

Abstract
During embryogenesis, stem cells are set aside to fuel the postnatal hair cycle and repair the epidermis after injury. To define how hair follicle stem cells are specified and maintained in an undifferentiated state, we developed a strategy to isolate and transcriptionally profile embryonic hair progenitors in mice. We identified Lhx2 as a transcription factor positioned downstream of signals necessary to specify hair follicle stem cells, but upstream from signals required to drive activated stem cells to terminally differentiate. Using gain- and loss-of-function studies, we uncovered a role for Lhx2 in maintaining the growth and undifferentiated properties of hair follicle progenitors.
Hair follicle morphogenesis involves a temporal series of reciprocal interactions between the ectoderm and its underlying mesenchyme (fig. S1) (1-3). In response to an inductive Wnt and an inhibitory Bmp signal (Noggin), small hair placodes bud from the epithelium, giving rise to larger hair germs (4-7). In the presence of the mitogen Shh, these hair germs develop further and grow downward to form a mature follicle that actively produces hair (8-10). Although the molecular details of bud formation are still poorly defined, the general features of this process are repeated at the start of each postnatal hair cycle when multipotent stem cells in the hair follicle bulge become activated to initiate a new round of hair growth. In addition, the early epithelial remodeling to form the hair germ shares many features with the development of other epithelial tissues and organs, including feathers, teeth, and mammary glands (11-13). Understanding how tissues form buds that then progress along different lineages is predicated on elucidating the molecular mechanisms that funnel these early signaling pathways into a transcriptional program that drives morphogenesis.
To examine the genetic changes that occur during epithelial bud formation, we developed a strategy to isolate embryonic hair progenitors. To this end, we generated mice doubly transgenic for a Keratin 14–GFP gene expressed in skin keratinocytes and the Wnt reporter gene TOPGAL, transcribed in hair placodes and germs where β-catenin/Lef1 complexes are active (4, 14). In these early hair progenitors, E-cadherin is down-regulated and P-cadherin is up-regulated (Fig. 1A) (7). By embryonic day 17 (E17), we could use dispase to separate the epidermis, including hair placodes and germs, from the underlying dermis, which harbored more mature hair pegs and follicles (fig. S2). With the use of fluorescence-activated cell sorting (FACS) on the epidermal fraction, the early “PCAD+” hair progenitors (K14-GFP+, α6-integrin+, P-cadherin+) were then separated from the “PCAD−” interfollicular epidermis (K14-GFP+, α6-integrin+, P-cadherin−) on the basis of their differential surface P-cadherin expression (fig. S3). Characterization of these two cell populations confirmed that they had similarities in the expression of K5 and β4-integrin but distinct activities of TOPGAL and the expression of known hair-placode markers (Fig. 1, B to D, and fig. S4).
Fig. 1.
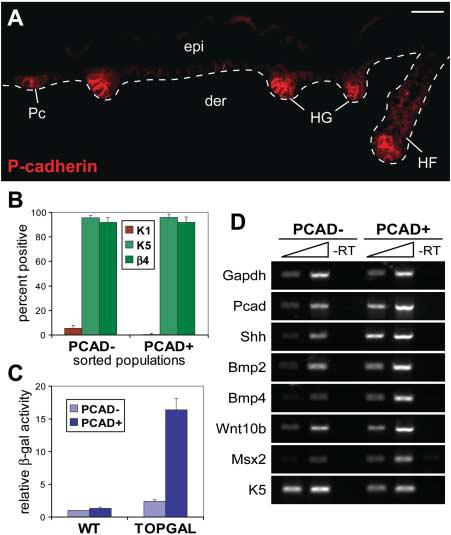
Isolation of embryonic hair follicle progenitors. (A) P-cadherin is up-regulated at sites of hair follicle morphogenesis. This differential expression was used to isolate PCAD+ hair progenitors and PCAD− interfollicular basal cells by FACS (figs. S2 and S3). Scale bar, 40 μm. (B) Summary of cytospin analyses for epidermal markers K1, K5, and β4-integrin. (C) The differential activity of the Wnt reporter gene TOPGAL was assessed by chemiluminescence. β-gal, β-galactosidase; WT, wild type. (D) Semi-quantitative RT-PCR shows differential expression of known placode mRNAs. K5 and K14 are known to be down-regulated, but still expressed, in hair germs (15). Abbreviations are as follows: epi, epidermis; der, dermis; Pc, hair placode; HG, hair germ; HF, hair follicle; β4, β4-integrin.
The gene expression profiles of purified PCAD+ hair progenitors and PCAD− interfollicular basal keratinocytes were further analyzed using oligonucleotide microarrays. Using fold differences of known hair-placode markers as a sensitivity gauge, a twofold cutoff was assigned as a genuine difference between the two populations. A total of 1394 probes (660 in PCAD+ and 734 in PCAD−) were preferentially expressed in one population relative to the other (table S1).
A short list of differentially expressed genes that are relevant to the present study is provided in table S2. As anticipated, a number of these genes have documented roles in either hair morphogenesis (PCAD+) or epidermal differentiation (PCAD−). The interfollicular epidermal population was typified by adhesive and cytoskeletal components, Notch signaling factors, c-Myc, Kruppel-like factors, and Bmp-responsive transcription factors (Grainyhead-like and Ovo1) previously implicated in epidermal differentiation (15-18). In contrast, the hair germ signature featured Wnts, Shh, Bmps, transforming growth factor–β's, and tyrosine kinase receptor signaling morphogens, as well as a number of different transcription factors. Although some of these transcription factors have not been previously implicated in the specification of skin progenitor fates, others have previously been associated with postnatal genetic hair disorders, including Cutl1, Gli1, Hoxc13, Sox9, Trps1, and Vdr (2, 3).
Several of the uncharacterized transcription factors on this list were also found to be differentially expressed in the postnatal hair follicle bulge (19, 20) (table S2), suggesting that the embryonic hair germ may exhibit functional properties similar to those of adult stem cells. Although the hair germ is committed to a follicular cell fate, it remains undifferentiated like bulge stem cells, yet capable of differentiating into all the lineages of the hair follicle, including the sebaceous gland (21, 22).
To explore the possibility that the early hair germs may reflect hair follicle stem cells and regulate key steps in progenitor cell differentiation, we focused on transcription factors emanating from our screen that are known to govern developmental cell fate specification in other tissues and organs. Lim-homeodomain transcription factor Lhx2 was particularly interesting because Lhx2 null mutant animals display defects in patterning and cell fate determination during brain development (23-25). In addition, they lack definitive erythropoeisis and conversely, hematopoetic progenitor cells can be maintained in vitro by forced expression of Lhx2 (26). Lhx2 null animals die between E15.5 and E16.5, and a possible role for Lhx2 in skin has not been examined.
Lhx2 was up-regulated 18-fold in the PCAD+ population relative to the PCAD− population, as determined by microarray analysis. Semi-quantitative reverse transcription polymerase chain reaction (RT-PCR) and in situ hybridization confirmed this marked differential expression (fig. S5). When examined by immunofluorescence, Lhx2 first appeared in early hair placodes, and as morphogenesis progressed, became prominent at the leading front of invaginating hair germs and pegs (Fig. 2A). As downgrowth neared completion and hair differentiation began, Lhx2 concentrated in the upper outer root sheath (ORS) at a presumptive site (bulge) of the developing postnatal follicle stem cell compartment (Fig. 2A and fig. S1). Concomitantly, expression diminished at the base of the follicle, where highly proliferative matrix cells give rise to the differentiating inner root sheath and hair shaft (Fig. 2B). In adult follicles, Lhx2 concentrated in the bulge, and as the new hair cycle began, Lhx2 extended to the emerging secondary hair germs (Fig. 2, C and D). Based on these patterns, we posit that Lhx2 functions in specifying the embryonic hair follicle progenitor cells that then persist as bulge stem cells in adult follicles.
Fig. 2.
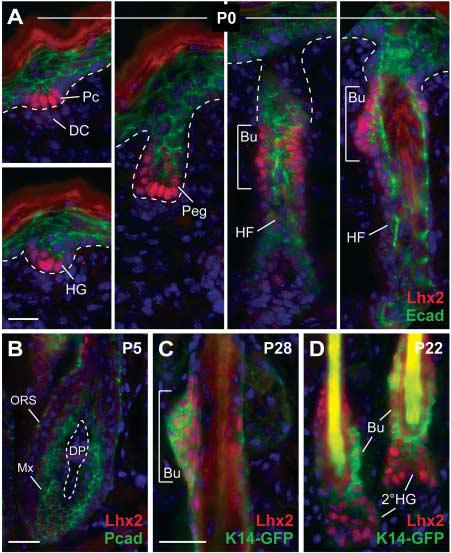
Lhx2 is expressed in early hair progenitors and postnatal stem cells. (A to D) Back skin sections from mice at indicated ages were stained with antibodies as color-coded and counterstained with DAPI (blue). Lhx2 is expressed in cells at the leading front of invaginating follicles and in the postnatal bulge compartment, but is diminished in mature proliferative hair progenitors (matrix). Abbreviations are as follows: Peg, hair peg; Bu, presumptive bulge; Mx, matrix cells; DC, dermal condensate; DP, dermal papilla; 2°HG, secondary hair germ emerging at the start of the postnatal hair cycle. Scale bars, 20 μm.
To more precisely define Lhx2's role in hair follicle stem cell specification and/or maintenance, we examined its status in various genetic mutant embryos that are defective in different aspects of hair morphogenesis. In the complete absence of hair follicle induction or bulge maintenance, as reflected in β-catenin conditionally null [conditional knockout (cKO)] skin, Lhx2 was not expressed (Fig. 3B). In Shh knockout embryos, where hair germs are specified but unable to progress, Lhx2 expression was dramatically reduced (Fig. 3C). This positioned Lhx2 downstream of Wnt and Shh, where it could play a role in establishing or expanding the early progenitors necessary for hair follicle morphogenesis.
Fig. 3.
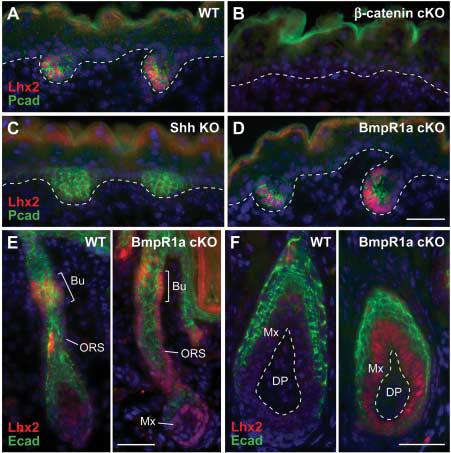
Lhx2 functions downstream of follicle stem cell specification but upstream of their differentiation. (A) Lhx2 is expressed in P-cadherin–positive hair germs. Shown is a representative wild-type littermate at E17.5. (B) Lhx2 is not expressed in the absence of hair follicle induction, as reflected in the β-catenin null skin epithelium (5). K14-Cre was used to conditionally target β-catenin. (C) Lhx2 is reduced in Shh null hair germs. Follicles are specified in the absence of Shh but fail to progress further (8, 10). (D) Lhx2 is expressed in BmpR1a conditional null hair germs. (E and F) In addition to expression in the presumptive bulge, Lhx2 persists in the lower ORS and matrix of neonatal and adult BmpR1a null follicles. Unable to undergo terminal differentiation, these BmpR1a null, Lhx2-expressing cells appear to be undifferentiated follicle stem cells (27, 28). Scale bars, 40 μm.
Bmp signaling is not required for hair follicle induction, even though Bmp ligands and receptors are expressed in embryonic hair germs and in postnatal follicle stem cells. Correspondingly, in BmpR1a cKO skin, Lhx2 was expressed in both embryonic hair germs and the presumptive bulge of developing follicles (Fig. 3, D and E). Conversely, Bmp signaling is required for differentiation, and in the absence of BmpR1a, proliferating undifferentiated hair progenitor cells accumulate at the follicle base (27, 28). Lhx2 was noticeably enhanced in these follicles, with strong staining throughout the ORS and matrix (Fig. 3, E and F). These cells were also positive for Shh and Lef1. Thus, in the absence of terminal hair differentiation, cells accumulating in postnatal BmpR1a null follicles resembled early embryonic hair follicle progenitors.
If Lhx2 governs the gene expression program of undifferentiated follicle stem cells or their early progenitors, then misexpression of Lhx2 in interfollicular epidermis might result in an induction of hair follicle progenitor genes. To test this possibility, we engineered K14-Lhx2 transgenic mice. Although more hair follicles were not induced, Lhx2 markedly suppressed morphological and biochemical signs of epidermal differentiation and failed to produce a functional lipid barrier (Fig. 4A). Most notable was the induction of Tcf3 and Sox9 (Fig. 4B), two key transcription factors of adult hair follicle stem cells (29, 30). Lhx2 also suppressed differentiation in tongue epithelium (fig. S6). These findings suggest that Lhx2 can maintain cells in an undifferentiated state, reinforcing the link between Lhx2 and stemness.
Fig. 4.
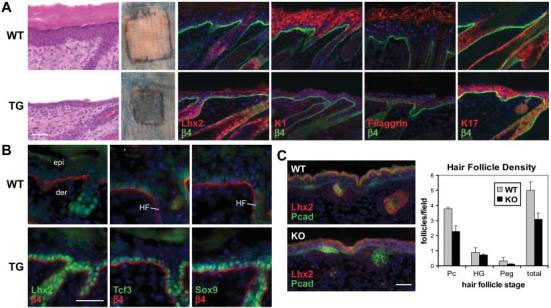
Gain- and loss-of-function studies reveal a role for Lhx2 in promoting follicle stem cell maintenance. (A) Transgenic expression of Lhx2 in the interfollicular epidermis suppresses terminal differentiation. Morphological and biochemical features of spinous, granular, and stratum corneum stages of epidermal differentiation are diminished in d8 (day 8 after graft) skin from K14-Lhx2 transgenic mice (TG) relative to wild-type littermates. Patches outlined in black denote skins grafted onto Nude mice and subjected to a β-galactosidase substrate exclusion assay to test for an intact epidermal barrier. The background absorption of dye in the surrounding Nude skin arises from defective hair follicle orifices. (B) Immunofluorescence reveals an induction of follicle stem cell markers in d4 K14-Lhx2 epidermis. (C) Loss of Lhx2 reduces hair morphogenesis. Representative skin sections from E16 littermates show comparable epidermal differentiation but fewer follicles at all stages in Lhx2 null embryos. The graph provides quantification from multiple sections of three embryos. Scale bars, 40 μm.
If Lhx2 is required for follicle stem cell maintenance, then its absence could alter the ability of hair follicles to form. In support of this notion, E16 Lhx2 null embryos displayed an ∼40% reduction in the overall density of P-cadherin–positive hair follicles (Fig. 4C), with no noticeable defect in the epidermis or embryo size. A marked reduction in follicle density is a feature of other mouse mutants in key hair follicle morphogenetic genes. Although Lhx2 KO follicle density was reduced, Shh, Wnt10b, Bmp2, Bmp4, and Lef1 expression appeared unaffected in those hair placodes and germs that developed (fig. S7). In Lhx2 null skin engraftments, follicles appeared morphologically and biochemically indistinguishable from their wild-type counterparts (fig. S8). Taken together, the gain- and loss-of-function studies suggest that Lhx2, reflecting its expression pattern, functions to specify and maintain hair follicle stem cells but does not function in their differentiation.
If Lhx2 maintains the undifferentiated state of embryonic and adult follicle stem cells, then Lhx2 null follicles might exhibit alterations in the transition of stem cells from the resting (telogen) to the growing (anagen) phase of the postnatal hair cycle. Using skin grafts, we compared the hair cycles of wild-type and Lhx2 KO follicles. The initial morphogenetic and first postnatal Lhx2 KO hair cycles progressed similarly to those in the wild type, and by 8 weeks, KO follicles had returned to telogen (fig. S9). By contrast, at 11 weeks, when most wild-type follicles were still in this extended telogen, KO follicles had precociously entered the next hair cycle (Fig. 5A). Moreover, after being shaved at 8 weeks, most wild-type hairs remained in telogen, whereas KO hairs consistently and uniformly grew back within 3 weeks, confirming their shortened resting phase (Fig. 5B).
Fig. 5.
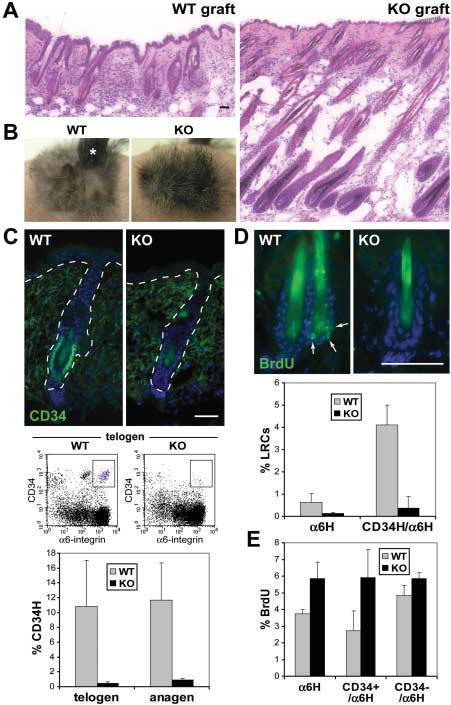
Lhx2 maintains follicle stem cells in a quiescent, inactive state. (A) Histology of wild-type and Lhx2 KO skins at 11 weeks after graft. Wild-type follicles were still in telogen, but KO follicles prematurely entered anagen. (B) Upon shaving at 8 weeks, wild-type hairs did not grow back, confirming their resting state. In contrast, KO hairs grew back within 3 weeks. A small portion (*) of the wild-type graft grew hairs because of a wound. (C) CD34 expression is dramatically reduced in KO follicle bulges, irrespective of whether they are in telogen or anagen. Shown are representative immunofluorescence images and FACS profiles of telogen follicles at 8 weeks, and CD34 quantification by flow cytometry in telogen and anagen follicles during the first post-natal cycle. (D) Loss of BrdU label retention in KO follicles. After a 3-day BrdU pulse on days 26 to 28 at the onset of anagen in both wild-type and KO skin grafts, and a 4-week chase when follicles had entered telogen (fig. S9), LRCs concentrated in the infrequently dividing bulge stem cells of wild-type follicles, but LRCs were diminished in Lhx2 KO skin. Shown are skin sections stained with antibody to BrdU and results of quantification by flow cytometry. (E) Increased BrdU incorporation by KO follicle stem cells. After a 4-hour BrdU pulse at day 40, when wild-type and KO follicles were in mid-anagen of their first postnatal hair cycle (fig. S9), cells were isolated and α6-integrin–expressing S-phase cells were quantified by flow cytometry. Scale bars, 40 μm.
Immunofluorescence and FACS analyses revealed that KO follicles exhibited diminished expression of CD34, a surface marker of bulge stem cells (Fig. 5C) (19). This reduction in CD34 was observed irrespective of hair cycle number or stage. Other stem cell markers that we examined were comparably expressed in wild-type and KO bulges (fig. S10).
Although CD34 marks adult stem cells, it is not found in embryonic skin progenitors, suggesting that its reduction could be an indication of enhanced proliferative activity within KO follicle stem cells. This hypothesis was supported by bromodeoxyuridine (BrdU) pulse-chase experiments conducted before marked deviations in hair cycling (Fig. 5D). Only the wild-type follicle bulge compartment retained appreciable BrdU label administered at the onset of anagen and chased for 4 weeks (19, 31). By contrast, KO hair follicles displayed very few label-retaining cells (LRCs), as confirmed and quantified by flow cytometry.
The reduction in label retention was accompanied by enhanced proliferation within the KO bulge. After a 4-hour BrdU pulse during full anagen, the percentage of S-phase labeled bulge cells was twice as high as normal (Fig. 5E and fig. S11). By contrast, the number of S-phase cells in the interfollicular epidermis/ORS of wild-type and KO skins was comparable, underscoring the specificity of this hyperproliferation. The elevated proliferative activity of the KO bulge did not appear to alter the overall size of the stem cell niche. We conclude that without Lhx2, follicle stem cells are more readily activated to proliferate and differentiate along the hair lineage. On the other hand, Lhx2 is not sufficient to induce quiescence, because transgenic expression did not suppress proliferation or induce CD34 in the skin epithelium.
Our ability to isolate and transcriptionally profile embryonic hair placodes and interfollicular epidermis has enabled us to substantiate genes previously implicated in hair and epidermal development and to uncover differences that could be important in orchestrating lineage specification of multipotent skin progenitors. Lhx2 has served as a paradigm for testing this premise, and our studies reveal that it functions as a molecular brake in regulating the switch between hair follicle stem cell maintenance and activation. Although follicles can be specified embryonically without Lhx2, their overall numbers are reduced, and Lhx2 null follicles that do form are not proficient in maintaining the resting state and precociously activate. Once committed, cells no longer require or express Lhx2 and progress along a normal program of terminal differentiation (fig. S12). Finally, Lhx2 is the first identified marker expressed specifically by both embryonic hair placodes and postnatal follicle stem cells of the bulge. Lhx2 now provides a means to dissect the transcriptional mechanisms that underlie stem cell maintenance within the hair follicle.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/312/5782/1946/DC1
Materials and Methods
Figs. S1 to S12
Tables S1 and S2
References
References and Notes
- 1.Hardy MH. Trends Genet. 1992;8:55. doi: 10.1016/0168-9525(92)90350-d. [DOI] [PubMed] [Google Scholar]
- 2.Millar SE. J. Invest. Dermatol. 2002;118:216. doi: 10.1046/j.0022-202x.2001.01670.x. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt-Ullrich R, Paus R. Bioessays. 2005;27:247. doi: 10.1002/bies.20184. [DOI] [PubMed] [Google Scholar]
- 4.DasGupta R, Fuchs E. Development. 1999;126:4557. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- 5.Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. Cell. 2001;105:533. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 6.Botchkarev VA, et al. Nat. Cell Biol. 1999;1:158. doi: 10.1038/11078. [DOI] [PubMed] [Google Scholar]
- 7.Jamora C, DasGupta R, Kocieniewski P, Fuchs E. Nature. 2003;422:317. doi: 10.1038/nature01458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiang C, et al. Dev. Biol. 1999;205:1. doi: 10.1006/dbio.1998.9103. [DOI] [PubMed] [Google Scholar]
- 9.Oro AE, Higgins K. Dev. Biol. 2003;255:238. doi: 10.1016/s0012-1606(02)00042-8. [DOI] [PubMed] [Google Scholar]
- 10.St-Jacques B, et al. Curr. Biol. 1998;8:1058. doi: 10.1016/s0960-9822(98)70443-9. [DOI] [PubMed] [Google Scholar]
- 11.Hogan BL. Cell. 1999;96:225. doi: 10.1016/s0092-8674(00)80562-0. [DOI] [PubMed] [Google Scholar]
- 12.Pispa J, Thesleff I. Dev. Biol. 2003;262:195. doi: 10.1016/s0012-1606(03)00325-7. [DOI] [PubMed] [Google Scholar]
- 13.Yue Z, Jiang TX, Widelitz RB, Chuong CM. Nature. 2005;438:1026. doi: 10.1038/nature04222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaezi A, Bauer C, Vasioukhin V, Fuchs E. Dev. Cell. 2002;3:367. doi: 10.1016/s1534-5807(02)00259-9. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs E, Raghavan S. Nat. Rev. Genet. 2002;3:199. doi: 10.1038/nrg758. [DOI] [PubMed] [Google Scholar]
- 16.Tao J, et al. Development. 2005;132:1021. doi: 10.1242/dev.01641. [DOI] [PubMed] [Google Scholar]
- 17.Ting SB, et al. Science. 2005;308:411. [Google Scholar]
- 18.Arnold I, Watt FM. Curr. Biol. 2001;11:558. doi: 10.1016/s0960-9822(01)00154-3. [DOI] [PubMed] [Google Scholar]
- 19.Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Cell. 2004;118:635. doi: 10.1016/j.cell.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Morris RJ, et al. Nat. Biotechnol. 2004;22:411. doi: 10.1038/nbt950. [DOI] [PubMed] [Google Scholar]
- 21.Ito M, et al. Nat. Med. 2005;11:1351. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- 22.Levy V, Lindon C, Harfe BD, Morgan BA. Dev. Cell. 2005;9:855. doi: 10.1016/j.devcel.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Porter FD, et al. Development. 1997;124:2935. doi: 10.1242/dev.124.15.2935. [DOI] [PubMed] [Google Scholar]
- 24.Bulchand S, Grove EA, Porter FD, Tole S. Mech. Dev. 2001;100:165. doi: 10.1016/s0925-4773(00)00515-3. [DOI] [PubMed] [Google Scholar]
- 25.Hirota J, Mombaerts P. Proc. Natl. Acad. Sci. U.S.A. 2004;101:8751. doi: 10.1073/pnas.0400940101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinto do O P, Richter K, Carlsson L. Blood. 2002;99:3939. doi: 10.1182/blood.v99.11.3939. [DOI] [PubMed] [Google Scholar]
- 27.Andl T, et al. Development. 2004;131:2257. doi: 10.1242/dev.01125. [DOI] [PubMed] [Google Scholar]
- 28.Kobielak K, Pasolli HA, Alonso L, Polak L, Fuchs E. J. Cell Biol. 2003;163:609. doi: 10.1083/jcb.200309042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merrill BJ, Gat U, DasGupta R, Fuchs E. Genes Dev. 2001;15:1688. doi: 10.1101/gad.891401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vidal VP, et al. Curr. Biol. 2005;15:1340. doi: 10.1016/j.cub.2005.06.064. [DOI] [PubMed] [Google Scholar]
- 31.Taylor G, Lehrer MS, Jensen PJ, Sun TT, Lavker RM. Cell. 2000;102:451. doi: 10.1016/s0092-8674(00)00050-7. [DOI] [PubMed] [Google Scholar]
- 32.In the supporting online materials and methods, we cite our many colleagues for their generous contributions of mice and reagents. We acknowledge colleagues in the Fuchs laboratory for constructive discussions and criticisms. We thank N. Stokes, J. Dela Cruz, S. Mazel, T. Shengelia, A. Viale, J. Li, and F. Berguido for invaluable technical assistance. E.F. is a Howard Hughes Medical Institute investigator. This work was supported in part by grant R01-AR050452 (E.F.) from NIH.