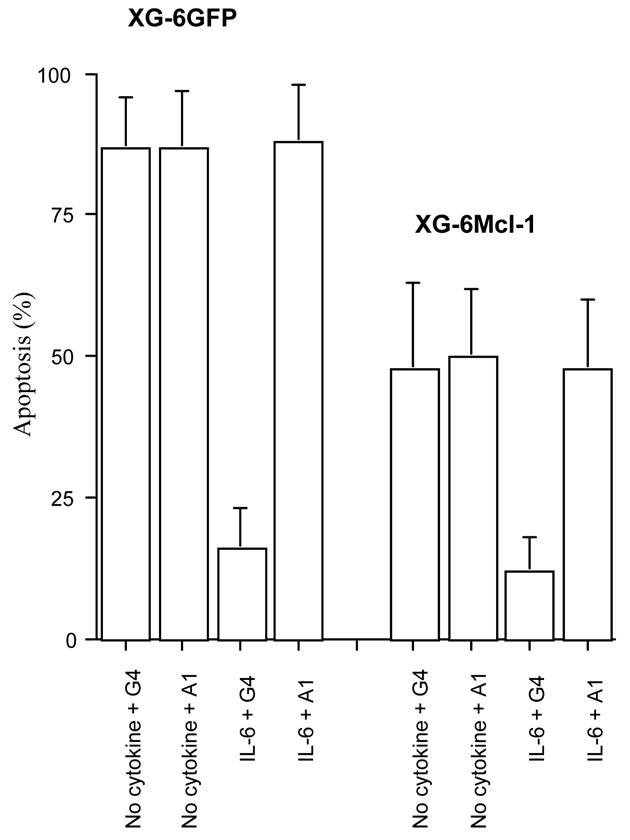
XG-6 cells or XG-6Mcl-1 cells were starved of IL-6 and cultured with or without 2 ng/μL IL-6 for 3 days in the presence of 150 μg/mL of the A1 neutralizing or the G4 nonneutralizing anti-gp130 mAb. The apoptosis was evaluated by annexin-V binding and FACS analysis. Results are means ± SD of the percentages of apoptotic cells determined in three separate experiments.
Discussion
In the present study, we demonstrate that a constitutive expression of the Mcl-1 anti-apoptotic protein makes it possible to reduce the IL-6 requirement for malignant plasma cell survival. We used cell lines whose survival is completely dependent upon addition of exogenous IL-6. After starvation of IL-6, virtually all cells underwent apoptosis within several days. Thus, these cell lines have kept the major characteristic of primary myeloma cells whose survival is strictly dependent on the addition of exogenous IL-6 (Klein et al., 1989; Gu et al., 2000). In patients with MM, IL-6 is largely produced by bone marrow stromal cells, indicating a paracrine stimulation of tumor cell survival by their environment (Portier et al., 1991; Uchiyama et al., 1993; Lokhorst et al., 1994).
A major role of Mcl-1 in IL-6-induced survival is supported by two observations. Firstly, among 10 genes coding for anti- or pro-apoptotic proteins of the Bcl-2 family, only the Mcl-1 gene was down-regulated upon IL-6 starvation and induced upon IL-6 stimulation. We initially reported that Mcl-1 protein was rapidly lost after starvation of IL-6, unlike Bcl-2 or Bcl-xL proteins (Jourdan et al., 2000). However, we did not know whether this Mcl-1 regulation occurred at a transcriptional level or posttranscriptional and translational levels since Mcl-1 expression is tightly regulated at these various levels. The present results indicate that IL-6 regulates primarily Mcl-1 gene expression in myeloma cells. Secondly, we demonstrated that a constitutive expression of Mcl-1 reduced the apoptosis induced by IL-6 starvation. This effect was not abrogated by neutralizing anti-gp130 IL-6 transducer antibodies. Accordingly, the myeloma cell lines transduced with a Mcl-1-GFP retrovirus expressed a high level of Mcl-1 that was unaffected by IL-6 starvation. On the contrary, IL-6 withdrawal resulted in Mcl-1 disappearance in cell lines transduced with the control GFP retrovirus. In addition, a constitutive Mcl-1 expression makes possible autonomous growth of myeloma cell lines whose growth has remained strictly dependent on IL-6 for 10 years. Altogether, these data indicate that an Mcl-1 constitutive expression can promote the survival of myeloma cells independently of gp130 IL-6 transducer activation and strongly argue that Mcl-1 is one of the major anti-apoptotic proteins involved in IL-6-induced myeloma cell survival.
To date, the mechanism of Mcl-1 anti-apoptotic activity has not been precisely established. Mcl-1 is able to form heterodimers with several pro-apoptotic proteins such as Bax, Bak, Bok, Bod, Bik and the unphosphorylated form of Bad (Hsu et al., 1997; Hsu et al., 1998; Bae et al., 2001; Epling-Burnette et al., 2001a; Epling-Burnette et al., 2001b). Thus, by forming heterodimers with these pro-apoptotic proteins, an excess of Mcl-1 induced by IL-6 could neutralize their activity and prevent apoptosis in myeloma cells. In the absence of IL-6, Mcl-1 disappears allowing the pro-apoptotic proteins to exercise their activity. The other anti-apoptotic proteins (Bcl-2, Bcl-xL and Bcl-w) that are also expressed by myeloma cells and are not donwnregulated after IL-6 starvation are not sufficient to promote alone myeloma cell survival. This may be explained by a too weak level of expression and/or by a different intracellular localization of these anti-apoptotic proteins. Bcl-2 and Bcl-xL are predominantly expressed to the outer mitochondrial membrane, unlike Mcl-1 which principally localizes to endoplasmic reticulum and nuclear envelope (Yang et al., 1995). This may also be explained by a different affinity with pro-apoptotic members. In particular, only Mcl-1, unlike Bcl-2, Bcl-xL or Bcl-w, is able to heterodimerize with the pro-apoptotic protein Bok (Hsu et al., 1997).
Our data suggest that other factors, besides Mcl-1, are involved in the IL-6-induced myeloma cell survival. Indeed, the survival induced by a constitutive Mcl-1 expression in myeloma cells is weaker than that induced by IL-6 and addition of IL-6 can further increase the survival of Mcl-1 transduced myeloma cells. Other members of the bcl-2 family (Boo/Diva, Bok, Bod, Hrk…) that were not presently studied might be regulated by IL-6. Beside the JAK/STAT pathway, IL-6 also activates the MAP kinase and PI3-kinase/AKT pathways in myeloma cells (reviewed in Klein et al., 2003). The two pathways are involved in myeloma cells survival through Mcl-1 unrelated mechanisms (Puthier et al., 1999a; Jourdan et al., 2000). The PI3-K/AKT pathway induces phosphorylation of Bad and Forkhead pro-apoptotic proteins and their sequestration by 14-3-3 protein (reviewed in Klein et al., 2003). The PI3-K/AKT pathway may also induce NF-KB activation and induction of inhibitor of apoptosis proteins (IAP) in myeloma cells. These IAP family members or their antagonist Smac/Diablo could be also involved, working downstream members of the Bcl-2 family as that was shown for the MM-1S myeloma cell line (Chauhan et al., 2001; Mitsiades et al., 2002).
Does Mcl-1 play a role in the emergence of myeloma disease? Mcl-1 is expressed by many cells but its expression is tightly regulated at the transcriptional and post-transcriptional levels and the protein has a short half-life. Mcl-1 is viewed as an early and transient survival factor that allows a cell to decide either to undergo survival by subsequently activating other anti-apoptotic genes or apoptosis (Craig, 2002). Overexpression of the Mcl-1 gene may result in long-term tumorigenesis. This is shown using Mcl-1 transgenic mice where the transgene is driven by the Mcl-1 promoter, yielding a high Mcl-1 expression in cells which naturally express Mcl-1 (Zhou et al., 2001). In these mice, overexpression of Mcl-1 results in the emergence of long-term lymphoma, in agreement with its expression at this stage of B cell differentiation. Mcl-1 expression in normal plasma cells has not been documented. It is noteworthy that no polyclonal plasmacytosis or plasma cell tumors were found in Mcl-1 transgenic mice whereas IL-6 transgenic mice develop massive polyclonal plasmacytosis (Suematsu et al., 1989) or plasmacytomas when crossed with Balb/c mice (Suematsu et al., 1992).
Presently, no mutations or translocations involving the Mcl-1 gene have been reported in myeloma disease. The Mcl-1 gene is located on chromosome 1q21 (Craig et al., 1994), a region frequently involved in cancer gene abnormalities and in particular in MM (Keung et al., 1998; Le Baccon et al., 2001), and close to recently identified myeloma genes (IRTA1 and IRTA2) (Hatzivassiliou et al., 2001). Given its major role in IL-6-induced myeloma survival, the Mcl-1 gene abnormalities should be closely evaluated using Fish analysis or microarrays. An increase in Mcl-1 protein may also occur without gene abnormalities, due to post-transcriptional or translational abnormalities. This should also be investigated using immunostaining of Mcl-1 protein in patients with MM at diagnosis. As an example, Mcl-1 protein overexpression was recently associated with poor prognosis in ovarian cancers and diffuse large cell lymphomas (Rassidakis et al., 2002; Shigemasa et al., 2002).
Materials and methods
Cytokines and antibodies
Purified human recombinant IL-6 was kindly provided by Sandoz Forschunginstitut (Vienna, Austria) and human recombinant IFN-α-2b by Schering Plough (Dardilly, France). Monoclonal anti-Bcl-2 antibody was purchased from Dako (Copenhagen, Denmark), anti-Stat3 from Transduction Laboratories (Lexington, KY, USA). Rabbit or goat polyclonal antibodies specific for Bcl-x and Mcl-1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and phospho-Stat3 from New England Biolabs (Beverly, MA, USA.). The neutralizing A1 anti-gp130 mAb and the G4 nonneutralizing anti-gp130 mAb, used as control mAb, were obtained in our laboratory (both are mouse IgG1) (Liautard et al., 1997).
Human myeloma cell lines (HMCL)
XG-1, XG-2, XG-3, XG-4, XG-5, XG-6, XG-7, XG-10, XG-12, XG-13, XG-14 and XG-16 myeloma cell lines were obtained from 12 patients with terminal disease as described previously (Zhang et al., 1994). These HMCLs are true myeloma cell lines: CD19−, CD20−, CD138+, CD38++, cytoplasmic immunoglobulin++. They were not infected with EBV. Their growth is dependent upon addition of exogenous IL-6, as does the growth of primary cells. U266 and RPMI8226 were obtained from ATCC (Rockville, MD, USA). Cells were free of mycoplasma contamination as assayed using the Boehringer Mannheim detection kit (Mannheim, Germany).
Culture of myeloma cells
The HMCLs, except U266 and RPMI8226, were routinely cultured with 2 ng/mL of IL-6 in RPMI 1640 supplemented with 10% fetal calf serum (FCS) and 5 × 10− 5 M 2-mercaptoethanol. U266 and RPMI8226 were cultured without IL-6. For apoptosis analysis, the HMCLs were starved of IL-6. Therefore, the cells were washed twice, cultured for 1 hour in RPMI1640 with FCS and washed once again. Cells were then cultured at a concentration of 2 × 105 cells/mL in RPMI1640, 10% FCS with either no cytokine or IL-6 (2 ng/mL) for 72 hours. For Western blot analysis, myeloma cells were starved of IL-6 for 1 hour in RPMI1640 supplemented with 1% bovine serum albumin (BSA, Sigma, St Louis, MO, USA), washed once again, then cultured with 1% BSA with or without IL-6 (2 ng/mL) for 3, 6, or 24 hours at a concentration of 2 × 105 cells/mL.
Assays for detection of apoptotic cells
As cell lines transduced with GFP retroviral vectors expressed GFP, apoptotic cells were detected by using biotin-conjugated annexin V (annexin-V-biotin, Boehringer Mannheim) and streptavidin-phycoerythrin. Annexin V has a high affinity for phosphatidylserine present on the outer cytoplasmic membrane of apoptotic cells. Cells were washed and incubated for 20 minutes with annexin-V-biotin according to the manufacturer’s recommendations. The cells were washed once and incubated for 15 minutes with streptavidin-phycoerythrin. Two-color immunofluorescence was analyzed with a FACScan flow cytometer using Cell Quest software (Becton Dickinson, Mountain View, CA, USA).
Western blot analysis
Cells were lysed in 10 mM Tris-HCl (pH 7.05), 50 mM NaCl, 50 mM NaF, 30 mM sodium pyrophosphate (NaPPi), 1% Triton X-100, 5 μM ZnCl2, 100 μM Na3VO4, 1 mM DTT, 20 mM β-glycerophosphate, 20 mM p-nitrophenolphosphate (PNPP), 2.5 μg/mL aprotinin, 2.5 μg/mL leupeptin, 0.5 mM PMSF, 0.5 mM benzamidine, 5 μg/mL pepstatin and 50 nM okadaik acid. The protein content of each lysate was quantified using the bicinchoninic acid mM NaCl, 3 mM KCl, 25 mM Tris-HCl (pH 7.4), 0.1% tween 20 (TBS-T), 5% non-fat milk), then incubated for 1 hour at room temperature with primary antibodies. The primary antibodies were visualized with goat anti-rabbit (Sigma) or goat anti-mouse (Biorad SA, Yvry Sur Seine, France) peroxidase-conjugated antibodies (at 1:10 000 dilution in TBS-T) using an enhanced chemiluminescence detection system. The membranes were stripped twice with 100 mM Glycin, pH 2.2, 0.1% NP 40 and 1% SDS for 30 minutes at room temperature. Blots were quantified by densitometry using acquisition into Adobe Photo Shop (Apple, Cupertino, CA, USA) and analyzing with the NIH Image software (National Institutes of Health, Bethesda, MD, USA).
RNA preparation and RNase protection analysis
Cell lines were harvested during the exponential growth phase (5–7 × 105 cells/mL) or after stimulation with or without cytokines, washed in cold phosphate-buffered saline (PBS), and lysed immediately in 5 M thiocyanate guanidium for RNA extraction. DNA was sheared with a 21-gauge needle and LiCl was added to a final concentration of 3 M. RNA was allowed to precipitate overnight at 4°C and then centrifuged at 10,000 g for 10 minutes. RNA was dissolved in TES (10 mM Tris HCl, pH7.5; 5 mM EDTA, 0.2% sodium dodecyl sulfate (SDS) and then subjected to phenol/chloroform extraction. The RNA quality was verified by electrophoresis on a formaldehyde 1.2% agarose gel. Total RNA (10 μg) was used in RiboQuant Multiprobe RNase Protection Assay System (Pharmingen, San Diego, CA, USA) with the hAPO-2b Human Apoptosis Multi-Probe Template Set (Pharmingen) according to the manufacturer’s instructions.
Retrovirus production and infection
Full-length Mcl-1 cDNA, kindly provided by Dr. Bingle (Bingle et al., 2000) (The University of Sheffield Medical School, Sheffield, UK), was subcloned in pBluescriptIISK+ vector (Stratagene, La Jolla, CA, USA) at the Sma I site. The Mcl-1 containing an EcoR I-BamH I fragment was then cloned in pEGN-MCS vector (a gift from Dr. Berberich (Kuss et al., 1999)) and transfected into the HEK293E17 packaging cell line (Transgene, Strasbourg, France). The packaging cell line was also transfected with the empty retroviral vector pEGN-MCS.
Supernatants derived from transfected HEK293E17 packaging cell line were cocultivated with target cells (XG-1 and XG-6 cell lines) in presence of Lipofectamine (Invitrogen-Life Technologies, Paisley, UK) for 18 hours. The cells were then washed twice and cultured with 0.7 mg/mL of G418 (Invitrogen-Life Technologies) to select transduced cells. Resistant clones obtained with the Mcl-1 retrovirus (named XG-1Mcl-1 and XG-6Mcl-1) or the empty retroviral vector (XG-1GFP and XG-6GFP) were analyzed for GFP expression and sorted using a Vantage cell sorter (Becton Dickinson). The stable transfected HMCLs obtained were subsequently maintained with 0.7 mg/mL of G418.
Statistical analysis
The variations in the levels of expression of the various Bcl-2 family genes in IL-6-stimulated or -starved myeloma cells were compared with a non-parametric Wilcoxon test for pairs. The mean percentages of apoptotic cells in the culture groups with IL-6 were compared to corresponding control groups and the statistical significance was evaluated with a Student t test for pairs.
Acknowledgments
This work was supported by a grant from La Ligue contre le Cancer (Equipe Labellisée) (Paris, France)
References
- Bae J, Hsu SY, Leo CP, Zell K, Hsueh AJ. Apoptosis. 2001;6:319–330. doi: 10.1023/a:1011319901057. [DOI] [PubMed] [Google Scholar]
- Bingle CD, Craig RW, Swales BM, Singleton V, Zhou P, Whyte MK. J Biol Chem. 2000;275:22136–22146. doi: 10.1074/jbc.M909572199. [DOI] [PubMed] [Google Scholar]
- Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, Dalton WS, Jove R. Immunity. 1999;10:105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- Chauhan D, Hideshima T, Rosen S, Reed JC, Kharbanda S, Anderson KC. J Biol Chem. 2001;276:24453–24456. doi: 10.1074/jbc.C100074200. [DOI] [PubMed] [Google Scholar]
- Craig RW. Leukemia. 2002;16:444–454. doi: 10.1038/sj.leu.2402416. [DOI] [PubMed] [Google Scholar]
- Craig RW, Jabs EW, Zhou P, Kozopas KM, Hawkins AL, Rochelle JM, Seldin MF, Griffin CA. Genomics. 1994;23:457–463. doi: 10.1006/geno.1994.1523. [DOI] [PubMed] [Google Scholar]
- De Vos J, Jourdan M, Tarte K, Jasmin C, Klein B. Br J Haematol. 2000;109:823–828. doi: 10.1046/j.1365-2141.2000.02127.x. [DOI] [PubMed] [Google Scholar]
- Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Blood. 2002;100:194–199. doi: 10.1182/blood.v100.1.194. [DOI] [PubMed] [Google Scholar]
- Epling-Burnette PK, Liu JH, Catlett-Falcone R, Turkson J, Oshiro M, Kothapalli R, Li Y, Wang JM, Yang-Yen HF, Karras J, Jove R, Loughran TP., Jr J Clin Invest. 2001a;107:351–362. doi: 10.1172/JCI9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epling-Burnette PK, Zhong B, Bai F, Jiang K, Bailey RD, Garcia R, Jove R, Djeu JY, Loughran TP, Jr, Wei S. J Immunol. 2001b;166:7486–7495. doi: 10.4049/jimmunol.166.12.7486. [DOI] [PubMed] [Google Scholar]
- Gu ZJ, Vos JD, Rebouissou C, Jourdan M, Zhang XG, Rossi JF, Wijdenes J, Klein B. Leukemia. 2000;14:188–197. doi: 10.1038/sj.leu.2401632. [DOI] [PubMed] [Google Scholar]
- Hamilton MS, Barker HF, Ball J, Drew M, Abbot SD, Franklin IM. Leukemia. 1991;5:768–771. [PubMed] [Google Scholar]
- Harada N, Hata H, Yoshida M, Soniki T, Nagasaki A, Kuribayashi N, Kimura T, Matsuzaki H, Mitsuya H. Leukemia. 1998;12:1817–1820. doi: 10.1038/sj.leu.2401168. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Miller I, Takizawa J, Palanisamy N, Rao PH, Iida S, Tagawa S, Taniwaki M, Russo J, Neri A, Cattoretti G, Clynes R, Mendelsohn C, Chaganti RS, Dalla-Favera R. Immunity. 2001;14:277–289. doi: 10.1016/s1074-7613(01)00109-1. [DOI] [PubMed] [Google Scholar]
- Hsu SY, Kaipia A, McGee E, Lomeli M, Hsueh AJ. Proc Natl Acad Sci U S A. 1997;94:12401–12406. doi: 10.1073/pnas.94.23.12401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SY, Lin P, Hsueh AJ. Mol Endocrinol. 1998;12:1432–1440. doi: 10.1210/mend.12.9.0166. [DOI] [PubMed] [Google Scholar]
- Jourdan M, De Vos J, Mechti N, Klein B. Cell Death Differ. 2000;7:1244–1252. doi: 10.1038/sj.cdd.4400758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keung YK, Yung C, Wong JW, Shah F, Cobos E, Tonk V. Cancer Genet Cytogenet. 1998;106:135–139. doi: 10.1016/s0165-4608(97)00316-6. [DOI] [PubMed] [Google Scholar]
- Klein B, Zhang XG, Jourdan M, Content J, Houssiau F, Aarden L, Piechaczyk M, Bataille R. Blood. 1989;73:517–526. [PubMed] [Google Scholar]
- Klein B, Jourdan M, De Vos J, Mathouk K, Tarte K, Rossi JF. In: Biology of Multiple Myeloma. Bersagel PL, Kuehl M, editors. Humana Press; Totowa: 2003. [Google Scholar]
- Kuss AW, Knodel M, Berberich-Siebelt F, Lindemann D, Schimpl A, Berberich I. Eur J Immunol. 1999;29:3077–3088. doi: 10.1002/(SICI)1521-4141(199910)29:10<3077::AID-IMMU3077>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Le Baccon P, Leroux D, Dascalescu C, Duley S, Marais D, Esmenjaud E, Sotto JJ, Callanan M. Genes Chromosomes Cancer. 2001;32:250–264. doi: 10.1002/gcc.1189. [DOI] [PubMed] [Google Scholar]
- Liautard J, Sun RX, Cotte N, Gaillard JP, Mani JC, Klein B, Brochier J. Cytokine. 1997;9:233–241. doi: 10.1006/cyto.1996.0159. [DOI] [PubMed] [Google Scholar]
- Lokhorst HM, Lamme T, de Smet M, Klein S, de Weger RA, van Oers R, Bloem AC. Blood. 1994;84:2269–2277. [PubMed] [Google Scholar]
- Mitsiades CS, Mitsiades N, Poulaki V, Schlossman R, Akiyama M, Chauhan D, Hideshima T, Treon SP, Munshi NC, Richardson PG, Anderson KC. Oncogene. 2002;21:5673–5683. doi: 10.1038/sj.onc.1205664. [DOI] [PubMed] [Google Scholar]
- Pettersson M, Jernberg-Wiklund H, Larsson LG, Sundstrom C, Givol I, Tsujimoto Y, Nilsson K. Blood. 1992;79:495–502. [PubMed] [Google Scholar]
- Portier M, Rajzbaum G, Zhang XG, Attal M, Rusalen C, Wijdenes J, Mannoni P, Maraninchi D, Piechaczyk M, Bataille R, Klein B. Eur J Immunol. 1991;21:1759–1762. doi: 10.1002/eji.1830210727. [DOI] [PubMed] [Google Scholar]
- Puthier D, Bataille R, Amiot M. Eur J Immunol. 1999a;29:3945–3950. doi: 10.1002/(SICI)1521-4141(199912)29:12<3945::AID-IMMU3945>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Puthier D, Derenne S, Barille S, Moreau P, Harousseau JL, Bataille R, Amiot M. Br J Haematol. 1999b;107:392–395. doi: 10.1046/j.1365-2141.1999.01705.x. [DOI] [PubMed] [Google Scholar]
- Rassidakis GZ, Lai R, McDonnell TJ, Cabanillas F, Sarris AH, Medeiros LJ. Am J Pathol. 2002;160:2309–2310. doi: 10.1016/S0002-9440(10)61178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemasa K, Katoh O, Shiroyama Y, Mihara S, Mukai K, Nagai N, Ohama K. Jpn J Cancer Res. 2002;93:542–550. doi: 10.1111/j.1349-7006.2002.tb01289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Yoshioka R, Hirose Y, Susumu S, Tachibana J, Konda S. J Exp Med. 1989;169:339–344. doi: 10.1084/jem.169.1.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spets H, Jernberg-Wiklund H, Sambade C, Soderberg O, Nilsson K. Br J Haematol. 1997;98:126–133. doi: 10.1046/j.1365-2141.1997.1903004.x. [DOI] [PubMed] [Google Scholar]
- Suematsu S, Matsuda T, Aozasa K, Akira S, Nakano N, Ohno S, Miyasaki JI, Yamamura KI, Hirano T, Kishimoto T. Proc Natl Acad Sci USA. 1989;86:7547–7551. doi: 10.1073/pnas.86.19.7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suematsu S, Matsusaka T, Matsuda T, Ohno S, Miyazaki J, Yamamura K, Hirano T, Kishimoto T. Proc Natl Acad Sci USA. 1992;89:232–235. doi: 10.1073/pnas.89.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarte K, De Vos J, Thykjaer T, Zhan F, Fiol G, Costes V, Reme T, Legouffe E, Rossi JF, Shaughnessy J, Jr, Orntoft TF, Klein B. Blood. 2002;100:1113–1122. [PubMed] [Google Scholar]
- Uchiyama H, Barut BA, Mohrbacher AF, Chauhan D, Anderson KC. Blood. 1993;82:3712–3720. [PubMed] [Google Scholar]
- Westendorf JJ, Ahmann GJ, Greipp PR, Witzig TE, Lust JA, Jelinek DF. Leukemia. 1996;10:866–876. [PubMed] [Google Scholar]
- Yang T, Kozopas KM, Craig RW. J Cell Biol. 1995;128:1173–1184. doi: 10.1083/jcb.128.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gojo I, Fenton RG. Blood. 2002;99:1885–1893. doi: 10.1182/blood.v99.6.1885. [DOI] [PubMed] [Google Scholar]
- Zhang XG, Gaillard JP, Robillard N, Lu ZY, Gu ZJ, Jourdan M, Boiron JM, Bataille R, Klein B. Blood. 1994;83:3654–3663. [PubMed] [Google Scholar]
- Zhou P, Levy NB, Xie H, Qian L, Lee CY, Gascoyne RD, Craig RW. Blood. 2001;97:3902–3909. doi: 10.1182/blood.v97.12.3902. [DOI] [PubMed] [Google Scholar]