

Abstract
Elimination of a DSB flanked by direct repeat sequences is mediated by single strand annealing (SSA), which relies on a distinct set of gene products involving recombination, mismatch repair and nucleotide excision repair. Here, we screened for yeast mutants defective in SSA using a plasmid based SSA assay coupled to a bar-code microarray readout. The screen identified Yal027Wp/Saw1 (single-strand annealing weakened 1) and Slx4 besides other known SSA proteins. Saw1 interacts physically with Rad1/Rad10, Msh2/Msh3, and Rad52 proteins and cells lacking SLX4 or SAW1 accumulate recombination intermediates blocked at the Rad1/Rad10-dependent 3’-flap cleavage step. Slx4 and Saw1 also contribute to integrity of ribosomal DNA arrays. Saw1 mutants that fail to interact with Rad1, but retain interaction with Rad52 and Msh2 are defective in 3’-flap removal and SSA repair. Deletion of SAW1 abolished association of Rad1 at SSA intermediates in vivo. We propose that Saw1 targets Rad1/Rad10 to Rad52-coated recombination intermediates.
Introduction
DNA double strand breaks (DSBs) form as a result of exposure to a variety of genotoxins such as ionizing radiation, radiomimetic chemicals, endogenous cellular metabolic byproducts or stalled replication forks, and pose significant challenges to a cell and the organism’s genetic integrity (Wyman and Kanaar, 2006). Under unique circumstances (such as meiosis or V(D)J recombination), cells also induce DSBs to initiate chromosome rearrangements for increases in genetic diversity (Lieber et al., 2006). In all these instances, however, efficient healing of these breaks should follow to prevent cell death or accumulation of mutations.
The two most common mechanisms for repairing DSBs are homologous recombination (HR), which uses an undamaged homologous template to guide repair synthesis, and non-homologous end joining (NHEJ) that mediates direct ligation of broken DNA ends regardless of sequence identity at or near DNA breaks (Paques and Haber, 1999). Three distinct subtypes of HR: gene conversion, break-induced replication (BIR) and single-strand annealing (SSA) have been described, all of which begin with exonucleolytic processing of DNA ends and formation of recombinogenic 3’ single-stranded DNA (ssDNA) (Paques and Haber, 1999; Symington, 2002). However, unlike gene conversion or BIR in which ssDNA coated with Rad51 proteins searches for and invades a homologous sequence, SSA simply anneals complementary single stranded sequences flanking either side of a DSB and the resulting recombination intermediates are processed by trimming of 3’-flaps and gap-fill-in synthesis prior to ligation, yielding repair products always associated with deletion of intervening sequences and one of the repeats (Paques and Haber, 1999).
SSA relies on a unique set of genes and their protein products; these are substantially different from those used in gene conversion. The biggest difference lies in the requirement for Rad51 and Rad59 protein: Rad51 is dispensable for SSA (SSA is rather inhibited by Rad51) whereas Rad59 is essential (Sugawara et al., 2000; Symington, 2002). SSA also does not depend on Rad54, Rad55, and Rad57 proteins but absolutely requires Rad52 proteins (Ivanov et al., 1996). Rad59 is a Rad52 protein paralog and has an affinity to single- and double-stranded DNA for binding (Bai and Symington, 1996; Petukhova et al., 1999). Rad59 also has strand-annealing activity for complementary ssDNA (Petukhova et al., 1999). SSA also depends on the Mre11/Rad50/Xrs2 complex, Exo1, and potentially as yet identified protein(s) for 5’ end resection (Ivanov et al., 1996; Mantiero et al., 2007). The Rad1/Rad10 structure specific endonuclease and the Msh2/Msh3 mismatch repair proteins then remove 3’-flaps from a SSA intermediate that forms after annealing of complementary ssDNA (Fishman-Lobell and Haber, 1992; Ivanov and Haber, 1995; Sugawara et al., 1997). The same Rad1/Rad10 nuclease is recruited to UV-induced DNA lesions to incise 5’ to initiate nucleotide excision repair, apparently responding to physical interactions between Rad1/Rad10 and Rad14 (Guzder et al., 2006). In contrast, how Rad1/Rad10 are targeted to DSB damage is yet unknown. A mutation in the single strand DNA binding protein (rfa1-t11) or deletion of SRS2 also reduces SSA frequency (Paques and Haber, 1997; Umezu et al., 1998), but the precise role of these proteins in SSA is not yet established.
SSA was initially proposed as a mechanism for mammalian DSB repair (Lin and Sternberg, 1984). Subsequently, the equivalent SSA process was identified in yeast as well as in some prokaryotes (Fishman-Lobell and Haber, 1992; Stahl et al., 1997). The ubiquitous presence of Alu and other repetitive elements in the human genome may make SSA a more significant pathway in repairing DSBs in human cells (Stenger et al., 2001). Additionally, ribosomal DNA and telomeric and centromeric DNAs are also made up of tandem repeats of DNA sequence ranging from a few nucleotides to a kilobase (Csink and Henikoff, 1998; de Lange, 2005; Kobayashi, 2006). Therefore, the integrity of these repeats likely depends on SSA because a wide range of disorders including multiple genetic diseases, cancers and premature aging are the consequences of unstable rDNA, centromeres or telomere sequences (Bitterman et al., 2002; Hug and Lingner, 2006; Johnson et al., 1999; Nigg, 2006; Pierce et al., 2001). Available evidence also suggests that targeted gene replacement is achieved by a mechanism highly similar to SSA (Langston and Symington, 2005; Niedernhofer et al., 2001; Storici et al., 2006). Consistent with this view, gene products that function in SSA are critical for targeted gene replacement (Langston and Symington, 2005). Since efficient gene targeting is a prerequisite to successful gene therapy, understanding the molecular mechanism of SSA may lead to improvements in the efficiency of gene targeting in mammalian cells and help pave the way for successful gene therapy in humans.
At least 10 genes (RAD52, RAD59, RFA1, RFA2, RFA3, RAD1, RAD10, MSH2, MSH3, SRS2) have been shown to play a role in SSA (Paques and Haber, 1999). However, a genome-wide screen for genes involved in SSA has not yet been performed. Using a novel plasmid-based SSA assay and the available collection of yeast deletion strains, we screened for novel SSA genes and identified YAL27W/SAW1 as a new SSA gene. Based on biochemical characterizations, we propose that Yal027w/Saw1 recruits the Rad1/Rad10 endonuclease complex to SSA intermediates for 3’-flap cleavage by physically linking the Rad1/Rad10 complex to Rad52 proteins.
Results
Microarray based screen for novel SSA genes using a plasmid-based SSA assay
We developed a plasmid-based assay system that monitors re-circularization of a centromeric plasmid (pNSU208) linearized by Bsu36I restriction enzyme cleavage by SSA between 240 bp LacZ direct repeat sequences flanking the break in a yeast strain lacking selection marker LEU2 (Supplemental Fig. 1A)(Sugawara et al., 1995). Yeast cells cannot retain a linear plasmid without telomeres (Boulton and Jackson, 1996). Integration of plasmids into the genome results in a dicentric chromosome, which compromises chromosome segregation (Thrower et al., 2003). Therefore, plasmid re-circularization is required for pNSU208 maintenance. Re-circularization may occur by two distinct mechanisms: Ku-dependent end joining or Rad52-dependent recombination between repeats. To examine the relative contributions of these two mechanisms on re-circularization of linearized pNSU208, we determined the Leu+ transformation efficiency after introduction of the linear plasmid into strains lacking YKU70 or RAD52, and normalized by their general transformation efficiency with a mock-digested circular plasmid. We found that SSA between LacZ repeats is the primary means to circularize the plasmid; less than 20% of re-circularization relies on Ku-dependent end joining (Supplemental Fig. 1B).
Using this plasmid-based SSA assay, we then developed a strategy to screen all nonessential gene deletion mutants for defects in SSA repair of a DNA break (Supplemental Fig. 2). A pool of yeast mutant strains deleted for every nonessential gene was transformed with either Bsu36I-linearized pNSU208 or mock-treated circular plasmid (Ooi et al., 2001). Transformed cells were selected for their leucine prototrophy by growing them in SC–Leu media. Genomic DNA was isolated and the “barcodes” uniquely assigned to each deletion mutant were PCR amplified by UPTAG and DOWNTAG universal primers. The amplified PCR products were hybridized to a microarray containing oligonucleotide barcodes (Ooi et al., 2001). Mutants deleted for a gene necessary for recombination between repeats should maintain the linearized plasmid poorly and fail to become Leu2+. Transformation with the circular (undigested) plasmid normalizes differences in transformation efficiency. We performed this transformation-based plasmid repair assay screen twice independently in haploid and diploid deletion pools to determine the ploidy- or mating type-dependent alteration in genetic requirements for SSA. Overall, we performed 4 microarray analyses in total (2 with haploids; 2 UPTAGs and 2 DOWNTAGs, and 2 with diploids; 2 UPTAGs and 2 DOWNTAGs, Supplemental Table 2). Transformation ratios were calculated by dividing the signal obtained with the circular plasmid by that for the linearized plasmid for each mutant. A high ratio indicates deficiency in recombination between repeats.
Identification of new genes involved in recombination between repeat sequences
To confirm the data set obtained from the microarray-based screen, we arbitrarily limited our analysis to the mutants >3-fold reduction in linear vs. circular plasmid transformation efficiency. All of these mutants (68 for haploid and 21 for diploids) were individually re-tested for their ability to repair linearized plasmids using the same plasmid-based transformation assay used in our screen (Table 1 and data not shown).
Table 1.
Summary of various SSA assay results.
| Genes | Microarray based assay
|
Plasmid DSB repair (%)2 | Chromosome DSB repair (%)3 | |||
|---|---|---|---|---|---|---|
| Haploid | Diploid | |||||
| UPTAG | DOWNTAG | UPTAG | DOWNTAG | |||
| Wildtype1 | 1.00 | 1.00 | 1.00 | 1.00 | 100 | 100 |
| rad1Δ | N.D.4 | 19.70 | 10.7 | 50.8 | 4.0 ± 0.9 | 0.7 |
| slx4 Δ | 12.3 | 11.28 | 42.7 | 50.5 | 1.1 ± 1.0 | 1.0 |
| rad10Δ | 10.5 | 10.70 | 21.4 | 44.4 | 3.3 ± 3.7 | N.D |
| yal027WΔ | 6.8 | N.D. | 11.5 | N.D. | 4.4 ± 3.5 | 0.8 |
| msh2Δ | 2.8 | 4.70 | 5.9 | 12.3 | 8.7 ± 9.5 | N.D |
| msh3Δ | 4.9 | 3.2 | 7.4 | 4.8 | 5.3 ± 2.1 | 2.5 |
| rad52Δ | 1.5 | N.D. | 1.0 | N.D. | 5.1 ± 0.1 | <0.01 |
| rad59Δ | 2.5 | 4.2 | N.D. | N.D. | 10.4 ± 4.4 | 3.8 |
hoΔ::KANrMX for a microarray-based SSA assay; BY4741 for a plasmid-based SSA assay; tNS1379 for chromosome-based SSA assay.
Percent plasmid DSB repair is shown as transformation efficiency of Bsu36I-linearized pNSU208 plasmid divided by that of circular plasmid, and normalized by the repair efficiency of wild type as 100%.
Percent chromosome DSB repair is shown as the level of single strand annealing product (see Fig. 2) quantified by phosphorimager, normalized by loading control. The tNS1379 after 4 h post galactose addition is set to 100%.
not defined.
Since we used the plasmid-based assay for a screen, deficiency in the linearized plasmid transformation assay may not entirely result from recombination deficiency, but also from defects associated with plasmid maintenance. To rule out any such effects in the plasmid-based assay, we secondarily tested each mutant candidate for SSA activity using a chromosome-based SSA assay that monitors repair of an HO endonuclease-induced DSB by annealing 205-bp of ura3 direct repeat sequences flanking the break (Sugawara et al., 2000). Only 7 mutants exhibited defects in recombination between repeats in both haploid and diploid strains and were deficient in SSA between chromosomal repeats (Table 1 and Fig. 1). Five of them represented well-known factors in SSA: Msh2, Msh3, Rad1, Rad10, and Rad59. The role of Rad52 in SSA was confirmed by using a chromosome-based assay despite its absence among the list of SSA mutants by the microarray-based screen (see discussion for further details). A role for SLX4 in SSA was recently described (Flott et al., 2007). We also identified YAL027W (SAW1, single strand annealing weakened 1) as a new factor required for SSA in yeast.
Figure 1. Slx4 and Saw1 are needed for efficient repair of a DSB flanked by direct repeat sequences on a chromosome.

A. SSA between 205 bp ura3 repeats at yeast chromosome V. Survival rate after HO endonuclease induced DSB (C) and Southern blot hybridization of BglII digested genomic DNA (E) with radio-labeled probe next to the URA3 gene to detect SSA product formation are shown for wild type tNS1379 strain and its mutant derivatives. B. SSA between 1.3 kb leu2 repeats that located 24 kb apart on chromosome III. Survival rate of wild type and the SLX4, SAW1, or RAD1 gene deletion derivatives upon HO induction (D) were shown. Data represent means ± standard deviation (s.d.) from three independent experiments.
slx4 and yal027w/saw1 are genuine mutants defective in recombination between repeats
Slx4 forms a heterodimeric endonuclease complex with Slx1 and catalyzes processing of recombination intermediates (Fricke and Brill, 2003; Mullen et al., 2001). The same complex was previously identified for its synthetic interaction with the Sgs1 helicase that plays a key role in a variety of DNA transactions including recombination of repetitive sequences and resolution of tangled replication intermediates (Ira et al., 2003; Mullen et al., 2001). We therefore tested whether deletion of SLX1, like deletion of SLX4, leads to defects in SSA. Consistent with our microarray screen which indicated that a slx1Δ mutant can repair a DSB in a plasmid by SSA with an efficiency identical to SLX1 strains, we confirmed the full functionality of the slx1 deletion mutant for recombination between repeats using our plasmid- and chromosome-based recombination assays (Table 1). The results suggest that Slx4 mediates SSA independent of Slx1 and such function is apparently distinct from the role of the Slx1-Slx4 complex in recombination and damage signaling (Flott et al., 2007; Roberts et al., 2006). Our results also confirm the recently proposed role of Slx4 in the SSA pathway of DSB repair (Flott et al., 2007).
To further assess the role of Slx4 and Saw1 in a wider range of genotoxic damage resistance, we challenged strains deleted for SLX1, SLX4 or SAW1 and those deleted for RAD1, RAD52, or RAD59 with 80 Jm−2 UV, 0.02 % MMS, 200 mM HU, and 20 μg/ml phleomycin. Unlike RAD52- or RAD59-deleted cells, SLX1 or SAW1 deletion mutants were insensitive to MMS, HU or phleomycin treatment (Fig. 2). Deletion of SLX4 or SAW1 did not sensitize cells to UV lesions like the RAD1-deleted strain. Deletion of both RAD1 and SAW1 did not further sensitize cells to any of the drugs described above more than a RAD1 deletion alone, suggesting that these two proteins function in the same pathway. Consistent with previous findings, deletion of SLX4, but not SLX1, sensitizes cells to DNA damaging agents (Fig. 2) (Roberts et al., 2006). The result reaffirms the role of Slx4 in the Slx1-independent process of repair of MMS damage. To rule out strain-specific components in the drug sensitivity profile, we repeated the experiments in the S288c strain background and obtained essentially identical results (data not shown).
Figure 2. DNA damage sensitivity profiles of SSA mutants.

Serial dilutions of each strain were plated onto YEPD with or without indicated concentration of MMS, HU, and phleomycin. To measure UV sensitivity, strains were irradiated immediately after plating onto YEPD using a Stratalinker UV source. Plates were incubated for 3 to 4 days at 30 °C.
In yeast, the ribosomal DNA (rDNA) locus comprises ~150 repeats of rDNA coding 35S and 5S rRNA genes (Fig. 3A) (Kaliraman and Brill, 2002; Kobayashi, 2006). We reasoned that the spontaneous recombination rate at the rDNA locus may depend on Slx4- or Saw1-mediated recombination between repeat sequences. To test this idea, we measured the rDNA recombination rate by scoring the loss of an ADE2 gene inserted at the rDNA locus of yeast chromosome XII (Gottlieb and Esposito, 1989). To improve the sensitivity of this assay, we also performed the experiments in sir2 deletion strains because deletion of SIR2 enhances the recombination rate (i.e. Ade2- rate) up to 20-fold (Bitterman et al., 2002; Gottlieb and Esposito, 1989; Smith and Boeke, 1997). We found that deletion of SLX4 or RAD52 markedly reduced rDNA recombination by 3- and 8-fold, respectively, supporting a role for these two factors in recombination between rDNA repeat sequences (Fig. 3B). In contrast, a saw1Δmutation substantially increased (~ 3-fold) the rDNA recombination rate and the effect is additive to the sir2Δ mutation (Fig. 3B). The results support opposing roles for SLX4 and SAW1 in controlling recombination between rDNA repeats.
Figure 3. Deletion of SLX4 or SAW1 reduced integrity of ribosomal DNA (rDNA) arrays.

A. rDNA recombination assay. A circle is a centromere of chromosome XII. Gray boxes are telomeres. B. Wild type and SIR2 gene deletion derivatives carrying an ADE2 marker integrated into the rDNA array were grown overnight and then plated onto YEPD plate. Colonies were allowed to grow for 48 h at 30°C then placed at 4°C for additional 48 h. The number of half-red half-white colonies that represent a marker loss event during the first cell division after plating were divided by the total number of colonies to calculate rDNA recombination rate. The t-test was used for statistical analysis.
Slx4 and Saw1 catalyze 3’ non-homologous tail removal during recombination
SSA entails multiple steps: end resection and ssDNA formation; annealing of complementary ssDNAs; removal of 3’ single-stranded non-homologous tails; gap fill-in synthesis and ligation. To define which step(s) Slx4 or Saw1 catalyzes during SSA, we first examined the effect of deleting SLX4 or SAW1 on end resection and ssDNA formation in a yeast strain bearing a DSB site flanked by leu2 repeats located 24 kb apart on chromosome III. Specifically, we measured RPA binding to the break site by a ChIP assay using an anti-Rfa1 antibody (Lee and Lee, 2007). Deletion of SLX4 or SAW1 dramatically (9.8- or 10.8-fold, respectively) reduced recombination between the widely spaced leu2 repeat sequences (Fig. 1B), but RPA binding at 5 different regions located 0.2, 0.7, 1.5, 10.0, and 20.0 kb from the centromere distal or proximal side of the break showed no indication of a resection defect (Supplemental Fig. 3). Deletion of MRE11, which is known to delay end resection (Ivanov et al., 1994), was used as a control in this assay and clearly showed a dramatic reduction in RPA binding throughout these regions.
To test the role of Slx4 and Saw1 in the 3’ non-homologous tail removal step of SSA, we employed the well established plasmid-based flap removal assay that monitors the gene conversion frequency between two inverted LacZ repeat sequences, one of which contains the HO recognition sequence (Supplemental Fig. 4)(Colaiacovo et al., 1999). Six different plasmids containing differently sized 3’-flaps at one or both sides of the break were used to define the role of Slx4 and Saw1 in 3’ non-homologous tail removal and compared to that of Rad1. We found that deletion of either SLX4 or SAW1 reduced the 3’ non-homologous tail removal to the level identical to that in a RAD1 deletion (Table 2). Furthermore, slx4Δ rad1Δ or saw1Δ rad1Δ double mutant strains are as defective in 3’-flap removal in the gene conversion assay involving LacZ repeats as are rad1, slx4 or saw1 single gene deletion mutants, indicating these genes work in the same pathway of flap removal. Like the rad1Δ strain, slx4Δ or saw1Δ strains can partially perform gene conversion with either very small (10 to 20 bp) flaps or flaps at only one side of the break (Colaiacovo et al., 1999; Paques and Haber, 1997). Together, the results strongly suggest that Slx4 and Saw1 are involved in the Rad1-dependent 3’-flap removal step during SSA and gene conversion.
Table 2.
Double strand break repair DSB repair (DSBR) efficiency with 3’ non-homologous ends of various sizes in wild type and mutant cells.
| Plasmid retention levels (%)2 |
|||||||
|---|---|---|---|---|---|---|---|
| Plasmid | Wild type | rad1Δ | saw1Δ | slx4Δ | saw1Δ rad1Δ | slx4 Δrad1Δ | slx4Δ saw1Δ |
| pFP122(C→T)1 | 85 | 75 | 84 | 86 | 86 | 81 | 81 |
| pFP121(0/10) | 78 | 65 | 70 | 62 | 58 | 58.7 | 61 |
| pFP118(0/20) | 67 | 33 | 43 | 41 | 31 | 30 | 33 |
| pFP120(308/610) | 40 | 1.1 | 1.2 | 0.9 | 1.1 | 0.8 | 1.0 |
| pFP130 (0/610) | 43 | 13 | 14 | 15 | 16 | 13 | 14 |
| pFP131 (308/0) | 43 | 18 | 14 | 15 | 18 | 16 | 17 |
The numbers in bracket indicates the size of 3’ non-homologous sequence at left side and right side of the broken ends.
The percent plasmid retention was calculated as the ratio of colonies retaining assay plasmids on YEP-galactose media versus those on YEPD. The plasmid retention was calculated from more than 1000 colonies.
To further verify the role of Slx4 and Saw1 in 3’-flap removal, we developed another assay to monitor the kinetics of 3’-flap appearance and removal during SSA in vivo. In this assay, the amount of Sau3AI-resistant ssDNA, part of the 3’-flap after annealing of 205 bp ura3 direct repeats flanking the HO break was measured at several time points after HO expression by real time quantitative PCR (qPCR) using a set of appropriate primers (Fig. 4A). The assay exploits the inability of the restriction endonuclease Sau3AI to cleave the ssDNA that forms after end resection. We found that single-stranded flap DNA accumulated up to 2 h post-DSB induction and gradually disappeared to a level indistinguishable from the pre-induction by 5 h post-HO expression (Fig. 4B). The result is highly consistent with the kinetics of SSA product formation that begins appearing at 2 h post-HO induction (data not shown), suggesting that repair synthesis and ligation occurs quickly after removal of 3’-flaps to complete SSA. The rad1Δ, slx4Δ, and saw1Δ are all markedly deficient in 3’ non-homologous tail removal so that the Sau3AI-resistant single-stranded flaps persisted up to 5 h post-HO expression (Fig. 4B). These defects implicate Slx4 and Saw1 in either the annealing of complementary ssDNA or the 3’-flap removal step of SSA.
Figure 4. Kinetics of 3’ non-homologous tail appearance and removal in SSA.
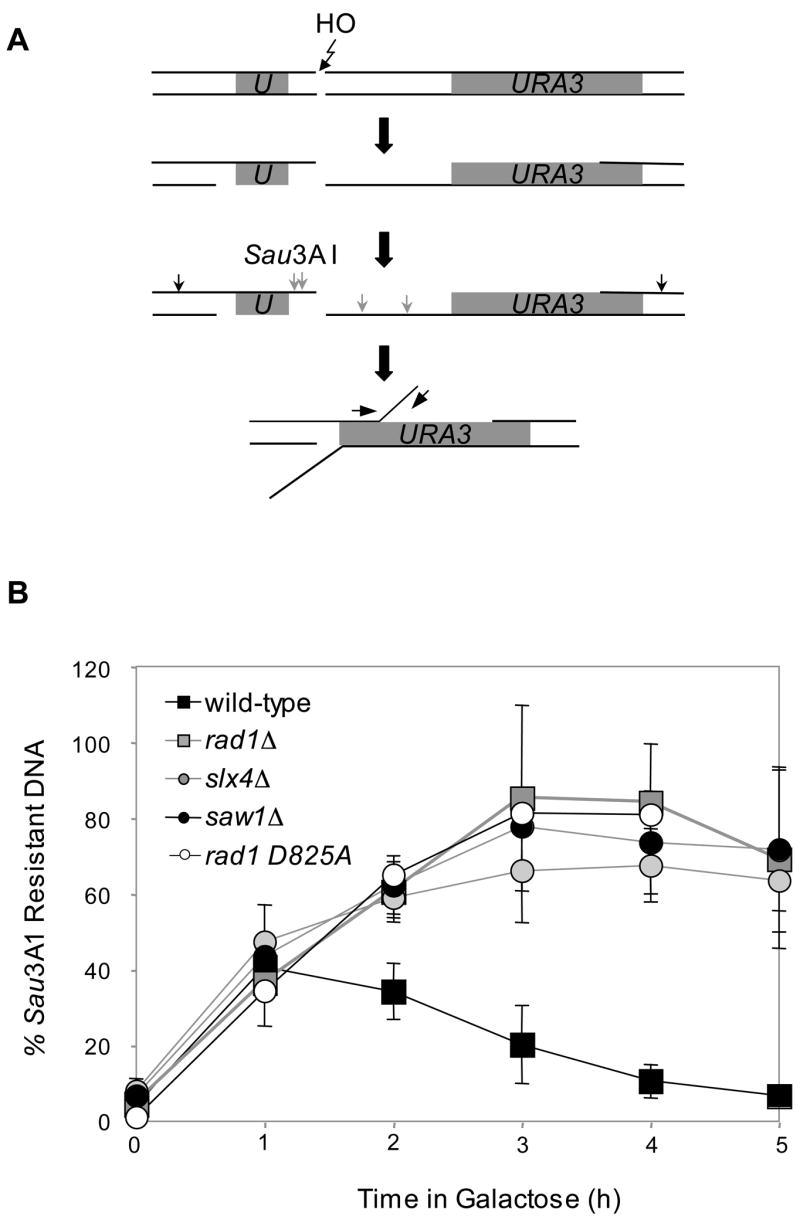
A. qPCR-based 3’-flap detection assay. Direct repeats (grey) and primers used for qPCR (arrows) are shown. B. Kinetics of 3’-flaps in tNS1379 (wild type) and various mutants. Genomic DNA from various mutants was digested by Sau3AI, and used for qPCR with primer sets that anneal to a 3’-flap. PCR results at multiple time intervals after HO expression using a primer set that anneal to a 3’-flap were normalized by control PCR with primers that anneal to ACT1 or PRE1, were shown to indicate kinetics of 3’-flap formation and removal. Data represent means ± s.d. from at least three independent experiments.
Slx4 and Saw1 interact with other proteins involved in 3’-flap removal during homologous recombination
Studies from several research groups previously demonstrated the role of Rad1/Rad10 and Msh2/Msh3 complexes in the removal of 3’ non-homologous flaps during SSA and gene conversion repair of DNA breaks (Fishman-Lobell and Haber, 1992; Ivanov and Haber, 1995; Schiestl and Prakash, 1988, 1990; Tomkinson et al., 1993). We examined whether Slx4 or Saw1 interact with factors involved in recombination and repair using a yeast two-hybrid assay. Slx4 or Saw1 was fused to both the Gal4 DNA binding domain (GBD) and activation domain (GAD) and expressed in yeast strains carrying Rad1, Rad10, Rad52, Rad59, Msh2, and Msh3 fused to the Gal4 activation domain and a HIS3 reporter. As predicted, Slx1-Slx4 and Rad1-Rad10 interactions were readily detected. In addition, we detected an interaction between Rad1 and Slx4 (Fig. 5A).
Figure 5. Physical interaction among SSA proteins.

A. Yeast two-hybrid assays were performed with GAL4 DNA binding domain (GBD) fusion of Slx4 and activation domain (GAD) fusion of Rad1 to detect interaction between these proteins. B. Pull-down assay with GST-Saw1. Yeast crude extract expressing TAP tagged proteins were incubated with GST-Saw1 coated beads for 2 h. After extensive washing with 20 volume of PBS + 0.2% Triton X-100, beads were treated with protein loading buffer and loaded on SDS-PAGE. TAP tagged proteins were detected by PAP reagent (Sigma). C. Saw1 interacts with Rad1 in vivo. FLAG Tagged Saw1 or Rad1 protein was pull down by anti FLAG or anti-Rad1 antibody and the immunoprecipitates were probed with anti-Rad1 or anti-FLAG antibody. D. Saw1 interacts with Msh2 and Rad52. Yeast strain expressing Saw1-TAP and Rad52-13Myc was treated first with 0.1 % MMS for 0.5 h and immunoprecipitated with IgG agarose. The pull-down proteins were hybridized with anti-Rad1, anti-Msh2 or anti-Myc antibodies. E, F, and G. 35S-labeled Rad1, C-terminal nuclease domain deleted rad1 mutant proteins (rad1ΔC), Msh2, and Rad52 obtained by coupled in vitro transcription-translation, were incubated with GST-Saw1 or GST. Proteins associated with GST-Saw1 or GST were isolated on glutathione-Sepharose beads, resolved by SDS-PAGE, and visualized by autoradiography.
Although we did not detect convincing yeast two-hybrid interaction between Saw1 and other recombination proteins, we were able to detect interactions between GST-Saw1 and TAP-tagged Rad1, Msh2, Msh3, Rad52 and Rad51 in a GST pull-down assay in yeast cells (Fig. 5B). The interaction seems highly specific because neither Rad14-TAP nor Ku70-TAP was pulled down by GST-Saw1. We also confirmed physical interactions between Saw1 and other recombination proteins by pull-down assays in yeast strains expressing Saw1-FLAG with anti-FLAG antibody by immunoprecipitation (Fig. 5C). The interactions between Saw1 and Rad1, Msh2 and Rad52-Myc were further confirmed by immunoprecipitation with IgG antibody and immunoblot assay with anti-Rad1, Msh2, and anti-Myc antibodies (Fig. 5D). Interestingly, exposure of cells to 0.1% MMS dramatically enhanced the interaction between Saw1 and Rad52 (Fig. 5D).
We also found that Rad1 likely interacts directly with Saw1, as we verified the interaction using the in vitro transcribed and translated 35S-labeled Rad1 and purified GST-Saw1 in a GST pull-down assay (Fig. 5E). The interaction does not require the intact nuclease domain of Rad1 and withstands ethidium bromide treatment, suggesting that the interaction is not mediated by residual DNA in the reaction. Similarly, we confirmed direct interaction between in vitro transcribed/translated Rad52 or Msh2 with GST-Saw1 (Fig. 5F, 5G).
saw1 mutations blocking Saw1/Rad1 interaction fail to remove the 3’-flap
Physical interaction between Saw1 and proteins known to catalyze 3’-flap removal during recombination prompted us to test the functional significance of the discovered interaction. To achieve this, we generated two mutant alleles of SAW1, saw1Δ18–24 and saw1Δ159–169, each of which bear deletions in highly conserved amino acid residues of Saw1 and other fungal homologs (Fig. 6A). Expression of these saw1 mutants caused a marked reduction in retention of a plasmid upon HO expression that causes a DSB at one of inverted LacZ repeats requiring gene conversion and the associated 3’-flap removal (Fig. 6B).
Figure 6. Interaction between Rad1 and Saw1 is critical for 3’-flap removal in recombination.

A. Positions of deletions in saw1 mutants. B. The effect of saw1 mutations on 3’-flap removal was tested using plasmid based flap removal assay with pFP120 carrying 308 and 610 bp of flaps. The percent plasmid retention were plotted for SAW1 deletion carrying wild type SAW1, saw1Δ18–24 or saw1Δ159–169 mutations. Data represent means ± s.d. from at least three independent experiments. C. Pull-down assay was performed to detect interaction between GST-Saw1 and GST-Saw1 mutants with Rad1 Msh2-TAP, Rad52-13Myc proteins. D. The effect of saw1 mutations on SSA between 205 bp ura3 direct repeat sequences flanked by a HO endonuclease-induced DSB. Southern blot hybridization of BglII digested genomic DNA with the radiolabeled probe next to the URA3 gene was performed to detect SSA product formation in SAW1 gene deletion strain expressing saw1 mutants from yeast centromeric plasmid.
Furthermore, we discovered that the GST-saw1Δ18–24 protein lost ability to interact with Rad1, although it fully retained interactions with Msh2-TAP and Rad52-TAP (Fig. 6C). Expression of these mutant proteins in the saw1Δ strain caused marked reduction in SSA between ura3 direct repeat sequences (Fig. 6D). The results suggest that the physical interaction between Saw1 and Rad1 plays a significant role in Rad1/Rad10-dependent 3’-flap removal during recombination.
Recruitment of Rad1 to 3’-flap depends on Saw1
Rad1/Rad10 is a structure-specific endonuclease that participates in two distinctly different repair pathways: nucleotide excision repair (NER) for removing UV-induced DNA lesions and recombinational repair of DNA breaks (Fishman-Lobell and Haber, 1992; Ivanov and Haber, 1995; Schiestl and Prakash, 1988, 1990; Tomkinson et al., 1993). In NER, Rad1/Rad10 is targeted to UV-induced DNA lesions by physical association with Rad14, the UV lesion recognition protein (Guzder et al., 2006). However, how Rad1/Rad10 is targeted to DNA breaks, is not yet clear. We hypothesized that Saw1 may target Rad1/Rad10 to the recombination intermediate carrying 3’-flaps through physical association with Rad1/Rad10.
To test this model, we monitored the recruitment of hemagglutinin (HA)-tagged Rad1 protein at the vicinity of DSB where 3’-flaps form after annealing of ura3 direct repeats after HO expression using chromatin immunoprecipitation (ChIP) assay (Fig. 7). Nevertheless, we were unable to detect Rad1 at the recombination intermediate carrying 3’-flaps by the ChIP assay. One possible reason for these results is that the Rad1/Rad10 complex may transiently associate with its substrates and then quickly dissociate from the DNA after the cleavage reaction. We thus constructed a rad1 mutant that retains the ability to bind DNA but is defective in DNA cleavage activity. The idea is that rad1 mutant proteins will bind DNA and then will not dissociate from the DNA due to its inability for DNA cleavage, enabling the detection of Rad1 association with the 3’-flap intermediate by ChIP. To generate a nuclease deficient rad1 variant, we substituted the aspartic acid residue at position 825 of Rad1 that shows strong conservation to the active site of XPF (human homologue of Rad1)(Enzlin and Scharer, 2002) to alanine by site-directed mutagenesis, fused the resulting mutant rad1 (rad1-D825A) in-frame with 3xHA tag at its carboxyl terminus, and integrated into the genomic RAD1 locus. The mutation made the cells hypersensitive to UV irradiation (Supplemental Fig. 7) and deficient in 3’-flap removal during SSA (Fig. 4B and Supplemental Fig. 8). Most importantly, we can readily detect the enrichment of rad1-D825A at the 3’-flaps (Fig. 7B, 7C), but not at a site 307 bp distal to the flap (Fig. 7D) from 1 h post HO expression that persists up to 4 h by ChIP. Deletion of SAW1 almost completely abolished the association of rad1-D825A at the recombination intermediates carrying 3’-flaps. Together, the results directly implicate that Saw1 plays a key role in targeting Rad1/Rad10 complex to 3’-flap cleavage substrate in recombination.
Figure 7. Recruitment of Rad1 at the recombination intermediate carrying 3’-flaps requires Saw1 proteins.

A. HO cleavage and the location of primers (arrows) used in the ChIP assay to detect Rad1 and the nuclease deficient rad1 derivative (rad1-D825A) at the recombination intermediate carrying 3’-flap. The levels of Rad1-3HA or rad1-D825A-3HA in wild type or saw1Δ at the DSB were detected by ChIP assay using an anti-HA antibody. Chromatin was isolated at the indicated time after galactose addition, crosslinked, and fragmented by sonication. After immunoprecipitation and reverse crosslinking, purified DNA was analyzed by qPCR using four sets of primers that anneal 0.2 kb (B, JC1/JC2), 2.4 kb (C, JC3/JC4), and 0.5 kb (D, JC5/JC6) to the DSB, as well as primers specific for the PRE1 gene situated on chromosome V as a control. PCR signals from each primer set at different durations of HO expression were quantified and plotted as a graph. IP represents the ratio of the Rad1 PCR signal before and after HO induction, normalized by the PCR signal of the PRE1 control. Each point is the average of two separate experiments.
Discussion
A substantial fraction of the eukaryotic genome contains repetitive DNA. Therefore, recombination between tandem repeat sequences likely provides an important opportunity for DNA DSB repair in eukaryotic cells. Furthermore, the same mechanism accounts for targeted gene replacement in yeast and mammals (Langston and Symington, 2005; Niedernhofer et al., 2001). Even though ten genes were identified for their contributions to SSA, no prior attempt has been made to systematically reveal genetic components involved in SSA. Here we report a novel genetic screen coupling a plasmid-based SSA assay with a microarray-based technique using yeast nonessential deletion pools. The screen uncovered the role of eight SSA genes, among which one is a new recombination gene: SAW1. Further analyses of the SAW1 and SLX4 gene products in different steps of SSA revealed that both Slx4 and Saw1 are needed for efficient Rad1/Rad10-dependent 3’-flap cleavage during homologous recombination. Saw1 is also essential in targeting Rad1 to the recombination intermediates in vivo. The results provide insights into how the Rad1/Rad10 endonuclease recognizes and processes recombination intermediates in a manner distinct from its function in the UV damage repair.
We developed a plasmid-based assay that quantitatively measure the recombination between direct repeat sequences and adapted it to a genome-wide screen that successfully revealed five known SSA genes (RAD1, RAD10, RAD59, MSH2 and MSH3), SLX4, a new SSA gene identified most recently (Flott et al., 2007), and the SAW1 gene previously not linked to SSA. Slx4 catalyzes SSA independently of Slx1, a well-known component for the Slx1-Slx4 endonuclease complex (Flott et al., 2007). The biochemical study showed that the Slx1-Slx4 complex catalyzes 5’-endonucleolytic cleavage of the replication intermediates, whereas Slx4 alone has a weak 3’-endonuclease activity (Fricke and Brill, 2003). The Slx1-independent role of Slx4 in the genotoxic damage repair has been proposed based on the hypersensitivity to MMS in the slx4Δ mutant but not in the slx1Δ mutant (Roberts et al., 2006). Slx4 but not Slx1 is post-translationally modified by Rad53 kinase and required for rDNA stability (Coulon et al., 2004; Flott and Rouse, 2005; Kaliraman and Brill, 2002).
We found that Slx4 is needed for an efficient removal of 3’-flap intermediates during homologous recombination. How does Slx4 stimulate 3’-flap intermediate processing? Slx4 may directly participate in removal of 3’-flaps through its weak 3’ endonuclease activity reported in biochemical study (Fricke and Brill, 2003). Alternatively, Slx4 may assist in formation or stabilization of 3’-flap intermediates and thus indirectly participate in the 3’-flap cleavage. As the analysis of 3’-flap cleavage in the slx4Δ, or rad1Δ slx4Δ strain implicated almost identical substrate specificity for 3’-flap removal with Rad1/Rad10 complex, contribution as a 3’ endonuclease to remove 3’-flap is unlikely. More biochemical studies are needed to elaborate either of these two models further to define the relevant enzymatic activity of Slx4 in homologous recombination.
We identified SAW1 as a new gene responsible for 3’-flap removal that operates in conjunction with Rad1/Rad10 endonuclease. The role of Saw1 in SSA and a subset of gene conversion processes was confirmed by five different recombination assays: the plasmid-based SSA assay, two chromosome-based SSA assays, a plasmid-based gene conversion assay with 3’-flap intermediates, and monitoring of 3’-flap intermediates in vivo. Saw1 likely performs this role by physically interacting with the Rad1/Rad10 endonuclease complex. Saw1 proteins also interact with Msh2/Msh3 and Rad52, and the interaction with Rad52 is induced by DNA damage. Size fractionation of cell extracts using sucrose density gradient ultracentrifugation suggests that Saw1 proteins form a large molecular weight complex and co-eluted at the same fractions with the Rad1 proteins (Supplemental Fig. 5). Furthermore, Saw1 is unstable in a rad1Δ strain, suggesting that all Saw1 likely exists as a complex with Rad1/Rad10 proteins (see Supplemental Fig. 6). Expression of saw1 mutants that disrupt physical interaction with Rad1, but retain interaction with Msh2 and Rad52 almost completely disabled SSA and gene conversion requiring Rad1/Rad10-dependent 3’-flap removal. Altogether, we propose that Saw1 is a new component of the 3’-flap removal complex comprising Rad1/Rad10, Msh2/Msh3 and Rad52 proteins.
Deletion of SAW1 causes a marked deficiency in the 3’-flap removal step during recombination. Saw1 apparently operates in the same pathway with the Rad1/Rad10 complex and Slx4 proteins because mutants deleting both SAW1 and RAD1 or SAW1 and SLX4 exhibited 3’-flap removal defect indistinguishable from each single gene deletion mutant. Furthermore, analysis of 3’-flap cleavage in the absence of SAW1 suggests identical substrate specificity with the Rad1/Rad10 complex and Slx4 proteins. However, the primary difference still exists between these proteins for their role in the repair of UV-induced DNA lesions: Rad1/Rad10 is essential for NER, but Saw1 is completely dispensable. Additionally, saw1 and slx4 mutations have opposing effects on the frequency of recombination at rDNA, despite their common molecular defect: lack of 3′-flap cleavage. We propose that one of these two proteins, besides its role in 3’-flap processing, has a distinct role in the regulation of recombination at the rDNA.
We showed that the rad1-D825A mutant with an amino acid substitution at the nuclease active site conserved between Rad1 and XPF fails to cleave 3’-flaps and produce SSA product but associates with 3’-flap intermediates in a Saw1-dependent manner in vivo, whereas wild type Rad1 is undetectable by ChIP assay at the 3’-flap intermediates. The results are highly congruent with the model that Rad1/Rad10 endonuclease only transiently associates with its substrates and that Saw1 is critical for this interaction. Upon successful cleavage of 3’-flaps from annealed intermediates, Rad1 may not sustain interaction with the recombination intermediates because Rad1 can no longer stably associate with the remaining duplex DNA or be removed along with the disjoined flaps due to tight association with ssDNA. Saw1 may target Rad1/Rad10 through physical association with Rad1/Rad10 and other recombination proteins (including RPA, Rad52 and/or Rad59) that bind to ssDNA after 5’ nucleolytic processing of the DNA break. In mammals, RPA and Rad52 are all implicated in modulating 3’-flap removal reaction (de Laat et al., 1998; Motycka et al., 2004). Saw1 and its interaction with Rad52 and Rad1 seem well suited for this purpose. Indeed, saw1 mutants that fail to interact with Rad1, but retain interaction with Msh2 and Rad52 are severely deficient in 3’-flap cleavage in vivo. Alternatively, Saw1 may target Rad1/Rad10 to the recombination intermediate by recognizing and binding directly to DNA substrates bearing distinct structural motif typical of 3’-flap cleavage.
We initially anticipated that our microarray-based genetic screen should reveal SSA factors in different steps of SSA: end resection, strand annealing, 3’-flap cleavage, gap synthesis, and ligation. However, the recombination defect from all identified SSA mutants except rad59Δ causes accumulation of 3’-flap recombination intermediates. Multiple explanations may account for this unexpected result. First, other recombination reactions prior to the 3’-flap cleavage may be carried out by redundant mechanisms and inactivation of one gene may not produce sufficient defects identifiable in the screen. In line with this premise, end resection depends on both Mre11/Rad50/Xrs2 complex and ExoI protein besides yet unidentified nuclease(s) (Moreau et al., 2001). Therefore, inactivation of either MRE11 or EXO1 did not reduce SSA to the level lower than those of false positives albeit the repair kinetics are significantly delayed (Ivanov et al., 1994). Secondly, HR factors involved in other steps of SSA may be essential for cell survival. Because we used nonessential gene deletion pools for the screen, we could not test the role of essential genes in SSA. An example of this category may include Cdc9, a likely ligase involved in ligation step of recombination but it is essential for DNA lagging strand synthesis (Game et al., 1979) and so mutants are not represented in the pool. Third, slow growth of certain deletion mutants may hamper a reliable assessment of their SSA deficiency by the screen employed here. For instance, the weak hybridization signals from RAD52 gene deletion mutants reflect their slow growth and likely account for the failure to detect a bigger effect on SSA in our screen. The inability to detect the effect of RAD52 deletion on SSA in the current screen leave open the possibility that additional factors important for efficient SSA may yet be undetected from our screen if the corresponding gene deletion mutants grow significantly slower than normal.
For a potentially simple biochemical reaction of the 3’-flap cleavage during recombination require at least 6 proteins among which two (Rad1 and Slx4) have a structure-specific endonuclease activity. Other proteins likely assist in recognition or stabilization of the 3’-flap structure recombination intermediates and position nuclease(s) to the proper target sequences. We propose that analogous biochemical activities are required for equivalent reactions in mammals. Additional molecular details how these proteins and complexes catalyze 3’-flap cleavage require further analysis on the biochemical properties of each SSA component and reconstitution of 3’-flap cleavage reaction using purified proteins and 3’-flap recombination intermediates.
Experimental Procedures
Strains
Strains used in these studies were listed in Supplemental Table 1. All single gene deletion mutants were constructed by using a PCR-derived KANMX module flanked by short terminal sequences homologous to the ends of each single gene open reading frame.
Chromatin immunoprecipitation
ChIP assays were performed as described previously (Lee and Lee, 2007) with both formaldehyde and ethylene glycol bis[succinimidylsuccinate] as crosslinkers (Zeng et al., 2006). Detailed Experimental Procedures for a microarray analysis of mutants and recombination assays and associated references are listed in the Supplemental Material.
Supplementary Material
Acknowledgments
We thank E. Alani, S. Brill, J. Haber, G. Ira, A. Malkova, K. Myung, N. Sugawara, P. Sung, and A. Tomkinson for gifts of strains, plasmids, and antibodies. We also thank J. Haber, P. Sung, D. Walther, and members of the S.E.L. laboratory for helpful discussion. This work is funded by grants from NIH (GM071011) to S.E.L. and HG02432 and RR020839 to J.D.B. S.E.L. is a scholar of the Leukemia and Lymphoma Society.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bai Y, Symington LS. A Rad52 homolog is required for RAD51-independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev. 1996;10:2025–2037. doi: 10.1101/gad.10.16.2025. [DOI] [PubMed] [Google Scholar]
- Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J. 1996;15:5093–5103. [PMC free article] [PubMed] [Google Scholar]
- Colaiacovo MP, Paques F, Haber JE. Removal of one nonhomologous DNA end during gene conversion by a RAD1- and MSH2-independent pathway. Genetics. 1999;151:1409–1423. doi: 10.1093/genetics/151.4.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon S, Gaillard PH, Chahwan C, McDonald WH, Yates JR, 3rd, Russell P. Slx1-Slx4 are subunits of a structure-specific endonuclease that maintains ribosomal DNA in fission yeast. Mol Biol Cell. 2004;15:71–80. doi: 10.1091/mbc.E03-08-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csink AK, Henikoff S. Something from nothing: the evolution and utility of satellite repeats. Trends Genet. 1998;14:200–204. doi: 10.1016/s0168-9525(98)01444-9. [DOI] [PubMed] [Google Scholar]
- de Laat WL, Appeldoorn E, Sugasawa K, Weterings E, Jaspers NG, Hoeijmakers JH. DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev. 1998;12:2598–2609. doi: 10.1101/gad.12.16.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- Enzlin JH, Scharer OD. The active site of the DNA repair endonuclease XPF-ERCC1 forms a highly conserved nuclease motif. EMBO J. 2002;21:2045–2053. doi: 10.1093/emboj/21.8.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman-Lobell J, Haber JE. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science. 1992;258:480–484. doi: 10.1126/science.1411547. [DOI] [PubMed] [Google Scholar]
- Flott S, Alabert C, Toh GW, Toth R, Sugawara N, Campbell DG, Haber JE, Pasero P, Rouse J. Phosphorylation of slx4 by mec1 and tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol Cell Biol. 2007;27:6433–6445. doi: 10.1128/MCB.00135-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flott S, Rouse J. Slx4 becomes phosphorylated after DNA damage in a Mec1/Tel1-dependent manner and is required for repair of DNA alkylation damage. Biochem J. 2005;391:325–333. doi: 10.1042/BJ20050768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke WM, Brill SJ. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 2003;17:1768–1778. doi: 10.1101/gad.1105203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Game JC, Johnston LH, von Borstel RC. Enhanced mitotic recombination in a ligase-defective mutant of the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1979;76:4589–4592. doi: 10.1073/pnas.76.9.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb S, Esposito RE. A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell. 1989;56:771–776. doi: 10.1016/0092-8674(89)90681-8. [DOI] [PubMed] [Google Scholar]
- Guzder SN, Sommers CH, Prakash L, Prakash S. Complex formation with damage recognition protein Rad14 is essential for Saccharomyces cerevisiae Rad1-Rad10 nuclease to perform its function in nucleotide excision repair in vivo. Mol Cell Biol. 2006;26:1135–1141. doi: 10.1128/MCB.26.3.1135-1141.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug N, Lingner J. Telomere length homeostasis. Chromosoma. 2006;115:413–425. doi: 10.1007/s00412-006-0067-3. [DOI] [PubMed] [Google Scholar]
- Ira G, Malkova A, Liberi G, Foiani M, Haber JE. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Haber JE. RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:2245–2251. doi: 10.1128/mcb.15.4.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, Fishman-Lobell J, Haber JE. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics. 1996;142:693–704. doi: 10.1093/genetics/142.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, White CI, Fabre F, Haber JE. Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:3414–3425. doi: 10.1128/mcb.14.5.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FB, Sinclair DA, Guarente L. Molecular biology of aging. Cell. 1999;96:291–302. doi: 10.1016/s0092-8674(00)80567-x. [DOI] [PubMed] [Google Scholar]
- Kaliraman V, Brill SJ. Role of SGS1 and SLX4 in maintaining rDNA structure in Saccharomyces cerevisiae. Curr Genet. 2002;41:389–400. doi: 10.1007/s00294-002-0319-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T. Strategies to maintain the stability of the ribosomal RNA gene repeats–collaboration of recombination, cohesion, and condensation. Genes Genet Syst. 2006;81:155–161. doi: 10.1266/ggs.81.155. [DOI] [PubMed] [Google Scholar]
- Langston LD, Symington LS. Opposing roles for DNA structure-specific proteins Rad1, Msh2, Msh3, and Sgs1 in yeast gene targeting. EMBO J. 2005;24:2214–2223. doi: 10.1038/sj.emboj.7600698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Lee SE. Saccharomyces cerevisiae Sae2- and Tel1-Dependent Single-Strand DNA Formation at DNA Break Promotes Microhomology-Mediated End Joining. Genetics. 2007;176:2003–2014. doi: 10.1534/genetics.107.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR, Yu K, Raghavan SC. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair (Amst) 2006;5:1234–1245. doi: 10.1016/j.dnarep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Lin FL, Sternberg N. Homologous recombination between overlapping thymidine kinase gene fragments stably inserted into a mouse cell genome. Mol Cell Biol. 1984;4:852–861. doi: 10.1128/mcb.4.5.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantiero D, Clerici M, Lucchini G, Longhese MP. Dual role for Saccharomyces cerevisiae Tel1 in the checkpoint response to double-strand breaks. EMBO Rep. 2007;8:380–387. doi: 10.1038/sj.embor.7400911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S, Morgan EA, Symington LS. Overlapping functions of the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNA metabolism. Genetics. 2001;159:1423–1433. doi: 10.1093/genetics/159.4.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motycka TA, Bessho T, Post SM, Sung P, Tomkinson AE. Physical and functional interaction between the XPF/ERCC1 endonuclease and hRad52. J Biol Chem. 2004;279:13634–13639. doi: 10.1074/jbc.M313779200. [DOI] [PubMed] [Google Scholar]
- Mullen JR, Kaliraman V, Ibrahim SS, Brill SJ. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics. 2001;157:103–118. doi: 10.1093/genetics/157.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Essers J, Weeda G, Beverloo B, de Wit J, Muijtjens M, Odijk H, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J. 2001;20:6540–6549. doi: 10.1093/emboj/20.22.6540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA. Origins and consequences of centrosome aberrations in human cancers. Int J Cancer. 2006;119:2717–2723. doi: 10.1002/ijc.22245. [DOI] [PubMed] [Google Scholar]
- Ooi SL, Shoemaker DD, Boeke JD. A DNA microarray-based genetic screen for nonhomologous end-joining mutants in Saccharomyces cerevisiae. Science. 2001;294:2552–2556. doi: 10.1126/science.1065672. [DOI] [PubMed] [Google Scholar]
- Paques F, Haber JE. Two pathways for removal of nonhomologous DNA ends during double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:6765–6771. doi: 10.1128/mcb.17.11.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhova G, Stratton SA, Sung P. Single strand DNA binding and annealing activities in the yeast recombination factor Rad59. J Biol Chem. 1999;274:33839–33842. doi: 10.1074/jbc.274.48.33839. [DOI] [PubMed] [Google Scholar]
- Pierce AJ, Stark JM, Araujo FD, Moynahan ME, Berwick M, Jasin M. Double-strand breaks and tumorigenesis. Trends Cell Biol. 2001;11:S52–59. doi: 10.1016/s0962-8924(01)02149-3. [DOI] [PubMed] [Google Scholar]
- Roberts TM, Kobor MS, Bastin-Shanower SA, Ii M, Horte SA, Gin JW, Emili A, Rine J, Brill SJ, Brown GW. Slx4 regulates DNA damage checkpoint-dependent phosphorylation of the BRCT domain protein Rtt107/Esc4. Mol Biol Cell. 2006;17:539–548. doi: 10.1091/mbc.E05-08-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Prakash S. RAD1, an excision repair gene of Saccharomyces cerevisiae, is also involved in recombination. Mol Cell Biol. 1988;8:3619–3626. doi: 10.1128/mcb.8.9.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiestl RH, Prakash S. RAD10, an excision repair gene of Saccharomyces cerevisiae, is involved in the RAD1 pathway of mitotic recombination. Mol Cell Biol. 1990;10:2485–2491. doi: 10.1128/mcb.10.6.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Boeke JD. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 1997;11:241–254. doi: 10.1101/gad.11.2.241. [DOI] [PubMed] [Google Scholar]
- Stahl MM, Thomason L, Poteete AR, Tarkowski T, Kuzminov A, Stahl FW. Annealing vs. invasion in phage lambda recombination. Genetics. 1997;147:961–977. doi: 10.1093/genetics/147.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenger JE, Lobachev KS, Gordenin D, Darden TA, Jurka J, Resnick MA. Biased distribution of inverted and direct Alus in the human genome: implications for insertion, exclusion, and genome stability. Genome Res. 2001;11:12–27. doi: 10.1101/gr.158801. [DOI] [PubMed] [Google Scholar]
- Storici F, Snipe JR, Chan GK, Gordenin DA, Resnick MA. Conservative repair of a chromosomal double-strand break by single-strand DNA through two steps of annealing. Mol Cell Biol. 2006;26:7645–7657. doi: 10.1128/MCB.00672-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Ira G, Haber JE. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol Cell Biol. 2000;20:5300–5309. doi: 10.1128/mcb.20.14.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Ivanov EL, Fishman-Lobell J, Ray BL, Wu X, Haber JE. DNA structure-dependent requirements for yeast RAD genes in gene conversion. Nature. 1995;373:84–86. doi: 10.1038/373084a0. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Paques F, Colaiacovo M, Haber JE. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc Natl Acad Sci U S A. 1997;94:9214–9219. doi: 10.1073/pnas.94.17.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrower DA, Stemple J, Yeh E, Bloom K. Nuclear oscillations and nuclear filament formation accompany single-strand annealing repair of a dicentric chromosome in Saccharomyces cerevisiae. J Cell Sci. 2003;116:561–569. doi: 10.1242/jcs.00251. [DOI] [PubMed] [Google Scholar]
- Tomkinson AE, Bardwell AJ, Bardwell L, Tappe NJ, Friedberg EC. Yeast DNA repair and recombination proteins Rad1 and Rad10 constitute a single-stranded-DNA endonuclease. Nature. 1993;362:860–862. doi: 10.1038/362860a0. [DOI] [PubMed] [Google Scholar]
- Umezu K, Sugawara N, Chen C, Haber JE, Kolodner RD. Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics. 1998;148:989–1005. doi: 10.1093/genetics/148.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman C, Kanaar R. DNA double-strand break repair: all's well that ends well. Annu Rev Genet. 2006;40:363–383. doi: 10.1146/annurev.genet.40.110405.090451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.