

Abstract
The action of calmodulin (CaM) on target proteins is important for a variety of cellular functions. We demonstrate here, however, that the presence of a CaM-binding site on a protein does not necessarily imply a functional effect. The α-subunit of the cGMP-gated cation channel of human retinal cones has a CaM-binding site on its cytoplasmic N-terminal region, but the homomeric channel that it forms is not functionally modulated by CaM. Mutational analysis based on comparison to the highly homologous olfactory cyclic nucleotide-gated channel α-subunit, which does form a CaM-modulated channel, indicates that residues downstream of the CaM-binding domain on these channels are also important for CaM to have an effect. These findings suggest that a CaM-binding site and complementary structural features in a protein probably evolve independently, and an effect caused by CaM occurs only in the presence of both elements. More generally, the same may be true for other recognized binding sites on proteins for modulators or activators, so that a demonstrated physical interaction does not necessarily imply functional consequence.
Calmodulin (CaM) plays an important role in a variety of cellular processes. Typically, it acts by first binding Ca2+, with the Ca2+–CaM complex then binding to diverse targets such as protein kinase and phosphatase, adenylyl cyclase, cyclic-nucleotide phosphodiesterase, nitric oxide synthase, cytoskeletal proteins, ion transporters, and ion channels (1–3). The direct effects of CaM on ion channels have been recognized only recently (4–18). One ion channel that shows very strong Ca2+–CaM modulation and has been studied quite extensively is the cyclic nucleotide-gated, nonselective cation channel mediating olfactory transduction in olfactory receptor neurons (OLF) (11, 12, 19). The opening of this channel generates the olfactory receptor potential (20–23). Because of its high Ca2+ permeability (24, 25), the opening of OLF leads to a rise in cytosolic Ca2+, which in turn strongly inhibits the channel via Ca2+–CaM to produce negative feedback (11), making the cell adapt to the olfactory stimulus (24, 26, 27). This inhibition by Ca2+–CaM consists of a favoring of the closed state of the channel, manifested as an increase in the half-activation constant, K1/2, of the channel for cyclic nucleotide (i.e., a shift of the dose–response relation to higher ligand concentrations) (11, 12). The binding site on OLF for Ca2+–CaM has been localized to the cytoplasmic N-terminal region of its α-subunit (OLFα) (12). A similar, but far less potent, inhibition by Ca2+–CaM exists for a homologous cGMP-gated channel mediating phototransduction in retinal rods (ROD) (7–10, 28), but in this case the binding site for Ca2+–CaM is localized to a corresponding region on the channel’s β-subunit (RODβ) (29, 30). Another cGMP-gated channel mediates phototransduction in retinal cones (CONE) (31–33). We report here that human CONE’s α-subunit (CONEα) also has a Ca2+–CaM binding site that is in roughly the same location as on OLFα. Surprisingly, the homomeric channel formed by CONEα does not show any functional modulation by Ca2+–CaM. However, a modulation can be observed after appropriate mutagenesis.
Because of the broad physiological importance of CaM, the presence of a CaM-binding site on a protein often has been interpreted to imply a functional modulation by CaM. This report indicates that this is not necessarily the case.¶
Materials and Methods
CaM-Overlay Experiments on Fusion Proteins.
Fusion-protein constructs containing the N terminus (amino acid residues 1–161) or the C terminus (residues 406–694) of human CONEα were made by PCR amplification using primers with flanking BamHI and EcoRI sites and subcloning the PCR fragments into pGEX-2T (Amersham Pharmacia). The resulting constructs were transformed into Escherichia coli BL21 cells, and the fusion proteins were isolated and purified by using the Bulk glutathione S-transferase (GST) purification module from Amersham Pharmacia. The fusion proteins from previous work (12) containing the N or C termini of rat OLFα, and their mutants, also were used. The fusion proteins were loaded on SDS/PAGE gels and transferred to nitrocellulose (TransBlot, Bio-Rad) in Towbin buffer containing 10% methanol (34). The membranes were blocked in a buffer containing 150 mM NaCl, 10 mM Tris⋅HCl (pH 7.5), 1 mM CaCl2 or 5 mM EGTA, 0.1% antifoam A, and 5% nonfat dry milk for 30 min. Biotinylated CaM (Biomedical Technologies, Stoughton, MA) was added to give a final concentration of 1 μg/ml, followed by an incubation for 1–2 h at room temperature. After extensive washing in the same buffer without additives, the membrane was incubated with avidin and horseradish peroxidase (ABC system, Vector Laboratories) and developed by using the enhanced chemiluminescence system (Amersham Pharmacia). For Western blotting after the CaM-overlay experiments to assess the amount of protein, the membranes were stripped by using TBS/1% SDS/1 mM EDTA, blocked in 2% nonfat dry milk in TBS (140 mM NaCl/10 mM Tris⋅HCl, pH 7.5) and incubated with an anti-GST antibody (Amrad, Melbourne, Australia) in 1:5,000 dilution. The bands were visualized by using a horseradish peroxidase-coupled secondary antibody (Amersham Pharmacia) or the ABC system (Vector Laboratories).
Gel-Shift Assay.
CaM (250 or 375 pmol) was incubated with different molar amounts of a synthetic peptide in a buffer containing 10 mM Na⋅Hepes (pH 7.2) and 2 mM CaCl2 or 5 mM EGTA for 30 min at room temperature. The CaM–peptide complexes then were resolved by nondenaturing gel electrophoresis on 15% gels according to standard procedures for SDS/PAGE, but omitting SDS and adding 2 mM CaCl2 or 5 mM EGTA. The bands were visualized by Coomassie blue staining.
Electrophysiological Recordings.
The cDNAs coding for human CONEα (33), rat OLFα (23), or their mutants and chimeras, all subcloned in the pCIS expression vector, were transfected into human embryonic kidney (HEK) 293 cells by using the calcium phosphate method (35). At 2–4 days after transfection, voltage-clamp recordings were carried out from inside-out membrane patches excised from the cells (12, 29). Membrane voltage was at −60 mV in all experiments. The signals were low-pass filtered at 2.9 kHz (four-pole Bessel filter). For zero-Ca2+ conditions, the pipette and bath solutions both contained 140 mM NaCl, 5 mM KCl, 2 mM EGTA, and 10 mM Hepes/NaOH, pH 7.4. In experiments involving Ca2+–CaM, the bath was perfused with a solution containing 250 nM CaM and 50 μM buffered free Ca2+ (achieved by substituting 2 mM nitrilotriacetic acid and 704 μM CaCl2 for the EGTA). cGMP was added to the bath solution as needed. A solenoid-controlled rotary valve system was used to change the bath solution, and the solution change around the membrane patch was complete within 1–2 s. All experiments were performed at room temperature.
Mutagenesis and Chimera Construction.
Point and deletion mutants were made with the Stratagene mutagenesis kit. Chimeras between rat OLFα and human CONEα were made by PCR using unique restriction sites, and the products were confirmed by sequencing.
Results
Inspection of amino acid sequence suggests that human CONEα has a conspicuous consensus CaM-binding site in position corresponding to that on OLFα (Fig. 1A). The authenticity of this site was demonstrated by gel-overlay experiments with a GST fusion protein containing the N terminus of CONEα (Fig. 1B) and by gel-shift experiments with a synthetic peptide (KY17) corresponding to the site (Fig. 1C). In a competition experiment (see ref. 12) in which the KY17 peptide was premixed with Ca2+–CaM, the peptide also was able to block the Ca2+–CaM effect on homomeric rat OLFα channels expressed in HEK 293 cells (data not shown), suggesting that the OLFα- and CONEα-binding sites interact with the same domain on CaM. Surprisingly, however, homomeric channels formed by human CONEα transfected into HEK 293 cells did not show any functional modulation by Ca2+–CaM (Fig. 1D), unlike the situation for OLFα (11, 12).
Figure 1.

Identification of a CaM-binding site on human CONEα. (A) Sequence of the CaM-binding site on human CONEα aligned with that on rat OLFα. The consensus motif of three aromatic/hydrophobic amino acids at positions 1, 8, and 14 is indicated. Boldface indicates identical residues between the two sequences. (B) Gel-overlay experiment with biotinylated CaM and GST–fusion proteins of the N and C termini of CONEα. N′, N-terminal fusion protein with the CaM-binding site deleted. As controls, the corresponding fusion proteins of OLFα were included in the experiment. After the CaM overlay, the blots were stripped and probed with an α-GST antibody, and the results indicated roughly the same amount of protein in each lane (data not shown). The calculated Mr of the OLFα and CONEα N-terminal fusion proteins are 44 and 47 kDa, respectively, and 61 and 62 kDa for the C-terminal fusion proteins. The additional bands probably represent degradation products. (C) Gel-shift experiment with a peptide (KY17) corresponding to the CaM-binding site on CONEα (residues 65–89). The peptide KY9, corresponding to the site on OLFα (residues 62–87), was included for comparison. CaM (375 pmol) and a peptide in peptide/CaM mole ratios of 1, 2, or 10 (indicated above the lanes), plus 2 mM Ca2+, was resolved on a 15% nondenaturing gel and visualized with Coomassie blue staining. The leftmost lane contains CaM but no peptide. The arrowhead indicates the position of free CaM. No shifts were observed without Ca2+ (data not shown). (D) Dose–response relation between activated current and cGMP concentration for wild-type CONEα expressed in HEK 293 cells in the presence (●) and absence (□) of 250 nM CaM, both with 50 μM Ca2+. Results from patch-clamp recordings from excised, inside-out membrane patches of the transfected cells. Membrane potential at −60 mV. Individual data points from three patches are plotted with the same symbols. Curve fits are according to the Hill equation, I/Imax = Cn/[Cn + K1/2n]. The K1/2 values in the absence or presence of CaM were 19.1 and 18.4 μM cGMP, respectively, both with n = 2.1.
It appears that the absence of CaM modulation on human CONEα is not caused by incompatibility of the protein’s C terminus. We constructed two chimeras: one with a rat OLFα C terminus in a human CONEα background (ChiMG1) and the other with a CONEα C terminus in a OLFα background (ChiMG4). Ca2+–CaM had an effect on ChiMG4 but not ChiMG1 (Fig. 2 and legend; see also Fig. 3A). Thus, the C terminus of CONEα is compatible with the rest of OLFα to produce a Ca2+–CaM effect. Previous results from chimeras between human RODα and rat OLFα (12) have indicated that the RODα C terminus is likewise compatible with the OLFα N terminus to produce a CaM effect.
Figure 2.

Effect of CaM on two rat OLFα/human CONEα chimeric channels.
Contribution from CONEα is in white and from OLFα in black. Numbers
indicate the last and first residues of the respective sequences across
the junction. Dashed lines indicate approximate borders between
N-terminal (N), transmembrane (TMDs), and C-terminal (C) domains.
Traces indicate patch-clamp recordings from excised, inside-out
membrane patches of transfected cells at low cGMP concentration.
Horizontal bars indicate times of application of the respective
treatments. CaM was at 250 nM; membrane potential at −60 mV. The cGMP
concentrations 10 μM and 5 μM correspond to roughly the
K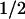 values of the two chimeras, respectively.
Another chimera similar to ChiMG4 but with the OLFα/CONEα
junction closer to the end of the transmembrane domains failed to give
functional channels.
values of the two chimeras, respectively.
Another chimera similar to ChiMG4 but with the OLFα/CONEα
junction closer to the end of the transmembrane domains failed to give
functional channels.
Figure 3.

Experiments to identify additional elements besides the CaM-binding site that are necessary to produce a CaM effect on human CONEα. (A) (Left) Schematic diagrams indicating the structures of various chimeras and mutants from rat OLFα and human CONEα. The locations of the CaM-binding site, the cyclic nucleotide-binding site, and the boundaries of the transmembrane domains (TMDs) are indicated. Numbers indicate the last and first residues of the respective sequences at the junctions or deletions. MG84 has the CaM-binding site deleted. MG92 has a stretch of residues (A117–T141) on CONEα deleted to match up the sequence length on OLFα downstream of the CaM-binding site. Hatched region indicates a region in which the CONEα sequence is modified. For MG75, the mutations A71Q, S74V, F78G, and R82D have been carried out in the CaM-binding site of CONEα to make it more resemble that on OLFα. For ChiMG11, the serial mutations are detailed in C. Two other chimeras were made but did not produce any cGMP-activated current when expressed: one with a CONEα N terminus in an OLFα background and the other with OLFα TMDs in a CONEα background. (Center) Presence (+) or absence (−) of a CaM effect, assayed electrophysiologically as in Fig. 2 with a nonsaturating concentration of cGMP and 50 μM Ca2+ and 250 nM CaM; the number in parentheses indicate number of experiments, some of which were in common with those at right. (Right) cGMP K1/2 values (at −60 mV) for the respective constructs in the absence of CaM and Ca2+, estimated by using three or more concentrations of cGMP. Averaged data with SDs and numbers of experiments are indicated. (B) Sequence alignments of the N-terminal regions of rat OLFα, human CONEα, and ChiMG11 by using the clustalw algorithm. The CaM-binding region is underlined. (C) Serially cumulative mutations (as indicated by flow arrows) to generate ChiMG11 from ChiMG10. The residues mutated are numbered according to the wild-type CONEα sequence. The depictions of CaM effect and cGMP K1/2 values in control conditions have the same meanings as in A.
To ask what elements besides the CaM-binding site are required for a CaM modulation, other chimeras and mutants were constructed and tested (Fig. 3A). The CaM effect was present when a chimera (ChiMG6 or -7) retained the CONEα transmembrane regions, indicating that these regions are probably irrelevant, further hinting that the N terminus is the critical region. Several mutations on CONEα did not bring about a CaM effect, including (i) mutating its CaM-binding site to make it almost identical to that on OLFα (MG75, see Fig. 3A, legend); (ii) shortening the stretch of residues downstream of its CaM-binding site to match the length of that on OLFα (MG92; see Fig. 3B and legend in Fig. 3A); (iii) replacing the region upstream of its CaM-binding site with the corresponding region from rat OLFα (ChiMG9); and (iv) combining the MG92 and ChiMG9 mutations (ChiMG10). However, with additional mutations using ChiMG10 as background to make the region immediately downstream of the CaM-binding site increasingly resemble OLFα, a construct eventually was obtained that showed a CaM effect, in particular after the P99L and A105P changes (ChiMG11; see flow diagram indicating sequential mutations in Fig. 3C). Associated with the CaM effect, the cGMP K1/2 in the absence of CaM also was much lower for ChiMG11 than for the CaM-insensitive constructs, resembling the behavior of wild-type OLFα. This observation suggests that a prerequisite for CaM sensitivity is an increase in the channel’s open probability under control conditions. This appearance of a CaM modulation concomitant with a sharp decrease in control K1/2 makes it unlikely that the serial mutations simply changed the protein conformation to render the CaM-binding site accessible to CaM. One or more of the serial mutations in Fig. 3C besides P99L and/or A105P appear to be also important for generating the CaM sensitivity, because P99L and A105P by themselves in a ChiMG10 background had no effect (data not shown). In view of the large number of possibilities, we did not attempt to track down these other residues further. Finally, we have carried out reciprocal mutations on the rat OLFα residues that correspond to P99 and A105 on human CONEα. One mutation (L97P) indeed produced a sharp increase in cGMP K1/2 and a simultaneous loss of CaM sensitivity, but the other (P103A) produced no changes (data not shown). The double mutant, L97P/P103A, behaved as L97P. Possibly, the position of a proline critically affects the protein’s secondary structure required for CaM modulation. The residue L97 on rat OLFα is immediately downstream of the CaM-binding site and happens to be within the region that we have found previously to influence gating and the CaM modulation (see results on Del 128 in ref. 12).
Besides residues downstream of the CaM-binding site described here, we have demonstrated previously (12) that the CaM-binding site on rat OLFα also contributes to gating. We have attempted to identify the relevant residues within the site. We began with the three aromatic/hydrophobic residues, F68, V75, and W81, that define the “1–8–14” motif for CaM binding (36–41). When all three residues were mutated to alanine (MG22; see Fig. 4A), the mutant OLFα homomeric channel was no longer sensitive to Ca2+–CaM and showed a concomitant increase in control cGMP and cAMP K1/2 values (Fig. 4 B and C), equivalent to having the entire binding site deleted (Del 86; see Fig. 4D and also ref. 12). When the three residues were mutated singly (MG48, MG56, and MG49), the cGMP K1/2 in the absence of Ca2+–CaM remained low (open bars in Fig. 4D), suggesting that these residues individually have only limited influence on gating. With pairwise mutations (MG30, MG34, MG55), it appears that F68 and W81 together are important, but not V75. The importance of F68 and W81 (separated by 12 residues) over V75 in gating is particularly interesting because, based on structural resolution of the complex formed by a highly homologous CaM-binding site (on skeletal-muscle myosin light-chain kinase) with CaM, these same residues are critical for CaM binding (37). For some of the mutants in Fig. 4D, the increase in cGMP K1/2 by Ca2+–CaM (dark bars) was only moderate when compared with wild-type OLFα, perhaps because these mutants had lower affinities for CaM.
Figure 4.

Analysis of the CaM-binding site on rat OLFα. (A) Amino acid sequence of the binding site and the introduced mutations. For each mutant, alanine replaced the wild-type residue at the indicated positions. (Right) Presence or absence of a CaM effect, assayed as in B; the number in parentheses indicates number of experiments. More detailed data on K1/2 in the absence and presence of CaM, assayed as in C, are shown in D. (B–D) Electrophysiological analysis of homomeric channels formed by each of the mutant proteins, using patch-clamp recordings from excised, inside-out membrane patches of transfected HEK 293 cells. Voltage was at −60 mV throughout. (B) Loss of the CaM effect for mutant MG22. CaM was at 250 nM. (C) Dose–response relations between activated current and concentration of cyclic nucleotide for MG22 in the presence of 50 μM Ca2+ and with (filled symbols) or without (open symbols) 250 nM CaM. Circles and squares, cGMP; triangles, cAMP. Averaged data from three patches for cGMP and two for cAMP; vertical bars are SDs. Curve fits are according to the Hill equation. Dashed lines represent curve fits for averaged data (not shown) from wild-type OLFα, for cGMP and cAMP, respectively, in control conditions (50 μM Ca2+ but no CaM). For cGMP, K1/2 = 12.7 μM and n = 2.2 without CaM, and K1/2 = 12.1 μM and n = 2.4 with CaM. For cAMP, K1/2 = 491 μM and n = 1.7 without CaM, and K1/2 = 481 μM and n = 1.8 with CaM. (D) Measured cGMP K1/2 values for the various mutant channels in the absence (open bars) and presence (filled bars) of 250 nM CaM. Averaged data and SDs, with the number above each bar indicating the number of experiments; for MG30, the SD for the open bar is too small to be depicted. The same procedure as in C was used. Del 86 lacks the entire CaM-binding site (12). In the presence of CaM, some of the mutants have lower K1/2 values compared with, e.g., wild-type OLFα, presumably because of a weaker affinity of the binding site for CaM, so that 250 nM CaM was unable to occupy all of the sites. (E) Gel-overlay experiment with biotinylated CaM and GST–fusion proteins of the N terminus of OLFα having the various mutations in the binding site. After the CaM overlay, the blots were stripped and probed with an α-GST antibody, and the results indicated roughly the same amount of protein in each lane (data not shown). (F) Gel-shift experiment with CaM and peptides corresponding to some of the mutants shown in A, D, and E. Peptide KY9 corresponding to amino acids 62–87 on wild-type OLFα. KY20 (MG48), KY21 (MG49), KY22 (MG63), KY23 (MG30), and KY27 (MG22) correspond to the same OLFα sequence except for the indicated mutations. Two hundred-fifty picomoles of CaM and a peptide in peptide/CaM mole ratios of 1 or 2 (indicated above each lane), plus 2 mM Ca2+, was used in each case. The leftmost lane contains CaM but no peptide. The arrowhead indicates the position of free CaM. No shifts were observed without Ca2+ (data not shown). Note that KY22 and KY27 gave the least shift of the CaM band, suggesting least affinity for CaM.
Substituting alanine for the residues Q69, L74, and D80 in the binding site, which are immediately adjacent to F68, V75, and W81 but presumably not critical for the CaM-binding motif (see refs. 36–41), produced the same properties as wild type (MG69, see Fig. 4 A and D). In another mutant (MG63), the positively charged arginines, also important for CaM binding (36–41), were replaced by alanine. This mutant behaved like wild type in the absence of Ca2+–CaM and showed a Ca2+–CaM effect broadly similar to wild type. One might have expected this mutant to show reduced CaM affinity, but this reduction could have been partly overcome by the sufficiently high CaM concentration used in the experiment. To check CaM binding directly, GST-fusion proteins of the OLFα N terminus containing the various point mutations were probed with biotinylated CaM in a gel-overlay assay. In agreement with electrical recordings, the fusion proteins corresponding to MG69 and MG56 gave strong binding signals (Fig. 4E). The other fusion proteins gave no obvious signals. As a more sensitive assay, gel-shift experiments were carried out with CaM and the synthesized peptides KY20, KY21, KY22, KY23, and KY27, which corresponded to the mutated CaM-binding sites on MG48, MG49, MG63, MG30, and MG22, respectively (Fig. 4F). The results showed that Ca2+–CaM still bound quite well to the site with F68 (KY20) and W81 (KY21) individually mutated (i.e., comparable to KY9, which corresponds to the wild-type site), but much less well when both residues were mutated together (KY23), and even worse when F68, V75, and W81 (KY27) or the three arginines (KY22) all were mutated (Fig. 4F). These binding results are broadly consistent with expectations based on the other experiments. Thus, the overall conclusion is that at least two residues on the OLFα CaM-binding site (F68 and W81) are important for both CaM binding and channel gating, several other residues (the arginines) are important for CaM binding but not channel gating, and still others are unimportant for either function.
Discussion
We have found that, even though the CaM-binding site on the N terminus of human CONEα is perfectly capable of binding CaM, the homomeric channel formed by this protein does not exhibit a CaM modulation, contrary to the highly homologous OLFα channel. It is possible that, although the CaM-binding site on human CONEα is accessible to CaM in the environment of the N-terminal fusion protein, this is not the case for the full-length protein. Such a scenario is unlikely, in view of a decrease in the control half-activation constant, K1/2, that is concomitant with the appearance of a CaM effect after appropriate mutations, suggesting that a prerequisite for CaM modulation is a preexisting condition in channel gating (see Results). Functionally speaking, it does not make any difference whether CaM binds to the protein and has no effect or does not bind at all.
Recent in vitro biochemical experiments by Varnum and Zagotta (19) have indicated that the N and C termini of OLFα directly interact with each other, and this interaction is disrupted by CaM. Presumably, this interaction promotes channel opening and accounts for the component of channel gating that is CaM-sensitive (12, 19). The biochemical experiments further showed that the domain on the N terminus corresponding to the CaM-binding site provides the interaction between the fusion proteins of OLFα N and C termini in solution (19). Our findings, on the other hand, suggest that additional residues downstream of the CaM-binding site are important for the CaM-sensitive component of gating, and hence presumably for the N/C-terminal interaction. The two results can be reconciled if, in the environment of the intact channel, a steric requirement for the N/C-terminal interaction is normally provided by the residues downstream of the CaM-binding site, but this requirement is unnecessary for the more flexible situation of fusion proteins in solution. Thus, substituting a proline (which disrupts α-helices) in residue position 97 on rat OLFα (the L97P mutant in this paper) or deleting a stretch of residues in the vicinity (the Del 128 mutant in ref. 12) may perturb the secondary structure sufficiently to interfere with the N/C-terminal interaction.
Besides the CaM-sensitive component of channel gating described here, there is another gating component that is independent of CaM but nonetheless also is conferred by the N terminus—this is true for both RODα and OLFα (42, 43) and presumably for CONEα as well. Some of the CONEα mutants described in this paper did not possess a CaM modulation but possibly showed some changes in K1/2 from wild-type CONEα (Fig. 3A). These changes could have resulted from mutational effects on this CaM-independent component of gating, although the limited number of experiments we have carried out does not allow a definite conclusion.
It may be coincidental that the two aromatic/hydrophobic residues (F68 and W81) being key for CaM binding on OLFα are also critical for gating and hence for the N/C-terminal interaction. On the other hand, protein–protein interactions often involve hydrophobic domains. Possibly, the C-terminal domain that interacts with the N-terminal CaM-binding site is a “CaM-like” domain—in other words, a domain resembling in functional topology the domain on CaM that interacts with the CaM-binding site and so would compete with CaM for the binding site. The existence of a CaM-like domain in target proteins has been proposed as a mechanism for the action of CaM (44).
In olfactory receptor cells, the Ca2+ modulation of the cyclic nucleotide-gated channel via CaM has a critical role in the cells’ adaptation to odorants (11, 24, 27). This modulation appears to be largely conferred by OLFα, although, presumably, a small component is also from a spliced variant of RODβ that is now known to be present in the native olfactory channel complex (45, 46). In rod photoreceptors, the Ca2+ modulation of the cGMP-gated channel is weak (refs. 47 and 48, but see also ref. 49) and apparently entirely because of RODβ (8). This modulation has only a minor role in light adaptation (50). As for cone photoreceptors, the results varied (48, 51, 52); in any case, however, the Ca2+ modulation has been reported to be either absent (51) or considerably weaker (48, 52) than observed for the olfactory channel. Even when a Ca2+ modulation was observed in the native cone channel, it was not clear which subunit (with the identity of any β-subunit in native CONE still being unknown) conferred this modulation and whether CaM or some other Ca2+-binding protein was involved (48, 52). In parallel, our findings suggest increasing divergence from OLFα to CONEα to RODα with respect to the action of CaM: OLFα has both a CaM-binding site and a CaM effect, human CONEα has a CaM-binding site but no CaM effect, and RODα does not even have a CaM-binding site. There is suggestion from alignments of visual-pigment sequences that cones arose earlier than rods in evolution (53, 54). If so, there does not appear to be any evolutionary pressure to select for a Ca2+–CaM modulation in phototransduction, as indicated by the progressive disappearance of the associated structural features. The fact that CaM does not modulate human CONEα functionally does not mean that the latter’s CaM-binding site is idle, if it indeed binds CaM. It can still serve as a physical docking site, and, as such, it provides a buffer for Ca2+–CaM. The significance of this buffer would have to depend on the abundance of the site and its relative affinity compared with CaM-binding sites on other target proteins. Finally, it should be mentioned that, interestingly, homomeric channels formed by chicken CONEα did show a weak modulation by CaM (55). Whether this represents a divergence or transitional situation in evolution is unclear.
Because of the important and diverse functions of CaM in Ca2+ signaling, a recognized CaM-binding site on a protein often has been interpreted to imply a functional effect on the protein by CaM. The example here indicates that this is not necessarily the case. Instead, additional structural elements have to be in place for CaM to have an effect, and these elements may not always coexist with the CaM-binding site on a target protein. As such, it appears as if there were a coincidence requirement built into the CaM mechanism, perhaps to provide more specificity than would arise from the mere presence of a CaM-binding site in a protein. The existence of structural elements that operate in concert with a CaM-binding site to produce a CaM effect is well known among CaM-activated or modulated proteins (see ref. 56, for example, for CaM kinase II). It remains to be seen how common it is to encounter proteins like human CONEα that have a CaM-binding site but fail to show a CaM effect because of lack of complementary structural elements. By extension, the same question can be asked about recognized binding sites on proteins for other modulators or activators.
Acknowledgments
We thank Drs. Peter Gillespie, Min Li, Jeremy Nathans, and David Yue, and also Drs. Robert D. Barber, Yingbin Fu, and Wei-Hong Xiong of the Yau laboratory for discussions and comments on the manuscript. This work is supported in part by a grant from the U.S. National Eye Institute.
Abbreviations
- CaM
calmodulin
- OLF
olfactory cyclic nucleotide-gated channel
- ROD
rod cGMP-gated channel
- CONE
cone cGMP-gated channel
- GST
glutathione S-transferase
- HEK
human embryonic kidney
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
An abstract describing some preliminary results has been published [Grunwald, M. E., Lai, J. & Yau, K.-W. (1998) Biophys. J. 74, A125].
References
- 1.Klee C B, Vanaman T C. Adv Protein Chem. 1982;35:213–321. doi: 10.1016/s0065-3233(08)60470-2. [DOI] [PubMed] [Google Scholar]
- 2.Means A R. Recent Prog Hormone Res. 1988;44:223–262. doi: 10.1016/b978-0-12-571144-9.50012-0. [DOI] [PubMed] [Google Scholar]
- 3.James P, Vorherr T, Carafoli E. Trends Biochem Sci. 1995;20:38–42. doi: 10.1016/s0968-0004(00)88949-5. [DOI] [PubMed] [Google Scholar]
- 4.Preston R R, Kink J A, Hinrichsen R D, Saimi Y, Kung C. Annu Rev Physiol. 1991;83:309–319. doi: 10.1146/annurev.ph.53.030191.001521. [DOI] [PubMed] [Google Scholar]
- 5.Smith J S, Rousseau E, Meissner G. Circ Res. 1989;64:352–359. doi: 10.1161/01.res.64.2.352. [DOI] [PubMed] [Google Scholar]
- 6.Klaerke D A, Karlish S J, Jorgensen P L. J Membrane Biol. 1987;95:105–112. doi: 10.1007/BF01869155. [DOI] [PubMed] [Google Scholar]
- 7.Hsu Y-T, Molday R S. Nature (London) 1993;361:76–79. doi: 10.1038/361076a0. [DOI] [PubMed] [Google Scholar]
- 8.Chen T-Y, Illing M, Molday L L, Hsu Y-T, Yau K-W, Molday R S. Proc Natl Acad Sci USA. 1994;91:11757–11761. doi: 10.1073/pnas.91.24.11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Körschen H G, Illing M, Seifert R, Sesti F, Williams A, Gotzes S, Colville C, Müller F, Dosé A, Godde M, et al. Neuron. 1995;15:627–636. doi: 10.1016/0896-6273(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 10.Gordon S E, Downing-Park J, Zimmerman A L. J Physiol (London) 1995;486:533–546. doi: 10.1113/jphysiol.1995.sp020832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen T-Y, Yau K-W. Nature (London) 1994;368:545–548. doi: 10.1038/368545a0. [DOI] [PubMed] [Google Scholar]
- 12.Liu M, Chen T-Y, Ahamed B, Li J, Yau K-W. Science. 1994;266:1348–1354. doi: 10.1126/science.266.5189.1348. [DOI] [PubMed] [Google Scholar]
- 13.Ehlers M D, Zhang S, Bernhardt J P, Huganir R L. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- 14.Xia X M, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen J E, Ishii T, Hirschberg B, Bond C T, Lutsenko S, et al. Nature (London) 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- 15.Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L. Proc Natl Acad Sci USA. 1999;96:2435–2438. doi: 10.1073/pnas.96.5.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson B Z, DeMaria C D, Adelman J P, Yue D T. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 17.Lee A, Wong S T, Gallagher D, Li B, Storm D R, Scheuer T, Catterall W A. Nature (London) 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- 18.Zühlke R D, Pitt G S, Deisseroth K, Tsien R W, Reuter H. Nature (London) 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 19.Varnum M D, Zagotta W N. Science. 1997;278:110–113. doi: 10.1126/science.278.5335.110. [DOI] [PubMed] [Google Scholar]
- 20.Kurahashi T. J Physiol (London) 1990;430:355–371. doi: 10.1113/jphysiol.1990.sp018295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Firestein S, Darrow B, Shepherd G M. Neuron. 1991;6:825–835. doi: 10.1016/0896-6273(91)90178-3. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura T, Gold G H. Nature (London) 1987;325:442–444. doi: 10.1038/325442a0. [DOI] [PubMed] [Google Scholar]
- 23.Dhallan R S, Yau K-W, Schrader K A, Reed R R. Nature (London) 1990;347:184–187. doi: 10.1038/347184a0. [DOI] [PubMed] [Google Scholar]
- 24.Kurahashi T, Shibuya T. Brain Res. 1990;515:261–268. doi: 10.1016/0006-8993(90)90605-b. [DOI] [PubMed] [Google Scholar]
- 25.Frings S, Seifert R, Godde M, Kaupp U B. Neuron. 1995;15:169–179. doi: 10.1016/0896-6273(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 26.Kramer R H, Siegelbaum S A. Neuron. 1992;9:897–906. doi: 10.1016/0896-6273(92)90242-6. [DOI] [PubMed] [Google Scholar]
- 27.Kurahashi T, Menini A. Nature (London) 1997;385:725–729. doi: 10.1038/385725a0. [DOI] [PubMed] [Google Scholar]
- 28.Bauer P J. J Physiol (London) 1996;494:675–685. doi: 10.1113/jphysiol.1996.sp021523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grunwald M E, Yu W-P, Yu H H, Yau K-W. J Biol Chem. 1998;273:9148–9157. doi: 10.1074/jbc.273.15.9148. [DOI] [PubMed] [Google Scholar]
- 30.Weitz D, Zoche D, Müller F, Beyermann M, Körschen H G, Kaupp U B, Koch K-W. EMBO J. 1998;17:101–112. doi: 10.1093/emboj/17.8.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weyand I, Godde M, Frings S, Weiner J, Müller F, Altenhofen W, Hatt H, Kaupp U B. Nature (London) 1994;368:859–863. doi: 10.1038/368859a0. [DOI] [PubMed] [Google Scholar]
- 32.Biel M, Zong X, Distler M, Bosse E, Klugbauer N, Murakami M, Flockerzi V, Hofmann F. Proc Natl Acad Sci USA. 1994;91:3505–3509. doi: 10.1073/pnas.91.9.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu W-P, Grunwald M E, Yau K-W. FEBS Lett. 1996;393:211–215. doi: 10.1016/0014-5793(96)00889-7. [DOI] [PubMed] [Google Scholar]
- 34.Towbin H, Staehelin T, Gordon J. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Okayama H. Mol Cell Biol. 1988;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Neil K T, DeGrado W F. Trends Biochem Sci. 1990;15:59–64. doi: 10.1016/0968-0004(90)90177-d. [DOI] [PubMed] [Google Scholar]
- 37.Ikura M, Clore G M, Gronenborn A M, Zhu G, Klee C B, Bax A. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- 38.Meador W E, Means A R, Quiocho F A. Science. 1992;257:1251–1255. doi: 10.1126/science.1519061. [DOI] [PubMed] [Google Scholar]
- 39.Meador W E, Means A R, Quiocho F A. Science. 1993;262:1718–1721. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]
- 40.Crivici A, Ikura M. Annu Rev Biophys Biomol Struct. 1995;24:85–116. doi: 10.1146/annurev.bb.24.060195.000505. [DOI] [PubMed] [Google Scholar]
- 41.Rhoads A R, Friedberg F. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 42.Goulding E H, Tibbs G R, Siegelbaum S A. Nature (London) 1994;372:369–374. doi: 10.1038/372369a0. [DOI] [PubMed] [Google Scholar]
- 43.Gordon S E, Zagotta W N. Neuron. 1995;14:857–864. doi: 10.1016/0896-6273(95)90229-5. [DOI] [PubMed] [Google Scholar]
- 44.Jarrett H W, Madhavan R. J Biol Chem. 1991;266:362–371. [PubMed] [Google Scholar]
- 45.Sautter A, Zong X, Hofmann F, Biel M. Proc Natl Acad Sci USA. 1998;95:4696–4701. doi: 10.1073/pnas.95.8.4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bönigk W, Bradley J, Müller F, Sesti F, Boekhoff I, Ronnett G V, Kaupp U B, Frings S. J Neurosci. 1999;19:5332–5347. doi: 10.1523/JNEUROSCI.19-13-05332.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakatani K, Koutalos Y, Yau K-W. J Physiol (London) 1995;484:69–76. doi: 10.1113/jphysiol.1995.sp020648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rebrik T I, Korenbrot J I. J Gen Physiol. 1998;112:537–548. doi: 10.1085/jgp.112.5.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sagoo M S, Lagnado L. J Physiol (London) 1996;497:309–319. doi: 10.1113/jphysiol.1996.sp021770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koutalos Y, Yau K-W. Trends Neurosci. 1996;19:73–81. doi: 10.1016/0166-2236(96)89624-x. [DOI] [PubMed] [Google Scholar]
- 51.Haynes L W, Stotz S C. Visual Neurosci. 1997;14:233–239. doi: 10.1017/s0952523800011378. [DOI] [PubMed] [Google Scholar]
- 52.Hackos D H, Korenbrot J I. J Gen Physiol. 1997;110:515–528. doi: 10.1085/jgp.110.5.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okano T, Kojima D, Fukada Y, Shichida Y, Yoshizawa T. Proc Natl Acad Sci USA. 1992;89:5932–5936. doi: 10.1073/pnas.89.13.5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bowmaker J K. Eye. 1998;12:541–547. doi: 10.1038/eye.1998.143. [DOI] [PubMed] [Google Scholar]
- 55.Bönigk W, Müller F, Middendorff R, Weyand I, Kaupp U B. J Neurosci. 1996;16:7458–7468. doi: 10.1523/JNEUROSCI.16-23-07458.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kennedy M B, Bennett M K, Erondu N E, Miller S G. In: Calcium and Cell Function. Cheung W Y, editor. Vol. 7. New York: Academic; 1987. pp. 62–107. [Google Scholar]