

Abstract
A hierarchical order of gene expression has been proposed to control developmental events in hematopoiesis, but direct demonstration of the temporal relationships between regulatory gene expression and differentiation has been difficult to achieve. We modified a single-cell PCR method to detect 2-fold changes in mRNA copies per cell (dynamic range, 250–250,000 copies/cell) and used it to sequentially quantitate gene expression levels as single primitive (CD34+,CD38−) progenitor cells underwent differentiation to become erythrocytes, granulocytes, or monocyte/macrophages. Markers of differentiation such as CD34 or cytokine receptor mRNAs and transcription factors associated with their regulation were assessed. All transcription factors tested were expressed in multipotent progenitors. During lineage-specific differentiation, however, distinct patterns of expression emerged. SCL, GATA-2, and GATA-1 expression sequentially extinguished during erythroid differentiation. PU.1, AML1B, and C/EBPα expression profiles and their relationship to cytokine receptor expression in maturing granulocytes could be distinguished from similar profiles in monocytic cells. These data characterize the dynamics of gene expression accompanying blood cell development and define a signature gene expression pattern for specific stages of hematopoietic differentiation.
Keywords: differentiation, hematopoiesis, hematopoietic stem cells, transcription factors, gene expression
Hematopoietic differentiation serves as a model by which events common to the developmental biology of other systems may be viewed. The features of blood cells along the continuum of differentiation have been extensively characterized and, while the contribution of gene expression to these events is self-evident, regulatory relationships governing the expression of particular transcripts remain ill-defined.
The cloning and knock-out deletion of lineage-specific transcription factors have proven extraordinarily powerful in demonstrating the necessary presence of specific factors in the development of hematopoietic cells (1). However, the role of these factors in the differentiation of normal blood elements cannot be directly deduced from these experiments since the knock-out phenotype reflects the earliest requirement for the factor in development and may be due to effects on supporting or accessory cells. Moreover, mouse strain-specific effects may obscure the factor’s more generalizable role. Other data for the relationship of transcription factors to differentiation have been generated by studies of differentiation-inducible tumor cell lines and of pools of primary cells, each system with significant limitations. Defining the differentiation-specific expression of transcription factors in isolated, single primary cells may avoid some of these limitations and provide insight into the events regulating the hematopoietic cascade in the mature organism. In addition, since the differentiation program of a cell is influenced by a complex pattern of expression of multiple transcription factors coupled to specific external signals and cell cycle regulators, the stoichiometry of gene products likely determines the differentiation outcome (2, 3). It is therefore necessary to quantitatively define the relationships of regulatory gene products to one another to better understand the mechanisms of differentiation. We sought to determine whether we could approach this issue through a combination of gene amplification techniques and cell micromanipulation. By sequentially isolating cells descending from a single primitive precursor, we have generated a depiction of the molecular accompaniments of human blood cell differentiation.
METHODS
Cell Culture.
Adult human bone marrow CD34+,CD38− cells were isolated by density-gradient centrifugation and cell sorting (4) and cultured in 0.8% methylcellulose, 30% fetal bovine serum, 1% BSA, 0.1 mM 2-mercaptoethanol, and 2.0 mM l-glutamine of Iscove’s modified Dulbecco’s medium semisolid matrix culture medium (StemCell Technologies, Vancouver, British Columbia) with cytokine combinations of 50 ng/ml stem cell factor (R & D Systems), 20 ng/ml interleukin 3 (R & D Systems), and 3 units/ml erythropoietin (Epo) (Amgen Biologicals) for erythroid burst-forming unit cells (BFU-E); 20 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (R & D Systems), and 50 ng/ml granulocyte colony-stimulating factor (G-CSF) (R & D Systems) for granulocyte colony-forming unit cells (CFU-G); or 20 ng/ml GM-CSF and 50 ng/ml macrophage colony-stimulating factor (M-CSF) (R & D Systems) for macrophage colony-forming unit cells (CFU-M). Cells were plated either at limiting dilution or by micromanipulator selection into 96-well plates (Costar). The location of the individual cells was marked and sequentially photomicrographed as single daughter cells differentiated into mature colonies. At 14 days, colonies were isolated by micropipetting and stained with Wright–Giemsa prior to photomicroscopic analysis.
Reverse Transcription (RT)–PCR.
Cells were lysed in 5.0 μl of buffer [100 mM Tris·HCl, pH 8.3/150 mM KCl/6 mM MgCl2/4 μM each dNTP (Pharmacia)/100 ng/ml oligo(dT)24/3360 units/ml RNA Guard (Pharmacia)/100 units/ml Prime inhibitor (5 Prime → 3 Prime, Inc.)/20 mM DTT/0.5% Nonidet P-40]. The samples were heated to 65°C for 1 min, cooled to 22°C for 3 min, and put on ice. Murine Moloney reverse transcriptase (100 units) (GIBCO/BRL) and 2 units of avian reverse transcriptase (Boehringer Mannheim) were added, and volume was made up to 10 μl with diethylpyrocarbonate (DEPC)-treated H2O. The samples were incubated at 37°C for 20 min and then heat inactivated at 65°C for 10 min. Resultant cDNA was then subjected to polyadenylate tailing in 20 μl total volume [50 mM Tris·HCl, pH 8.3/0.2 M potassium cacodylate/5 mM CoCl2/5 mM DTT/250 μg/ml BSA/0.2 mM dATP/10 units of terminal transferase (Boehringer Mannheim)] at 37°C for 15 min, heated at 65°C for 10 min, and placed on ice. PCR was performed in a final volume of 50 μl {30 mM Tris·HCl, pH 8.3/65 mM KCl/3.1 mM MgCl2/1 mM of each dNTP/100 μg/ml BSA/0.05% Triton X-100/10 μM (dT)24 primer [ATG TCG TCC AGG CCG CTC TGG ACA AAA TAT GAA TTC (dT)24]/5 units of AmpliTaq polymerase (Perkin–Elmer)}. Cell-free and reverse transcriptase-free samples were used as negative controls. Samples were initially amplified for 25 cycles of 30 sec at 94°C, 1 min at 42°C, and 3 min (plus 5-sec extension cycle) at 72°C in a thermal cycler (Perkin–Elmer/Cetus model 9600). An additional 5 units of Taq polymerase were then added and another 25 cycles were carried out.
Southern Blot Analysis.
The PCR product (4 μl/lane) was electrophoresed through 1.2% agarose, stained with ethidium bromide and photographed, transferred to a nylon membrane with vacuum blotter (Pharmacia), immobilized by UV (Stratagene), and hybridized to the indicated (32)P-radiolabeled probes (2 × 106 cpm/ml) with the use of either cDNA clones [human CD34, CD11b, erythropoietin receptor (Epo-R), G-CSF-R, M-CSF-R, GATA-1, GATA-2, PU.1, AML1B, and C/EBPα] or 3′-oligodeoxyribonucleotides derived from the following cDNA sequences: human SCL/TAL-1 (ATG TAT CGG GAA GAT TTC TAA ATA AAA GTT TTA CAA AGG G) (5) and NF-E2 (CCT CCA CCT TGT CTA AGC TTT GGT CTA TAA AGT GCG CTA C) (6) in express hybridization solution (Clontech). The hybridization temperature for cDNA probes was 68°C, and final wash condition was 0.1× standard saline citrate (SSC)/0.1% SDS at 50°C. For the oligonucleotide probes, the hybridization temperature was 37°C and the final wash temperature was 37°C. The hybridized blots were analyzed by PhosphorImager (Molecular Dynamics) quantitation and autoradiography. Eight southern blots were performed for each single cell used in RT-PCR. Fresh blots were used for each transcript shown to avoid loss of substrate during probe stripping and rehybridization. Equivalence between blots was confirmed by sequential hybridization with indicated and control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probes.
In Vitro Transcription and Poly(A) Tailing.
The mutated HIV-1 gag fragment (K4) (7) was cloned into the plasmid pGEM-3Z (Promega) and in vitro transcribed using T7 polymerase (MEGAscript; Ambion). The resultant K4 cRNA was purified by ultracentrifugation through a 5.7 M CsCl cushion and was polyadenylylated by poly(A) polymerase (Pharmacia) in buffer (40 mM Tris·HCl, pH 8.0/10 mM MgCl2/2.5 mM MnCl2/250 mM NaCl/250 μM ATP/50 μg/ml BSA). The poly(A)-tailed K4 cRNA was purified with oligo(dT)-cellulose (FastTrack, Invitrogen), precipitated, resuspended in DEPC-treated H2O, and quantitated by spectrophotometry.
RESULTS AND DISCUSSION
Using combined RT-PCR to achieve quantitative or semiquantitative estimates of input nucleic acid generally requires the presence of a competitive target template that utilizes similar primer sequences (8, 9). We reasoned that the poly(A) technique developed by Brady and coworkers (10–12), which depends on poly(A) sequences for priming in both the 5′ and 3′ directions, could be adapted to provide a semiquantitative estimate of input amounts of a range of specific mRNAs. Briefly, this method uses oligo(dT)-primed reverse transcription followed by poly(A)-tailing via a terminal transferase-catalyzed reaction to generate a 5′-oligo(dT)-transcript-poly(A)-3′ first-strand cDNA. This is followed by oligo(dT)-primed PCR to amplify a representative population of cDNAs. To evaluate the quantitative potential of this system, we tested our modifications on small numbers of cells with known differences in marker gene expression. That is, we diluted erythroid-derived cells, which express Epo-R message into a background of NIH 3T3 fibroblasts, and myeloid progenitor cells, which do not express this gene. Using dilutions of input cells and quantitating output Epo-R by phosphorimaging analysis of hybridized radiolabeled Epo-R-specific probe, we were able to discriminate 2-fold differences in cell number with a correlation coefficient of 0.800 (Fig. 1a). Further assessment was performed using known quantities of in vitro transcribed and polyadenylylated HIV-1 gag-derived message, K4, added to single hematopoietic cells (Fig. 1b). As few as 250 transcripts per cell could be detected with a linear coefficient for 2-fold dilutions of input K4 of 0.946. The reproducibility of this system was demonstrated through multiple, independent experiments (n = 5) that yielded a mean linear coefficient of 0.862 (95% confidence interval, 0.099). Thus, using two independent transcripts, linearity of the yield from 2-fold changes in input mRNA dosing was demonstrated. Furthermore, the method was reproducible with a sensitivity permissive for evaluating in vivo transcript levels of multiple specificities.
Figure 1.
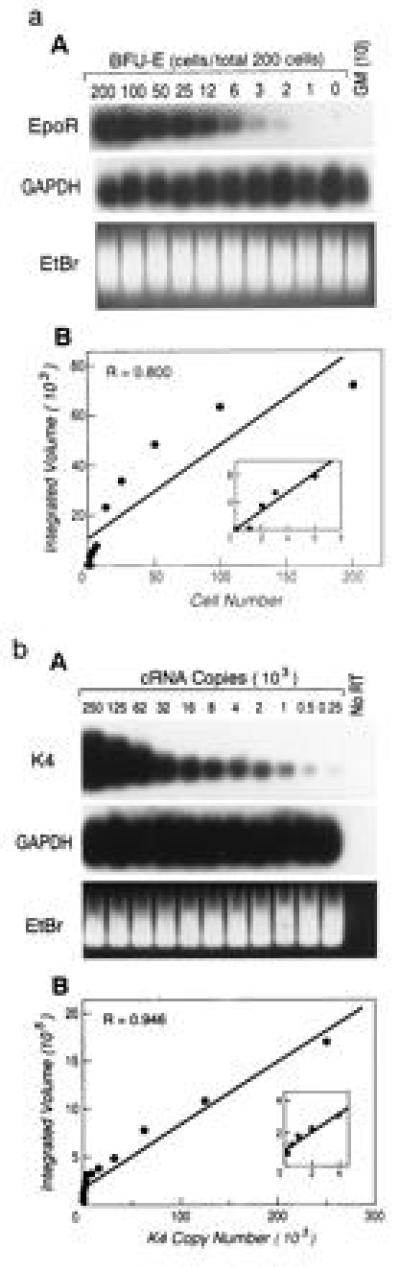
(a) Titration of input cell dose confirms the semi-quantitative nature of the modified RT-PCR technique. (A) The indicated number of cells extracted from a maturing BFU-E were diluted into cells extracted from a cell line (NIH 3T3) maintaining a constant total number of cells at 200. RT-PCR was performed as described and the resultant cDNA visualized by ethidium bromide (EtBr) staining prior to Southern transfer and hybridization with radiolabeled probes from the cDNAs from either the housekeeping gene GAPDH or the BFU-E-specific Epo-R gene. PhosphorImager quantitation of the Epo-R signal was plotted (B), and the indicated linear coefficient was determined. (Inset) Lower cell number values from the same line plot. Lanes in which 10 cells from a developing CFU-GM instead of BFU-E were tested by the RT-PCR reaction are designated GM(10). (b) Limiting dilution of known copy numbers of RNA to define the sensitivity and quantitative capacity of the single-cell RT-PCR technique. (A) An in vitro transcribed, mutated HIV-1 gag fragment, K4, was polyadenylylated, quantitated by spectrophotometry, and added in 2-fold dilutions as indicated to lysates from single CMK cells or (in independent experiments) primary quiescent bone marrow mononuclear cells prior to RT-PCR. The cDNA resulting from the RT-PCR technique was visualized following EtBr staining prior to hybridization with radiolabeled probes from the cDNAs from either GAPDH or K4 (a). PhosphorImager quantitation of the K4 signal was plotted (B) and the indicated linear coefficient was determined. (Inset) Lower K4 copy number values from the same line plot. Copy number values below 250/cell yielded a signal that was detectable but no longer linearly related to input. Lanes in which reverse transcriptase was not added to the RT-PCR mixture are designated No RT.
Micromanipulation of emerging hematopoietic colonies plated at limiting dilution in defined medium in a semisolid methylcellulose matrix was performed to isolate individual daughter cells. Direct microscopic visualization of cells expelled from a microcapillary pipette confirmed that a single cell could be mechanically eased away from an evolving colony, removed intact, and expelled into assay buffers (Fig. 2). The intact replating potential of the isolated cells confirmed the limited impact of micromanipulation on the homeostasis of the manipulated cells (data not shown).
Figure 2.
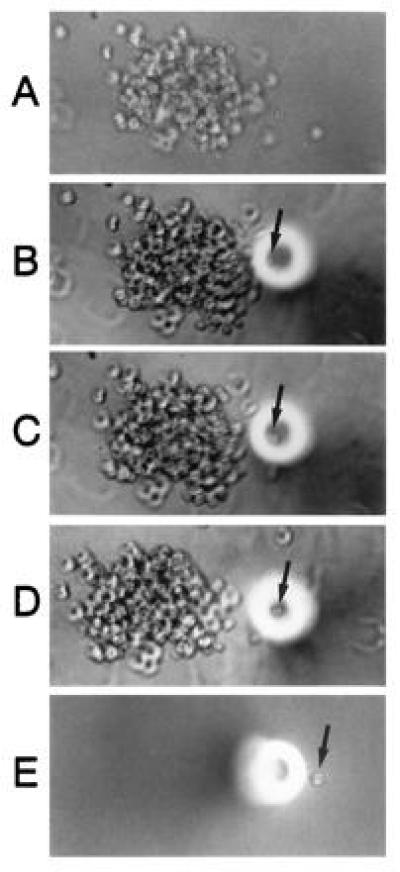
Single cells from an evolving colony can be isolated and analyzed in conjunction with single cell RT-PCR. A colony (A) was approached with a micromanipulation pipette (B), and a target cell gently isolated from the colony (C and D) was aspirated into the pipette and expelled directly into the assay buffer (E). Direct visualization confirmed the singularity of the isolated cell.
Adult human bone marrow cells of primitive immunophenotype (CD34+,CD38−) were plated at limiting dilution in medium known to enhance specific myeloid lineage development. The location of individual progenitor cells was marked and sequentially photomicrographed as single daughter cells were removed for analysis. Due to the predicted heterogeneity within an evolving hematopoietic colony, multiple representative cells were isolated from each colony every 2–4 days, and Southern blots were generated from the amplified cDNA of each cell. Samples were stored until the colony had matured and could be defined by morphologic analysis and Wright–Giemsa staining. Samples were then analyzed in bulk to ensure uniformity of hybridization, wash, and analysis conditions.
To test proof-of-principle, genes whose products are markers of lineage-specific differentiation were evaluated using specific radiolabeled cDNA hybridization (Fig. 3). Message levels for marker genes were normalized against the constitutively expressed gene, GAPDH, to account for variability due to mRNA quality or RT-PCR efficiency. Levels of the surface-marker characteristic of primitive cells (CD34) decreased while genes whose products accompany the differentiation of mature blood cells (Epo-R and CD11b for erythroid and myeloid cells, respectively) increased in detectable transcript levels. These patterns of change correspond to what would be anticipated from prior immunophenotypic characterization of cell expanding in cytokine-supplemented liquid culture in which CD34 antigen is lost and CD11b acquired as progenitor cells are terminally differentiated.
Figure 3.
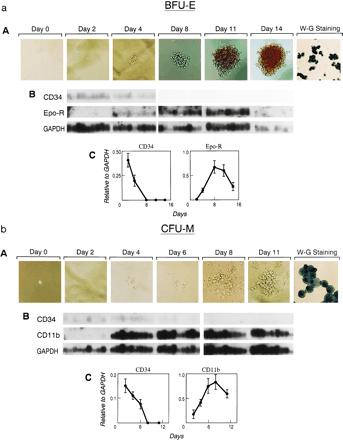
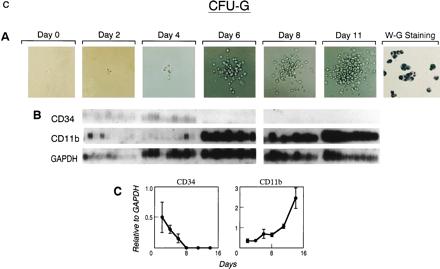
Defining the pattern of marker gene expression during the differentiation of a BFU-E (a), CFU-M (b), or CFU-G (c) from a single CD34+,CD38− progenitor cell. Individual cells were flow-cytometrically sorted, cultured under the stated conditions, and sequentially photomicrographed (A) at indicated time points. Single cells harvested from the emerging colony depicted or colonies of identical morphology were analyzed by RT-PCR and Southern blotting after hybridization of independent filters from the same RT-PCR to the indicated cDNA probes (B). Quantitative PhosphorImager analysis of the indicated probes was normalized to GAPDH signals and used to derive mean and standard error values for each time point plotted in the graph (C).
The events that trigger differentiation are complex and are likely altered by a combination of external stimuli and an internal state of readiness for signal processing (13). Levels of cytokine and adhesion receptors and their downstream signaling partners that mediate blood cell differentiation have been reported to be influenced by transcription factors otherwise implicated in hematopoiesis (14). A role for transcription factor genes in the development of blood cells has been determined through a range of experimental systems (13, 15) and the timing of their expression in hematopoietic development inferred through the phenotype of emerging blood cells when the genes were absent or diminished. To investigate the relationship of gene expression to hematopoietic differentiation in adult human cells, we assessed the relative levels of specific relevant transcription factor genes during blood cell differentiation. The pattern of early expression of the transcription factors, SCL and GATA-2, in erythroid differentiation predicted by knock-out animals in which erythroid cell development was absent (16) or severely impaired (17) was recapitulated in the differentiation of single primitive human cells within our system (Fig. 4A). The sequential down-modulation of SCL, GATA-2, and GATA-1 (18) while NF-E2 (19) gradually increased as primitive cells became hemoglobinized erythrocytes, suggests that these transcription factors are independently regulated and may have stage-specific functions in the differentiation process. It has been shown that PU.1, C/EBPα, and AML1B are regulators of myeloid CSF receptor gene expression (3). In this system, we found that PU.1 and C/EBPα up-regulation precedes targets like CD11b and G-CSF receptor in developing granulocytic cells (CFU-G) (Fig. 4C), but that in developing monocytic cells (CFU-M), CD11b and M-CSF receptor mRNA levels can increase before these factors reach their maximum levels (Fig. 4B). These data imply that regulation of the levels of M-CSF receptor and CD11b is not exclusively related to the levels of the transcription factors PU.1, AML1B, or C/EBPα during differentiation, perhaps reflecting the fact that M-CSF receptor or CD11b mRNA up-regulation may be mediated by posttranscriptional (20, 21) as well as transcriptional mechanisms (22–25). In addition, PU.1 and AML1B appeared to be coordinately regulated whereas C/EBPα was affected independently in CFU-M. Unlike cells proceeding down a monocytic differentiation pathway (CFU-M), cells acquiring granulocytic characteristics (CFU-G) did not regulate AML1B levels in coordination with PU.1, suggesting lineage-specific control of mRNA levels of these transcription factors. The pattern of expression thus suggests regulatory relationships that may be mechanistically defined through other experimental systems.
Figure 4.
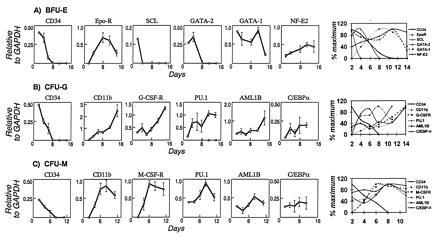
Quantitative portrayal of the expression of indicated marker and regulatory genes during the differentiation of individual CD34+,CD38− cells into either BFU-E (A), CFU-M (B), or CFU-G (C). Each data point shown represents the mean and standard error for up to nine individual cells isolated from the same emerging colony at the indicated time points. Earlier time points had fewer input cells due to small colony size. Hybridizations for each transcript indicated were performed on Southern blots generated from a single RT-PCR for each cell sample. Independent colonies of identical morphologic type yielded comparable patterns of gene expression. The last graph in each row represents the superimposition of the data normalized to maximal expression level and represented as curves.
Although these data indicate the sequential change in RNA levels in cells undergoing differentiation, the expression of regulatory genes in primitive cells prior to proliferation cannot be examined using these techniques, because analysis of the input cell would necessarily sacrifice subsequent time points. Yet examination of the input cells is of particular interest to define whether multipotential cells express multiple lineage-associated regulatory genes that are selectively altered in their expression during differentiation or whether there is a relative “blank slate” to which specific transcripts are added during differentiation. To evaluate this, we applied the single-cell RT-PCR technique to individual representative cells of the CD34+,CD38− phenotype used in the differentiation cultures described above. In addition, we assessed a population of quiescent, multipotent cells isolated by a functional, directed, suicide approach as described (26) that have characteristics of stem cells. The data from these analyses can be compared with those shown in Fig. 4, but only on a qualitative basis, since the relationships between the cells are indirect. However, when using probes for SCL, GATA-1, GATA-2, NF-E2, PU.1, or AML1B, abundant message was detectable in both the representative stem cells and CD34+,CD38− cells (Fig. 5). The presence of detectable message for multiple factors suggests that cell fate may be determined by modulation of a multilineage “ready” state. A complex array of transcription factors is expressed whose modification in expression level is associated with lineage-specific events. Whether alteration in expression of the genes is induced by external signals or intrinsic properties of the cells is unclear at present but possible to assess using these techniques.
Figure 5.
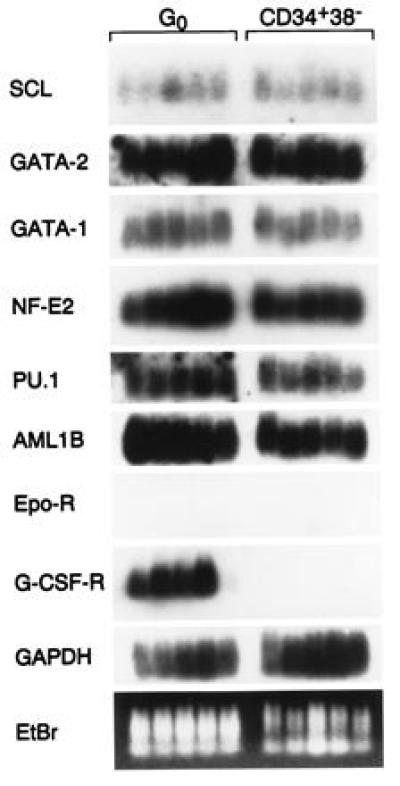
Gene expression of transcription factors in individual quiescent multipotent hematopoietic cells (G0) or CD34+,CD38− cells analyzed using the single cell RT-PCR technique. The ethidium bromide (EtBr)-stained cellular cDNA gel was subsequently transferred to a nylon filter, probed with the indicated cDNA probes, and subjected to autoradiography. Panels of five cells of each type were used to avoid individual cell artifacts.
An unanticipated finding was the presence of G-CSF-R expression in quiescent stem-like cells compared with primitive CD34+,CD38− cells. These data may explain the observations of other investigators that G-CSF is capable of influencing long-term bone marrow culture (27) or CFU-blast formation (28) in vitro, effects anticipated if G-CSF-R were expressed on stem cells rather than exclusively on mature myeloid elements. Yet clinical investigators generally have not seen expansion of primitive hematopoietic elements in vivo, perhaps due to the tight regulation of stem cell proliferation in vivo and the absence of receptor message in the more mature and highly proliferative CD34+,CD38− compartment.
The data presented here demonstrate that the genetic program expressed by cells undergoing specific lines of differentiation can be imaged during the differentiation process. By permitting the analysis of individual cells, a precision of definition is possible beyond that available through the use of populations of cells (15, 29–31), and the possibility of using primary cells is realized despite limited availability of such cell types. Through sequential and quantitative evaluation of daughter cells, a specific marker and regulatory gene expression pattern is demonstrated to define the stage of differentiation with a resolution beyond that possible with prior studies (11, 12) and to provide insight into the lineage-specific regulation of genes thought to play important roles in determining cell fate. Applied to disease states, it may be possible to evaluate the impact of an aberrant gene product (such as an oncogene, fusion gene, or viral gene) on the molecular program expressed during differentiation and thereby provide hypotheses as to the basis for phenotypic change. These techniques may be adaptable to other primary cell systems and may be particularly well suited for investigating the molecular biology of stem cells of various types.
Acknowledgments
We thank Ellen Kornell for her assistance in manuscript preparation. We gratefully acknowledge the support of the Richard Saltonstall Charitable Foundation and the National Institutes of Health (DK50234) (HL44851).
Footnotes
Abbreviations: Epo-R, erythropoietin receptor; BFU-E, erythroid burst-forming unit cells; GM-CSF, granulocyte-macrophage colony-stimulating factor; G-CSF, granulocyte colony-stimulating factor; CFU-G, granulocyte colony-forming unit cells; M-CSF, macrophage colony-stimulating factor; CFU-M, macrophage colony-forming unit cells; RT, reverse transcription; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
References
- 1.Shivdasani R A, Orkin S H. Blood. 1996;87:4025–4039. [PubMed] [Google Scholar]
- 2.Kulessa H, Frampton J, Graf T. Genes Dev. 1995;9275:1250–1262. doi: 10.1101/gad.9.10.1250. [DOI] [PubMed] [Google Scholar]
- 3.Zhang D E, Hohaus S, Voso M T, Chen H M, Smith L T, Hetherington C J, Tenen D G. Curr Top Microbiol Immunol. 1996;211:137–147. doi: 10.1007/978-3-642-85232-9_14. [DOI] [PubMed] [Google Scholar]
- 4.Huang S, Terstappen L W M M. Blood. 1994;83:1515–1526. [PubMed] [Google Scholar]
- 5.Begley C G, Aplan P D, Denning S M, Haynes B F, Waldmann T A, Kirsch I R. Proc Natl Acad Sci USA. 1989;86:10128–10132. doi: 10.1073/pnas.86.24.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan J Y, Han X L, Kan Y W. Proc Natl Acad Sci USA. 1993;90:11366–11370. doi: 10.1073/pnas.90.23.11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scadden D T, Wang Z, Groopman J E. J Infect Dis. 1992;165:1119–1123. doi: 10.1093/infdis/165.6.1119. [DOI] [PubMed] [Google Scholar]
- 8.Wang A M, Doyle M V, Mark D F. Proc Natl Acad Sci USA. 1989;86:9717–9721. doi: 10.1073/pnas.86.24.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilliland G, Perrin S, Blanchard K, Bunn H F. Proc Natl Acad Sci USA. 1990;87:2725–2729. doi: 10.1073/pnas.87.7.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brady G, Barbara M, Iscove N N. Methods Mol Cell Biol. 1990;2:17–25. [Google Scholar]
- 11.Brady G, Billia F, Knox J, Hoang T, Kirsch I R, Voura E B, Hawley R G, Cumming R, Buchwald M, Siminovitch K, Miyamoto N, Boehmelt G, Iscove N N. Curr Biol. 1995;5:909–922. doi: 10.1016/S0960-9822(95)00181-3. [DOI] [PubMed] [Google Scholar]
- 12.Hoang T, Paradis E, Brady G, Billia F, Nakahara K, Iscove N N, Kirsch I R. Blood. 1996;87:102–111. [PubMed] [Google Scholar]
- 13.Kehrl J H. Stem Cells (Dayton) 1995;13:223–241. doi: 10.1002/stem.5530130304. [DOI] [PubMed] [Google Scholar]
- 14.Melotti P, Calabretta B. Proc Natl Acad Sci USA. 1996;93:5313–5318. doi: 10.1073/pnas.93.11.5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voso M T, Burn T C, Wulf G, Lim B, Leone G, Tenen D G. Proc Natl Acad Sci USA. 1994;91:7932–7936. doi: 10.1073/pnas.91.17.7932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shivdasani R A, Mayer E L, Orkin S H. Nature (London) 1995;373:432–434. doi: 10.1038/373432a0. [DOI] [PubMed] [Google Scholar]
- 17.Tsai F Y, Keller G, Kuo F C, Weiss M, Chen J, Rosenblatt M, Alt F W, Orkin S H. Nature (London) 1994;371:221–226. doi: 10.1038/371221a0. [DOI] [PubMed] [Google Scholar]
- 18.Pevny L, Simon M C, Robertson E, Klein W H, Tsai S F, D’Agati V, Orkin S H, Costantini F. Nature (London) 1991;349:257–260. doi: 10.1038/349257a0. [DOI] [PubMed] [Google Scholar]
- 19.Peters L L, Andrews N C, Eicher E M, Davidson M B, Orkin S H, Lux SEM. Nature (London) 1993;362:768–770. doi: 10.1038/362768a0. [DOI] [PubMed] [Google Scholar]
- 20.Yue X, Favot P, Dunn T L, Cassady A I, Hume D A. Mol Cell Biol. 1993;13:3191–3201. doi: 10.1128/mcb.13.6.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hicksteim D D, Bach A L, Collins S J. J Biol Chem. 1989;264:21812–21817. [PubMed] [Google Scholar]
- 22.Zhang D E, Hetherington C J, Chen H M, Tenen D G. Mol Cell Biol. 1994;14:373–381. doi: 10.1128/mcb.14.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang D E, Fujioka K, Hetherington C J, Shapiro L H, Chen H M, Look A T, Tenen D G. Mol Cell Biol. 1994;14:8085–8095. doi: 10.1128/mcb.14.12.8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H M, Pahl H L, Scheibe R J, Zhang D E, Tenen D G. J Biol Chem. 1993;268:8230–8239. [PubMed] [Google Scholar]
- 25.Pahl H L, Scheibe R J, Zhang D E, Chen H M, Galson D L, Maki R A, Tenen D G. J Biol Chem. 1993;268:5014–5020. [PubMed] [Google Scholar]
- 26.Berardi A C, Wang A, Levine J D, Lopez P, Scadden D T. Science. 1995;267:104–108. doi: 10.1126/science.7528940. [DOI] [PubMed] [Google Scholar]
- 27.Sutherland H J, Eaves C J, Lansdorp P M, Thacker J D, Hogee D E. Blood. 1991;73:666–672. [PubMed] [Google Scholar]
- 28.Ikebuchi K, Clark S C, Ihl J N, Souza L M, Ogawa M. Proc Natl Acad Sci USA. 1988;85:3445–3449. doi: 10.1073/pnas.85.10.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orlic D, Anderson S, Biesecker L G, Sorrentino B P, Bodine D M. Proc Natl Acad Sci USA. 1995;92:4601–4605. doi: 10.1073/pnas.92.10.4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Labbaye C, Valtieri M, Barberi T, Meccia E, Masella B, Pelosi E, Condorelli G L, Testa U, Peschle C. J Clin Invest. 1995;95:2346–2358. doi: 10.1172/JCI117927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H M, Zhang P, Voso M T, Hohaus S, Gonzalez D A, Glass C K, Zhang D E, Tenen D G. Blood. 1995;85:2918–2928. [PubMed] [Google Scholar]