

Abstract
Members of the transforming growth factor β (TGF-β) superfamily are involved in diverse physiological activities including development, tissue repair, hormone regulation, bone formation, cell growth, and differentiation. At the cellular level, these functions are initiated by the interaction of ligands with specific transmembrane receptors with intrinsic serine/threonine kinase activity. The signaling pathway that links receptor activation to the transcriptional regulation of the target genes is largely unknown. Recent work in Drosophila and Xenopus signaling suggested that Mad (Mothers against dpp) functions downstream of the receptors of the TGF-β family. Mammalian Mad1 has been reported to respond to bone morphogenetic protein (BMP), but not to TGF-β or activin. We report here the cloning and functional studies of a novel mammalian Mad molecule, Mad3, as well as a rat Mad1 homologue. Overexpression of Mad3 in a variety of cells stimulated basal transcriptional activity of the TGF-β/activin-responsive reporter construct, p3TP-Lux. Furthermore, expression of Mad3 could potentiate the TGF-β- and activin-induced transcriptional stimulation of p3TP-Lux. By contrast, overexpression of Mad1 inhibited the basal as well as the TGF-β/activin induced p3TP-Lux activity. These findings, therefore, support the hypothesis that Mad3 may serve as a mediator linking TGF-β/activin receptors to transcriptional regulation.
Keywords: signal transduction, activin, bone morphogenetic factor, cloning, serine kinase
The transforming growth factor β (TGF-β) superfamily comprises a large group of growth and differentiation factors identified in a wide variety of species from insects to mammals. Among the most studied members are TGF-βs, activins, and bone morphogenetic proteins (BMPs). These polypeptides have been reported to mediate numerous physiological processes including cell differentiation, tissue repair, bone formation, regulation of hormone secretion, immune response, and various developmental functions in many organisms (1, 2). For example, the decapentaplegic (dpp) product in Drosophila, a BMP homologue, is found to play a role in the determination of cell fate during development (3, 4). In Xenopus, BMP mediates ventralization of mesoderm and timely removal of this signal is required for neural induction (5–7). On the other hand, activin induces formation of dorsal mesoderm (5).
Members of the TGF-β superfamily exert their biological effects by interacting with two types of transmembrane receptors (type I and II) with serine/threonine kinase activity (1, 2, 8). The type II receptors of TGF-β and activin are involved in initial ligand binding. The ligand-bound type II receptors recognize and recruit type I receptors and a heteromeric complex is formed. In this complex, the type I receptors are transphosphorylated by the kinase domain of the type II receptors, eliciting the propagation of downstream signals (9, 10). The ligand interaction is different for BMP receptors in that either the type I or the type II receptors are capable of low affinity interaction with the BMP ligands. However, both receptors together enable high affinity binding with the ligand and the formation of the heteromeric ligand–receptor complex is required for BMP signaling. Although the initial steps in the activation of the serine kinase receptors have been defined, little is known of their downstream mediators. Several proteins were identified on the basis of their interaction with either type I or type II receptors—e.g., farnesyl transferase-α (11, 12), FKBP-12 (13, 14), TRIP-1 (15), and a protein involved in the mitogen-activated protein kinase pathway (16, 17). However, functional links between these proteins and the receptors of TGF-β family remain to be established.
The search for mediators of the signal transduction pathways of TGF-β family members has been recently expedited by genetic studies in Drosophila. A protein called Mad (Mothers against dpp) was isolated by its involvement in the signaling of dpp, as the loss-of-function of Mad could interact with dpp alleles in embryonic dorsal–ventral patterning and adult appendage defects (18, 19). Based on their sequence similarities to Mad and their involvement in the formation of dauer larvae, three Mad homologues (Sma-2, Sma-3, and Sma-4) in Caenorhabditis elegans were also identified (20). More recently, mammalian homologues of Mad1 and Mad2 have been isolated (21–24). Mad1 was reported to mediate the BMP signaling (21, 25). This mammalian Mad1 was shown to be phosphorylated and translocated into the nucleus upon BMP stimulation. Interestingly, fusion of the C terminus of Mad1 with the yeast DNA binding protein GAL4 conferred transcriptional activation on a GAL4 reporter gene (25). This transactivating activity was not specific for Mad1, because the same phenomenon was also observed with the C-terminal domain of DPC4, another Mad-related protein that may function as a tumor suppressor in pancreatic cancers (26). The putative tumor suppressor function of Mad proteins was further supported by the finding that mutations of a human Mad2 homologue have been detected in some colorectal cancers. Two Xenopus Mad proteins were also identified and reported to mediate differential mesoderm induction in the Xenopus embryos (22). Injection of the Xenopus Mad1 mRNA induced the formation of ventral mesoderm, characteristic of a signal from BMP (22). On the other hand, injection of the Xenopus Mad2 mRNA mediated the induction of dorsal mesoderm, reminiscent of the signals downstream of activin, Vg1, and nodal (22).
We describe here the cloning and transcriptional regulation of two rat Mad proteins, Mad1 and Mad3. Sequence analysis reveals that the rat Mad1 is a rat homologue of human and Xenopus Mad1. The sequence of the rat Mad3 is similar to but distinct from the reported mammalian and Xenopus Mad2s. Overexpression of Mad3 leads to ligand-independent stimulation of p3TP-Lux transcription, whereas overexpression of the rat Mad1 blocks it. Immunofluorescence staining with a myc-tagged Mad3 reveals that Mad3 is predominantly localized in the nucleus independent by ligand stimulation. Furthermore, Mad3 can potentiate the transcriptional response of TGF-β and activin. These findings suggest that Mad3 differs from Mad1 in transcriptional regulation and that Mad3 may play a role in the signal transduction of TGF-β/activin receptors.
MATERIALS AND METHODS
Cell Culture.
The mink lung epithelial cell (Mv1Lu) and its derivative (R-1B) were maintained in minimal essential medium (MEM) containing 10% fetal bovine serum (FBS) and nonessential amino acids. The Chinese hamster cell line (CHO-K1) was cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS. A clone of the human erythroleukemia cell line (K562) that stably overexpresses isopropyl β-d-thiogalactoside (IPTG)-inducible activin type IB and type II receptors (J.-J.L. and W.V., unpublished) was cultured in RPMI 1640 medium with 10% FBS.
Library Screening, Cloning, and Reverse Transcription (RT)–PCR.
The full-length cDNA insert of the Drosophila Mad (25) was used to screen a rat brain cDNA library (27) at low stringency. The hybridization was performed at 52°C in 5× Denhardt’s solution, 5× SSPE (1× SSPE = 150 mM NaCl/10 mM sodium phosphate/1 mM EDTA, pH 6.8), 0.5% SDS, and 100 μg/ml of denatured salmon sperm DNA. The last wash was performed at 50°C in 0.5× standard saline citrate (SSC) and 0.1% SDS. Two independent isolates of rat Mad1 were isolated and the cDNA insert of the rat Mad1 clone was used to screen the same library under identical condition. Several Mad-related clones were isolated and sequenced from both strands (Sequenase version 2; United States Biochemical). The full-length cDNA inserts of Mad1 and another Mad clone, named Mad3, were subcloned into the mammalian expression vector pcDNA3 (Invitrogen) for expression studies in cell culture. For RT–PCR, two primers designed from the nucleotide sequences of mouse and human Mad2 (CTTGCCATTCACNCCGCCAG and CGGCAATATATAACATGTGGCAA) were used in a PCR with the cDNA transcribed from poly(A) RNA of the rat whole brain (first strand cDNA synthesis kit; Boehringer Mannheim). The PCR product was cloned into pBluescript (Stratagene) and subjected to sequencing.
Generation of a myc Epitope Tagged Form of Mad3.
To generate the N-terminal myc-epitope tagged Mad3, an oligonucleotide containing the myc tag sequence (CCGCTCGAGTCGACATGGCAGAGGAGCAAAAGCTCATTTCTGAAGAGGACTTGTTGTCGTCCATCCTGCCCTTCACCCC) was used in PCR with another primer that is located about 400 bp downstream of the putative initiation codon of Mad3 (CTGGAGGTAGCACTGGTGTCTCTA). The PCR product was swapped with the N-terminal region of the Wild-type Mad3 and confirmed by sequencing.
Luciferase and β-Galactosidase (β-gal) Assays.
Transfection for promoter assays was performed by calcium phosphate method. In brief, about 5 × 104 Mv1Lu, R-1B, and CHO cells were plated in 60-mm dishes a day before transfection. The cells were transfected with 1 μg of p3TP-Lux, 1 μg pCMV-β-gal, and 5 μg of other DNAs as indicated for each experiment. The transfected cells were washed the next day and allowed to grow for 24 hr in a medium containing 10% FBS. The medium was then switched to the low-serum one (0.2% FBS) and the cells were incubated for 2 hr. In the ligand treated groups, cells were incubated for 8 hr in the same medium that contained activin (25 ng/ml) or TGF-β1 (2.5 ng/ml). The cells were harvested by scraping with a rubber policeman and used for luciferase assays as described (28). Total light emission during the initial 10 sec was measured in a luminometer. The β-gal assay was performed with the same samples as described (28). The luciferase assay results were normalized for β-gal activity for all data described.
Immunofluorescence Staining.
Transfection for immunofluorescence studies was performed by the lipofectamine method (GIBCO/BRL). About 5 × 104 R-1B and CHO cells were transfected with 1 μg of myc-tagged Mad3 in pcDNA3 and split 1 day later into a 6-well plate that had glass coverslips on the bottom. The cells were allowed to attach and grow for 24 hr. The medium was switched to DME with 0.2% FBS for 1 hr and then treated with activin (25 ng/ml) or TGF-β1 (2.5 ng/ml) for 1 hr before fixation. The coverslips were washed three times with Hepes dissociation buffer (HDB) and fixed with 4% paraformaldehyde/0.1% Triton X-100 for 40 min. Following fixation, the coverslips were incubated with a monoclonal anti-myc antibody (9E10) at 2 μg/ml for 1 hr in the presence of 5% FBS. The coverslips were washed with HDB and incubated with a BODYPY-tetramethylrhodamine-conjugated goat anti-mouse antibody (1:100 dilution; Molecular Probes) for 30 min. A Hoechst stain (no. 33258; Sigma) was used for nuclear staining. The fluorescence was visualized and photographed on a Leitz Dialux 22 microscope with appropriate filters.
RESULTS AND DISCUSSION
Cloning of Rat Mad1 and Mad3.
To identify mammalian Mad homologues, we screened a rat brain cDNA library with a probe made from the full-length cDNA of Drosophila Mad (25). The initial screening yielded two independent clones with the same sequence in their putative protein coding region. This clone was later found to be a rat homologue of the human and Xenopus Mad1 (Fig. 1). Using the cloned rat Mad1 cDNA as a probe, we screened the same library and isolated several new clones.
Figure 1.
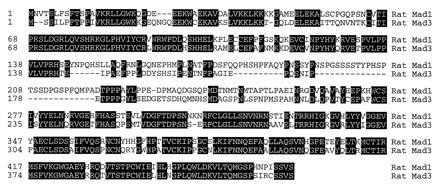
Amino acid sequences of rat Mad1 and Mad3. The putative amino acid sequences of rat Mad1 and Mad3 were aligned with the megalign program (DNAstar, Madison, WI). The conserved residues are highlighted in black boxes, and the positions of the amino acid residues are labeled on the left.
One of these clones, which we named Mad3, possesses distinctive sequence features when compared with reported Mad1 and Mad2 proteins (Figs. 1 and 2). The sequence of the rat Mad3 is more similar to Mad2 than to Mad1. Its amino acid sequence shares 89% identity to mouse, human and Xenopus Mad2 homologues (22–24), but only 61% to human or Xenopus Mad1 (21, 22, 26). Differences between Mad2 and -3 are scattered throughout the proteins. In addition, Mad3 is missing two stretches of 10 (residues 21–30 of Mad2) and 30 (residues 79–109 of Mad2) amino acids. To confirm that Mad2 and Mad3 are from different genes, we used a RT–PCR strategy to isolate the rat Mad2 homologue. Two primers that cover the partial N-terminal region of Mad2 were designed on the basis of the human and mouse Mad2 sequences. RT–PCR with the cDNA made from the rat brain RNA generated a rat Mad2 clone that revealed a nucleotide sequence 98% identical to mouse Mad2, but only 78% homologous to rat Mad3 (Fig. 2A). The amino acid sequences of the two rat Mads were further compared with other Mad members and are shown as a phylogenetic tree (Fig. 2B). The rat Mad1 is highly homologous to the human and Xenopus Mad1. Its putative protein sequence is 97% identical to human Mad1 (21, 25) and 93% to Xenopus Mad1 (22). The Drosophila Mad (19) is closely related to the mammalian and Xenopus Mad1 proteins (21, 22, 25).
Figure 2.
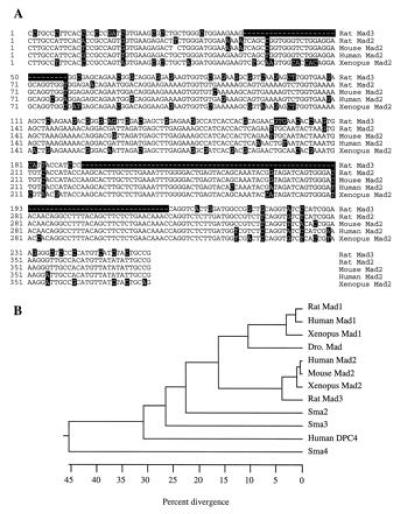
Sequence comparison of different Mads. (A) Comparison of the nucleotide sequence of rat Mad3 with other Mad2 clones of different species. The rat Mad2 sequence was derived from RT–PCR. Only the region that covers the RT–PCR is compared. The nucleotides that are different from the majority are highlighted in black boxes. (B) Phylogenetic tree of the Mad proteins. The numbers below the tree indicate the percent divergence among the amino acid sequences. The sequences used here include the two rat Mads, the human Mad1 (21, 25), the mouse Mad2 (24), the human Mad2 or JV18-1 (23), the human DPC4 (26), the Xenopus Mad1 and Mad2 (22), the Drosophila Mad (19), and the three C. elegans Mads, Sma2 to -4 (20).
The Drosophila Mad, which is homologous to mammalian Mad1, was isolated through genetic interaction with dpp, a BMP homologue in Drosophila. The mammalian Mad1 has been shown to respond to BMP and the Xenopus homologue could induce mesoderm ventralization (22), a downstream event of BMP stimulation. Both rat Mad3 and Mad1 are distantly related to the three Mad proteins (20) in C. elegans (Sma2, -3, and -4), as well as to DPC4 (26), a putative tumor suppressor in human pancreatic cancers. Similar to other Mad related proteins, rat Mad1 and Mad3 contain no significant sequence motifs—e.g., signal sequence, transmembrane domain, and DNA binding domain.
Transcriptional Stimulation by Mad3.
To determine if the cloned rat Mad3 functions downstream of TGF-β/activin, we tested the effect of expressing Mad3 protein on the transcriptional activity of a TGF-β/activin-responsive promoter construct, p3TP-Lux (29–32). The p3TP-Lux contains 3 repeats of a 12-O-tetradecanoylphorbol 13-acetate (TPA)-responsive element (TRE) plus the plasminogen activator inhibitor (PAI) promoter. This promoter construct has been shown to be stimulated by both TGF-β and activin through the mediation of their respective receptors (10, 29–32). The Mad3 cDNA sequence was cloned into a mammalian expression vector, pcDNA3, and was transiently transfected with p3TP-Lux into a variety of cell lines. As shown in Fig. 3A, overexpression of Mad3 could stimulate the basal p3TP-Lux activity almost 30-fold in the R-1B cell line, a variant of Mv1Lu cells. Similar transient expression experiments were also carried out in other cell lines including Mv1Lu, CHO, and K562 cells. Overexpression of the rat Mad3 (1–5 μg DNA) in these cells increased the basal p3TP-Lux activity by 3- to 6-fold (data not shown), lower than the stimulation observed in R-1B cells. The mechanisms underlying the different transcriptional stimulation by Mad3 in different cells remains to be determined. Interestingly, overexpression of Xenopus Mad2 in R-1B cells had no effect on the basal p3TP-Lux activity in R-1B cells (data not shown). This functional difference in transcriptional stimulation between Mad2 and Mad3 further suggests that the rat Mad3 is not a Mad2 analogue.
Figure 3.
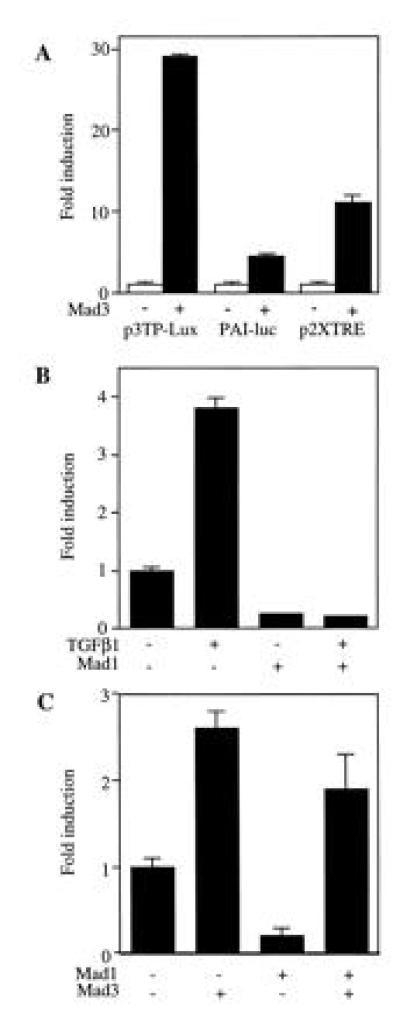
Differential regulation of transcription by Mad3 and Mad1. (A) Transactivation activity of Mad3. R-1B cells were transfected with Mad3 or vector (2 μg) plus 1 μg of p3TP-Lux, p2XTRE-luc or PAI-luc. The luciferase activity was normalized with β-gal activity. The fold change of the luciferase activity as compared with the value of vector-transfected cell (set to 1) is presented. Data from one representative experiment was shown as mean ± SEM, and the same experiment was repeated at least twice with similar results. (B) Inhibition of p3TP-Lux activity by Mad1. Mad1 DNA (5 μg) and p3TP-Lux (1 μg) were transfected into Mv1Lu cells and the luciferase activity of p3TP-Lux was detected after TGF-β1 treatment. The fold change of luciferase activity were compared with the value of unstimulated and vector-transfected cells. (C) Mad3 is dominant over Mad1 in regulating p3TP-luc activity. Mad1 (5 μg) was cotransfected with Mad3 (0.5 μg) and p3TP-Lux (1 μg) in Mv1Lu cells and the basal p3TP-Lux activity was determined.
To further dissect the promoter elements that are responsible for the Mad3 stimulation of p3TP-Lux, we tested two other promoter constructs in R-1B cells. Mad3 was cotransfected with p2XTRE or PAI-luc. The p2XTRE luciferase construct contains multiple repeats of the core consensus binding site for the activator protein AP-1 (33, 34). The PAI-luc possesses a TGF-β responsive plasminogen activator inhibitor gene element without the three TRE repeats added in p3TP-Lux (35). Overexpression of rat Mad3 in R-1B cells stimulated p2XTRE activity by 8-fold and the PAI-luc activity by 4-fold (Fig. 3A). Therefore the overall stimulation of p3TP-Lux by Mad3 (about 30-fold) could be accounted for by the addition of the individual effect of Mad3 on the TRE and PAI elements. These data implicate the TRE in the transcriptional regulation by Mad3. An AP-1 site has been shown to be involved in the TGF-β-mediated transcriptional regulation of the human pro-collagen type II gene promoter (36, 37) and monocyte chemoattractant promoter (38).
The finding that Mad3 could stimulate transcriptional activity provided an insight into the possible roles that Mad proteins may play during transcriptional regulation. The C-terminal regions of mammalian Mad1 and DPC4 have been reported to stimulate the transcriptional activity of a GAL4 reporter construct when fused with the DNA binding domain of yeast GAL4 (25). Interestingly, full-length Mad1 had only weak transactivating ability. It has been postulated that BMP stimulation could relieve Mad1 of the inhibitory effect of its N-terminal domain. Our findings with Mad3 further raise the possibility that Mad proteins might function as transcription factors or coactivators. Genetics studies in Drosophila have identified a putative zinc finger containing transcription factor, schnurri, that functions downstream of dpp (39–41). It will be interesting to determine the possible functional link between Mad and schnurri or the mammalian schnurri homologues.
Inhibition of p3TP-Lux Activity by Mad1.
Overexpression of rat Mad1 inhibited p3TP-Lux activity in a variety of cells. Expression of rat Mad1 in Mv1Lu cells reduced basal p3TP-Lux activity, as well as the TGF-β1-induced activity (Fig. 3B). Mad1 also inhibited the basal and activin stimulated p3TP-Lux activities in CHO and K562 cells (data not shown). Interestingly, the stimulatory effect of Mad3 on p3TP-Lux activity was dominant over the inhibitory effect of Mad1. As shown in Fig. 3C, coexpression of Mad1 with Mad3 at a 10:1 ratio could not abolish the stimulatory effect of Mad3 on p3TP-Lux, although it remains to be determined if the proteins are made in proportion to the input of DNA.
Mad3 Is Predominantly Localized in Nucleus.
To aid in understanding the nuclear function of Mad3, we studied its subcellular localization in R-1B and CHO cells. A myc epitope tagged Mad3 was generated and used for transient transfection experiment. Expression of the myc-tagged Mad3 in these cells gave rise to a single protein band with molecular weight of about 60 kDa in Western blotting (data not shown). This tagged protein was fully functional in stimulation of basal p3TP-Lux (data not shown). The myc-tagged Mad3 was transfected into R-1B and CHO cells and detected by immunofluorescence staining with an anti-myc antibody. The location of the nucleus was visualized by Hoechst staining. In the absence of ligand stimulation, the ectopically expressed Mad3 was predominantly located inside the nucleus in R-1B cells (Fig. 4 A and B) and CHO cells (Fig. 4 C and D). Since expression of Mad3 in these cells was accomplished by transient transfection which was only 10–20% efficient, many cells did not express the tagged Mad3 protein, as demonstrated by a positive nuclear staining but a negative anti-myc staining. Coexpression of the rat Mad3 with TGF-β/activin type I receptors, or treatment of these cells with activin or TGF-β1, had no effect on the subcellular localization of Mad3 (data not shown). Serum starvation of these cells for 24 hr did not change the localization as well (data not shown).
Figure 4.

Mad3 is predominantly localized in nucleus. R-1B and CHO cells were transfected with 1 μg of myc-tagged Mad3 DNA construct and used in immunofluorescence staining with a monoclonal anti-myc antibody. Localization of Mad3 was visualized with a BODYPY-tetramethylrhodamine-conjugated anti-mouse antibody (A, R-1B cells; C, CHO cells). Locations of nuclei of the same cells were visualized by Hoechst staining and shown next to the corresponding micrographs (B, R-1B cells; D, CHO cells).
The subcellular localization of Mad3 was different from that reported for mammalian Mad1 (21, 25). The transfected human Mad1 is localized in the cytoplasmic compartment of unstimulated cells and BMP treatment leads to its phosphorylation and nuclear translocation (21, 25). The nuclear localization of Mad3 in unstimulated cells may explain its ability to stimulate transactivation in the absence of ligand. However, it remains to be documented if the endogenous Mads have the same localization patterns as the ectopically expressed proteins.
It is presently unclear how the serine kinase receptors of TGF-β family transmit their signals to Mad3 and if phosphorylation of this protein plays a role in its nuclear localization. Since the serine kinase receptors are localized in the plasma membrane, it is unlikely that Mad3 is a direct target of the these receptors.
Mad3 Potentiates the Transcriptional Response of TGF-β and Activin Receptors.
To determine if Mad3 is involved in TGF-β and activin signaling, we tested the ability of Mad3 to potentiate a TGF-β-dependent transcriptional response. Different amounts of Mad3 were expressed in Mv1Lu cells and the p3TP-Lux activity was assayed after TGF-β1 stimulation. As shown in Fig. 5A, transfection with Mad3 led to a dose-dependent increase of basal p3TP-Lux activity. Furthermore, expression of Mad3 potentiated the transcriptional response to TGF-β. The TGF-β1-induced fold increase of p3TP-Lux activity was elevated from 5 times (in the absence of Mad3) to 10–12 times (in the presence of Mad3). A similar result was also observed in R-1B cells that were cotransfected with a plasmid expressing the type I receptor of TGF-β. Expression of Mad3 in these cells enhanced the TGF-β mediated p3TP-Lux stimulation (data not shown). These data are consistent with the hypothesis that Mad3 is involved in the signal transduction of TGF-β receptors.
Figure 5.
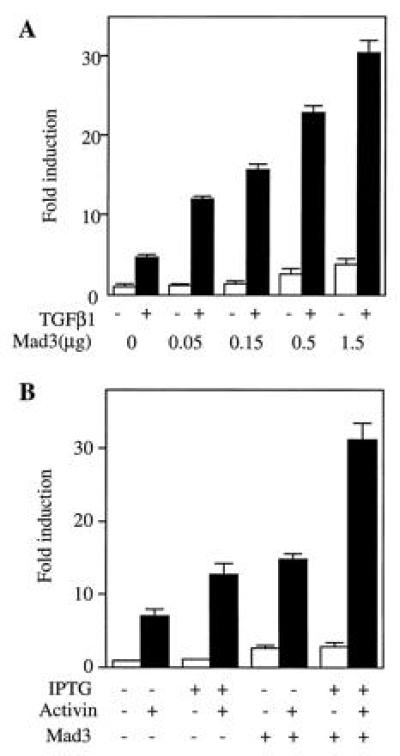
Mad3 potentiates the TGF-β and activin induced p3TP-Lux activities. (A) Mad3 potentiates transcriptional response of TGF-β1 in Mv1Lu cells. Different amounts of Mad3 DNA (0.05 to 1.5 μg) were transiently expressed in Mv1Lu cells, and the basal and TGF-β1-stimulated p3TP-Lux activities were determined and compared with the value of the vector-transfected cells. (B) Mad3 DNA (1 μg/million cells) was transfected into K562 cells containing IPTG-inducible type I and type II activin receptors. The transfected cells were treated with activin in the presence or absence of IPTG, and the fold changes of p3TP-Lux activity were determined by comparison with the value of vector-transfected and unstimulated cells.
Many aspects of the signal transduction pathways of activin receptors are similar to those of TGF-β receptors. To test if Mad3 could potentiate activin mediated p3TP-Lux stimulation, we expressed rat Mad3 in a clone of the human erythroleukemia cell line (K562) that stably expresses IPTG inducible activin type IB and type II receptors (J.-J.L. and W.V., unpublished work). IPTG treatment of these cells enhanced their responsiveness to activin (Fig. 5B) as a result of the inducible expression of both type I and type II activin receptors. Transient transfection of rat Mad3 elevated the basal p3TP-Lux activity by 2- to 3-fold (Fig. 5B). Furthermore, Mad3 expression increased activin-dependent p3TP-Lux stimulation to more than 30-fold when both type I and type II activin receptors were induced to overexpress (Fig. 5B).
In conclusion, two rat Mad proteins were cloned from a rat brain cDNA library. One is a rat Mad1 homologue. The second one, named Mad3, had unique sequence features distinct from Mad1 and Mad2. Expression of rat Mad1 and Mad3 in a variety of cells revealed different effects on the transcriptional activity of p3TP-Lux. Overexpression of Mad3 stimulated basal p3TP-Lux activity, whereas expression of Mad1 inhibited it. Furthermore, Mad3 was found to enhance both TGF-β and activin induced transcriptional responses. These findings suggest that Mad3 may serve as a mediator that links the TGF-β/activin receptors to the transcriptional regulation of their target genes.
Note Added in Proof.
Since submission of this manuscript, a human Mad3 homologue was reported by Zhang et al. (42) and its amino acid sequence is 99% identical to the rat Mad3 described here.
Acknowledgments
We thank W. M. Gelbart for providing the Drosophila Mad clone. We thank T. Hunter for critical reading of the manuscript. We also thank D. Gaddy-Kurten, K. Pickham, and E. Wiater for discussions and reading of the manuscript. This work was supported by National Institutes of Health Grant PO1 HD3527 and the Foundation for Medical Research, Inc. (to W.V.) and a American Cancer Society postdoctoral fellowship (to Y.C.) French Cancer Research Asso. and Adler Fdn (to J.-J. Lebron). W.V. is a Foundation for Medical Research Senior Investigator.
Footnotes
Abbreviations: TGF-β, transforming growth factor β; BMP, bone morphogenetic protein; dpp, decapentaplegic; Mad, Mothers against dpp; TRE, TPA responsive element; PAI, plasminogen activator inhibitor; IPTG, isopropyl β-d-thiogalactoside; RT–PCR, reverse transcription–PCR; β-gal, β-galactosidase; FBS, fetal bovine serum.
Data deposition: The sequences reported in this paper have been deposited in the GenBank data base [accession nos. U66478U66478 (Mad1) and U66479U66479 (Mad3)].
References
- 1.Massague J, Attisano L, Wrana J L. Trends Cell Biol. 1994;4:172–178. doi: 10.1016/0962-8924(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 2.Gaddy-Kurten D, Tsuchida K, Vale W. Recent Prog Horm Res. 1995;50:109–129. doi: 10.1016/b978-0-12-571150-0.50010-x. [DOI] [PubMed] [Google Scholar]
- 3.Padgett R W, St. Johnston R D, Gelbart W M. Nature (London) 1987;325:81–84. doi: 10.1038/325081a0. [DOI] [PubMed] [Google Scholar]
- 4.Ferguson E L, Anderson K V. Cell. 1992;71:451–461. doi: 10.1016/0092-8674(92)90514-d. [DOI] [PubMed] [Google Scholar]
- 5.Smith J C. Curr Opin Cell Biol. 1995;7:856–861. doi: 10.1016/0955-0674(95)80070-0. [DOI] [PubMed] [Google Scholar]
- 6.Hawley S H, Wunnenberg-Stapleton K, Hashimoto C, Laurent M N, Watabe T, Blumberg B W, Cho K W. Genes Dev. 1995;9:2923–2935. doi: 10.1101/gad.9.23.2923. [DOI] [PubMed] [Google Scholar]
- 7.Wilson P A, Hemmati-Brivanlou A. Nature (London) 1995;376:331–333. doi: 10.1038/376331a0. [DOI] [PubMed] [Google Scholar]
- 8.Mathews L S, Vale W W. Cell. 1991;65:973–982. doi: 10.1016/0092-8674(91)90549-e. [DOI] [PubMed] [Google Scholar]
- 9.Wrana J L, Attisano L, Wieser R, Ventura F, Massague J. Nature (London) 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 10.Attisano L, Wrana J L, Montalvo E, Massague J. Mol Cell Biol. 1996;16:1066–1073. doi: 10.1128/mcb.16.3.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawabata M, Imamura T, Miyazono K, Engel M E, Moses H L. J Biol Chem. 1995;270:29628–29631. doi: 10.1074/jbc.270.50.29628. [DOI] [PubMed] [Google Scholar]
- 12.Wang T, Danielson P D, Li B, Shah P C, Kim S D, Donahoe P K. Science. 1996;271:1120–1122. doi: 10.1126/science.271.5252.1120. [DOI] [PubMed] [Google Scholar]
- 13.Wang T, Donahoe P K, Zervos A S. Science. 1994;265:674–676. doi: 10.1126/science.7518616. [DOI] [PubMed] [Google Scholar]
- 14.Wang T, Li B, Danielson P D, Shah P C, Rockwell S, Lechleider R L, Martin J, Manganaro T, Donahoe P K. Cell. 1996;86:435–444. doi: 10.1016/s0092-8674(00)80116-6. [DOI] [PubMed] [Google Scholar]
- 15.Chen R H, Miettinen P J, Maruoka E M, Choy L, Derynck R. Nature (London) 1995;377:548–552. doi: 10.1038/377548a0. [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 17.Shibuya H, Yamaguchi K, Shirakabe K, Tonegawa A, Gotoh Y, Ueno N, Irie K, Nishida E, Matsumoto K. Science. 1996;272:1179–1182. doi: 10.1126/science.272.5265.1179. [DOI] [PubMed] [Google Scholar]
- 18.Raftery L A, Twombly V, Wharton K, Gelbart W M. Genetics. 1995;139:241–254. doi: 10.1093/genetics/139.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sekelsky J J, Newfeld S J, Raftery L A, Chartoff E H, Gelbart W M. Genetics. 1995;139:1347–1358. doi: 10.1093/genetics/139.3.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savage C, Das P, Finelli A L, Townsend S R, Sun C Y, Baird S E, Padgett R W. Proc Natl Acad Sci USA. 1996;93:790–794. doi: 10.1073/pnas.93.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoodless P A, Haerry T, Abdollah S, Stapleton M, O’Connor M B, Attisano L, Wrana J L. Cell. 1996;85:489–500. doi: 10.1016/s0092-8674(00)81250-7. [DOI] [PubMed] [Google Scholar]
- 22.Graff J M, Bansal A, Melton D A. Cell. 1996;85:479–487. doi: 10.1016/s0092-8674(00)81249-0. [DOI] [PubMed] [Google Scholar]
- 23.Riggins G, J, Thiagalingam S, Rosenblum E, Weinstein C L, Kern S E, Hamilton S R, Willson J K V, Markowitz S D, Kinzler K W, Vogelstein B. Nat Genet. 1996;13:347–349. doi: 10.1038/ng0796-347. [DOI] [PubMed] [Google Scholar]
- 24.Baker J, Harland R M. Genes Dev. 1996;10:1880–1889. doi: 10.1101/gad.10.15.1880. [DOI] [PubMed] [Google Scholar]
- 25.Liu F, Hata A, Baker J C, Doody J, Carcamo J, Harland R M, Massague J. Nature (London) 1996;381:620–623. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- 26.Hahn S A, Schutte M, Hoque A T, Moskaluk C A, da Costa L T, Rosenblum E, Weinstein C L, Fischer A, Yeo C J, Hruban R H, Kern S E. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 27.Snutch T P, Leonard J P, Gilbert M M, Lester H A, Davidson N. Proc Natl Acad Sci USA. 1990;87:3391–3395. doi: 10.1073/pnas.87.9.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, Knudsen E S, Wang J Y J. J Biol Chem. 1996;271:19637–19640. doi: 10.1074/jbc.271.33.19637. [DOI] [PubMed] [Google Scholar]
- 29.Wrana J L, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang X F, Massague J. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 30.Bassing C H, Yingling J M, Howe D J, Wang T, He W W, Gustafson M L, Shah P, Donahoe P K, Wang X F. Science. 1994;263:87–89. doi: 10.1126/science.8272871. [DOI] [PubMed] [Google Scholar]
- 31.Carcamo J, Weis F M, Ventura F, Wieser R, Wrana J L, Attisano L, Massague J. Mol Cell Biol. 1994;14:3810–3821. doi: 10.1128/mcb.14.6.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wieser R, Attisano L, Wrana J L, Massague J. Mol Cell Biol. 1993;13:7239–7247. doi: 10.1128/mcb.13.12.7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Angel P, Hattori K, Smeal T, Karin M. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 34.Renshaw M W, McWhirter J R, Wang J Y. Mol Cell Biol. 1995;15:1286–1293. doi: 10.1128/mcb.15.3.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keeton M R, Curriden S A, van Zonneveld A J, Loskutoff D J. J Biol Chem. 1991;266:23048–23052. [PubMed] [Google Scholar]
- 36.Chang E, Goldberg H. J Biol Chem. 1995;270:4473–4477. doi: 10.1074/jbc.270.9.4473. [DOI] [PubMed] [Google Scholar]
- 37.Chung K Y, Agarwal A, Uitto J, Mauviel A. J Biol Chem. 1996;271:3272–3278. doi: 10.1074/jbc.271.6.3272. [DOI] [PubMed] [Google Scholar]
- 38.Takeshita A, Chen Y, Watanabe A, Kitano S, Hanazawa S. J Immunol. 1995;155:419–426. [PubMed] [Google Scholar]
- 39.Staehling-Hampton K, Laughon A S, Hoffmann F M. Development (Cambridge, UK) 1995;121:3393–3403. doi: 10.1242/dev.121.10.3393. [DOI] [PubMed] [Google Scholar]
- 40.Arora K, Dai H, Kazuko S G, Jamal J, O’Connor M B, Letsou A, Warrior R. Cell. 1995;81:781–790. doi: 10.1016/0092-8674(95)90539-1. [DOI] [PubMed] [Google Scholar]
- 41.Grieder N C, Nellen D, Burke R, Basler K, Affolter M. Cell. 1995;81:791–800. doi: 10.1016/0092-8674(95)90540-5. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Feng X, Wu R, Derynck R. Nature (London) 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]