

Abstract
DNA topoisomerase II is a homodimeric molecular machine that couples ATP usage to the transport of one DNA segment through a transient break in another segment. In the presence of a nonhydrolyzable ATP analog, the enzyme is known to promote a single turnover of DNA transport. Current models for the enzyme’s mechanism based on this result have hydrolysis of two ATPs as the last step, used only to reset the enzyme for another round of reaction. Using rapid-quench techniques, topoisomerase II recently was shown to hydrolyze its two bound ATPs in a strictly sequential manner. This result is incongruous with the models based on the nonhydrolyzable ATP analog data. Here we present evidence that hydrolysis of one ATP by topoisomerase II precedes, and accelerates, DNA transport. These results indicate that important features of this enzyme’s mechanism previously have been overlooked because of the reliance on nonhydrolyzable analogs for studying a single reaction turnover. A model for the mechanism of topoisomerase II is presented to show how hydrolysis of one ATP could drive DNA transport.
Type II DNA topoisomerases are ubiquitous enzymes essential for the unlinking of intertwined chromosomes, chromosomal condensation/decondensation, and manipulation of DNA supercoiling (1, 2). These enzymes catalyze the ATP-dependent transport of one segment of DNA through a transient, enzyme-mediated break in a second DNA segment (for recent reviews, see refs. 3 and 4). The eukaryotic topoisomerase II enzymes are homodimers, with a monomer molecular mass of ≈160–180 kDa, making them relatively simple macromolecular machines for studying the coupling of ATP usage to complex protein movements. The N-terminal half of the enzyme is homologous to Escherichia coli gyrase B and contains the ATPase active site; the C-terminal half is homologous to gyrase A and contains the active site tyrosine for DNA cleavage and the primary dimerization interface. In addition to being mechanistically fascinating, the prokaryotic and eukaryotic members of this enzyme family are the targets of numerous antibiotic and anticancer drugs, respectively (5–7).
The mechanism of topoisomerase II is known to involve several steps and associated protein conformational changes (reviewed in ref. 4). The enzyme binds a segment of DNA, the gate or G segment. Both strands of this DNA segment are cleaved and religated by a pair of active site tyrosines, one in each monomer of the dimer. The cleavage reaction results in a four-base staggered break in the DNA, with an enzyme monomer covalently attached to each 5′ phosphate. Upon binding ATP, the amino-terminal ATPase domains of the enzyme dimerize, capturing a second segment of DNA, the transport or T segment, within the enzyme clamp. The ends of the cleaved G segment are separated, and the T segment is transported through the opening. The gate in the G segment is closed. The T segment is transported out of the topoisomerase, most likely through the C-terminal dimerization interface. The amino terminal dimerization interface reopens to complete the reaction cycle.
The generally accepted model of the topoisomerase II mechanism assumes that all but the last of these steps occur before the hydrolysis of ATP; hydrolysis of two ATPs and release of all four products, two Pi and two ADPs, are presumed to be coupled only to reopening the amino terminal dimerization interface (8, 9). These models are based on the fact that type II topoisomerases can perform a single round of DNA transport, such as decatenation of singly linked catenanes, in the presence of a nonhydrolyzed ATP analog (10–12).
Recent discoveries about the ATPase mechanism of yeast topoisomerase II have brought these models into question (13, 14). Because a single turnover of ATP hydrolysis and DNA transport is complete within ≈200 msec, rapid-quench techniques have been applied to resolve the individual steps of the reaction cycle. The results of pulse–chase experiments have shown that topoisomerase II, prebound to DNA, rapidly binds two ATPs and hydrolyzes them both before either can dissociate (14). However, the results of chemical quench studies showed that only one of the two ATPs is hydrolyzed rapidly, before the rate-determining step in the reaction cycle (13). Additionally, in the presence of vanadate, a phosphate mimic that binds and stabilizes the enzyme-ADP complex, topoisomerase II hydrolyzes the first ATP at the normal rate, but cannot hydrolyze the second ATP (13). These results, together with results from a rapid ADP trap experiment (13), indicate that topoisomerase II hydrolyzes one ATP and releases the Pi and ADP formed before hydrolyzing the second ATP. This sequential hydrolysis mechanism is difficult to explain in terms of the model described above, with hydrolysis of both ATPs occurring at the end of the reaction cycle. In the present study, rates of single turnover DNA decatenation by yeast topoisomerase II are determined under conditions where the enzyme can hydrolyze two ATPs, no ATPs, or one ATP. A model is presented for ATP-fueled DNA transport by type II topoisomerases based on these and previously published data.
Materials and Methods
Enzymes and DNA Substrates.
Expression vectors for all Saccharomyces cerevisiae topoisomerase II enzymes were derived from the plasmid YEpTOP2-PGAL1 (15). The expression plasmid pJEL236 encodes the wild-type (wt) enzyme fused to an intein chitin binding domain (16). The expression plasmid pCLB4 encodes yeast topoisomerase II with a 95-aa C-terminal truncation and a 6× histidine tag, wtΔC-his, on a TRP1 containing plasmid and was created by ligating the SalI–SmaI fragment from pTCH1 (17) into pRS424 (18). This truncated version of topoisomerase II is fully active (17). The expression plasmid pJEL245 is identical to pJEL236 except that it encodes the E66Q mutant enzyme, in which glutamate 66 has been changed to glutamine by using the Altered Sites II mutagenesis kit (Promega).
The enzyme-intein/chitin binding domain fusions were expressed to high levels from the plasmids pJEL236 and pJEL245 in the yeast strain BCY123 as described (17). In the same manner, heterodimeric enzymes were coexpressed in the same strain, doubly transformed with plasmids pCLB4 and pJEL236 (wt/wtΔC-his) or pCLB4 and pJEL245 (E66Q/wtΔC-his), in media lacking both uracil and tryptophan. The heterodimeric enzymes contained two different purification tags to allow their separation from the homodimeric enzymes also produced in the cells. SDS/PAGE analysis of the purified heterodimers showed that they were composed of essentially equal amounts of the two different polypeptides. The yield of the heterodimeric enzymes was approximately 30-fold less than that of a standard homodimeric topoisomerase II preparation.
The homodimeric enzymes were purified on a chitin bead column (New England Biolabs) essentially as described (16). To increase purity an additional wash step of buffer I (50 mM Hepes⋅KOH, pH 7.5/1 mM EDTA/1 mM EGTA/10% glycerol) plus 250 mM NaCl and 0.5% Triton X-100 was included. Highly purified enzyme was released from the column by inducing intein cleavage with 30 mM DTT. Heterodimeric enzymes were purified by nickel-nitrilotriacetic acid (Qiagen, Chatsworth, CA) chromatography, as recommended by the supplier, followed by chitin bead column chromatography, as described above.
A singly catenated DNA substrate was produced by reacting γδ resolvase with a 4.2-kb plasmid (pJEL257) containing two γδ resolvase sites separated by 2.1 kb, in a directly repeated orientation. This plasmid is a derivative of pR2d (a kind gift of the Cozzarelli lab, University of California, Berkeley) that was modified to contain a single BamHI site. An expression vector for γδ resolvase with a C-terminal histidine tag (pJEL267) was created by ligating a NdeI/BamHI cut PCR amplicon containing the resolvase gene into pET16b (Invitrogen). γδ resolvase expression was induced in BL21 E. coli cells grown to an OD600 of 0.6 before addition of 1 mM isopropyl β-d-thiogalactopyranoside for 2 hr. The histidine-tagged γδ resolvase was purified by nickel-nitrilotriacetic acid agarose chromatography. Singly catenated 2.1-kb DNA circles were produced by reacting 5 μM γδ resolvase with 100 nM pJEL257 for 1 hr at 37°C in 10 mM Tris⋅HCl (pH 7.0), 150 mM NaCl, 10 mM Mg(Oac)2, and 1 mM DTT. Recombination reactions were stopped by phenol/chloroform (1:1, vol/vol) extraction. The efficiency of the reactions was determined by agarose gel analysis of an aliquot digested with BamHI. A typical resolvase reaction produced approximately 50% catenanes. The DNA concentration used in the decatenation experiments was calculated with respect to the 2.1-kb catenanes and the 4.2-kb remaining pJEL257 plasmid, which have identical molecular weights.
Topoisomerase Assays.
Single turnover DNA decatenation experiments were performed by using the KinTek model RQF-3 rapid-quench apparatus (19). Topoisomerase II bound to the DNA substrate was rapidly mixed with ATP in reaction buffer [50 mM Hepes⋅KOH, pH 7.5/150 mM KOac/10 mM Mg(Oac)2]. The ATP concentration used ranged from 350 μM to 2 mM; however, the decatenation rate constants were equivalent for all of the concentrations. The reactions were allowed to proceed for times ranging from 40 msec to 15 sec before being quenched with 50 mM EDTA in 100 mM Tris⋅HCl (pH 8.0). SDS (final concentration, 1%) was present in the sample collection tubes. No topoisomerase-linearized DNA was detected when reactions were quenched in this manner. Gel loading buffer (5% glycerol/0.05% bromophenol blue/0.05% xylene cyanol/10 mM EDTA, final) was added to each quenched reaction before analysis by electrophoresis in a 0.8% agarose gel run at 35 V/cm for 16 hr in TBE (89 mM Tris-borate and 2 mM EDTA) and 0.5 μg/ml ethidium bromide. In this gel system linear 2.1-kb DNA migrates slightly faster than the catenated substrate. The gel was destained for 30 min in water and photographed over a UV transilluminator by using the Eagle Eye II still photo system (Stratagene). Band intensity was quantified by using Scion (Frederick, MD) image analysis software. The fluorescence intensity of the product band, measured in arbitrary units, was shown to always be within the linear range; no monomer circle could be detected at zero time. Aliquots from each reaction time point were analyzed on a minimum of two separate gels, photographed by using at least three different exposures. The data were fit to a single exponential equation, A(1 − e−Bt), by using kaleidagraph 3.0; the decatenation rate constant is the value for B in this equation. The errors for rate constants determined for a single reaction were always less than 15% of the determined rate constant. The reaction time courses all were repeated a minimum of three separate times, and the rate constants reported in Table 1 are the averages of these with the SDs.
Table 1.
Rate constants
| Experiment | Rate constant |
|---|---|
| Decatenation with hydrolysis of two ATP | |
| wt + ATP* | 7.1 ± 1.4 s−1 |
| Decatenation with no ATP hydrolysis | |
| wt + AMPPNP† | 0.2 ± 0.1 s−1 |
| E66Q + ATP† | 0.3 ± 0.1 s−1 |
| Decatenation with hydrolysis of one ATP | |
| E66Q/wtΔC-his + ATP† | 4.3 ± 0.4 s−1 |
| wt + ATP + vanadate† | 6.6 ± 1.3 s−1 |
| Presteady-state ATPase‡ | |
| ATP binding§ | >200 s−1 |
| Hydrolysis of first ATP | 40 ± 10 s−1 |
| Product release | 4 ± 1 s−1 |
*Averaged from seven separate experiments.
†Averaged from three separate experiments.
‡The ATPase rate constants are from previously published data (13).
§The apparent first-order binding rate constant at 1 mM ATP.
Steady-state ATPase rates were measured as described (20). Supercoiled DNA relaxation activity was determined by a published procedure (17).
Computer Modeling.
kinetics 1.0.4 (ARSoftware, Landover, MD) was used to model the steps of the topoisomerase II reaction pathway. To simulate the observed decatenation rate constants for the wt homodimer and the mutant/wt heterodimer reactions, the following simplified reaction scheme was used:
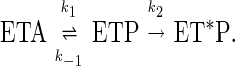 |
In this scheme, E represents the enzyme, T and T* represent the T segment of DNA before and after transport, respectively, A is ATP, and P is ADP/Pi; ET*P is the product observed in the decatenation assays. The value for k2 was determined to be 10 s−1 by simulation using the previously determined values of 40 s−1 and 4 s−1 for k1 and k-1 in the above scheme, respectively (13), and the observed average decatenation rate constant for the wt enzyme (7.1 s−1). The value of k1 for the heterodimer was assumed to be 20 s−1 because this enzyme has half the chances of hydrolyzing one ATP as the wt enzyme.
Results
ATP Hydrolysis by Topoisomerase II Accelerates DNA Transport as Measured by Single Turnover Decatenation.
To determine how topoisomerase II uses ATP to catalyze the transport of a T segment of DNA through a break in the G segment, the rate constants with which this enzyme unlinks two singly catenated DNA circles were determined under several conditions. The ATPase reaction of this enzyme has been extensively studied, and rate constants for many individual steps are known (13). If DNA transport occurs before any ATP is hydrolyzed, as has been previously proposed, then under conditions where ATP binding is rapid, the rate constant for decatenation should be faster than that for hydrolysis of the first ATP.
Single turnover decatenation experiments were performed by using a rapid mix apparatus that allows time points to be taken in the millisecond range. The reaction, unlinking of singly linked DNA catenanes, is illustrated in Fig. 1A. Topoisomerase II prebound to the catenated circles was mixed with ATP and quenched after the desired reaction time. The topoisomerase was at a 2:1 molar excess of the DNA to ensure that only a single turnover was being measured. The catenated circles were supercoiled, and topoisomerase II has been previously shown to preferentially decatenate before relaxing supercoils (12); this preferential decatenation is again seen in the present studies. Aliquots of the quenched reactions were analyzed by agarose gel electrophoresis, an example of which is shown in Fig. 1B. The amount of free DNA circle produced at each time point was determined as described in Materials and Methods. The substrate band does not appear to significantly decrease in intensity with time because a remnant of the reaction used to generate the catenanes comigrates with the substrate. The quantitation of data from Fig. 1B is shown in Fig. 1C. By fitting these data to a single exponential equation, the rate constant for this decatenation reaction was found to be 6.5 ± 0.8 s−1, with an average value of 7.1 ± 1.4 s−1 determined from seven separate experiments.
Figure 1.

Single turnover decatenation measured in the millisecond time scale. (A) Schematic of the reaction showing the three steps of decatenation: G segment cleavage, T segment transport, and G segment religation. The singly linked double-strand catenanes used in the reactions are supercoiled, but shown as relaxed for clarity. (B) A decatenation reaction time course with 50 nM wt topoisomerase II and 25 nM DNA was started by rapid addition of ATP (1 mM, final). After times varying from 50 to 800 msec, reactions were quenched and analyzed by ethidium bromide-stained agarose gel electrophoresis, as described in Materials and Methods. (C) The results shown in B were quantified and fit to the single exponential equation A(1 − e−Bt). Although the data shown were from the single integration shown in B, the rate constant of 6.5 ± 0.8 s−1 was determined for this reaction from three different integrations of the bands.
The rate constants for ATP binding, hydrolysis, and product release by topoisomerase II previously were determined by using rapid-quench kinetics (13). A lower estimate of the apparent first-order rate constant for ATP binding can be determined from the burst rate constant of pulse–chase experiments; at 1 mM ATP the apparent first-order rate constant is ≥200 s−1. The results of chemical-quench experiments showed that the first ATP is hydrolyzed with a rate constant of 40 ± 10 s−1. After hydrolysis of the first ATP, a slow, rate-determining step involving ADP dissociation occurs with a rate constant of 4 ± 1 s−1. Only after this product dissociation step is the second ATP hydrolyzed. The presently observed decatenation rate constant of ≈7 s−1, compared with the significantly more rapid rate constants of ATP binding and hydrolysis of the first ATP, suggests that topoisomerase II hydrolyzes at least one ATP before the products of decatenation are detectable.
The decatenation reaction involves the steps of G segment cleavage, T segment transport, and G segment religation, as illustrated in Fig. 1A. G segment cleavage can occur in the absence of ATP, during the preincubation of the enzyme and DNA, and therefore should not contribute significantly to the measured decatenation rate. G segment religation does not need to occur before quenching the reaction for free DNA products to be detected; the T segment product will be detected once transport has occurred and the EDTA quench will promote religation of the G segment, allowing this second circular product to form. Therefore, T segment transport, broadly defined to include separation of the cleaved G segment ends, is most likely the rate-determining step in the single turnover decatenation reaction. Although we cannot rule out the possibility that ATP hydrolysis accelerates G segment cleavage, for the reasons described above, decatenation rates most likely reflect T segment transport rates. It also should be noted that DNA products noncovalently bound or trapped by the topoisomerase will be released during the quench and detected as free products.
If in fact topoisomerase II normally hydrolyzes at least one ATP before transporting the T segment, then blocking this hydrolysis should slow DNA transport and, therefore, reduce the decatenation rate. This hypothesis was tested in two different ways. In the first experiment, single turnover decatenation was assayed by using wt topoisomerase II and a nonhydrolyzable ATP analog, 5′-adenylyl-β,γ-imidodiphosphate (AMPPNP). For the second experiment, a mutant of topoisomerase II that can bind but not hydrolyze ATP was used. Based on the crystal structure of the ATPase region of gyrase B, glutamate 42 (corresponding to Glu-66 in the yeast enzyme) was predicted to be the catalytic base involved in the activation of a water molecule for nucleophilic attack on the γ-phosphate of ATP (21, 22). Mutating this residue in gyrase B abolishes ATP hydrolysis but not binding (22). The similar mutant of the yeast enzyme (E66Q) also binds ATP (data not shown) and has a very low level of ATPase activity, with the kcat for ATP hydrolysis reduced at least 200-fold (<0.02 s−1 compared with 5 s−1 for the wt enzyme). The results of the single turnover decatenation assays containing wt topoisomerase II with AMPPNP and the E66Q mutant with ATP are shown in Fig. 2. Whereas the decatenation reactions occurred to a similar extent as with the wt enzyme and ATP, the rates of the reactions were much slower. The average decatenation rate constants determined from three separate experiments are 0.2 s−1 and 0.3 s−1 for reactions with AMPPNP and the E66Q mutant enzyme, respectively. AMPPNP binds slowly to topoisomerase II at concentrations below 50 μM; however, at higher concentrations binding is rapid (J.E.L., unpublished results). In the present studies, AMPPNP binding was apparently not rate determining because doubling its concentration from 1 mM to 2 mM had no effect on the decatenation rate. Therefore, as predicted above, topoisomerase II unlinks the single catenanes significantly faster when it hydrolyzes ATP compared with when it cannot.
Figure 2.

Topoisomerase II transports DNA with a reduced rate when it cannot hydrolyze ATP. Results from single turnover DNA decatenation time courses with either 50 nM E66Q enzyme and 1 mM ATP (○), or 50 nM wt enzyme and 1 mM AMPPNP (□) are shown. For these particular reactions, rate constants for decatenation are 0.39 ± 0.07 s−1 for E66Q and ATP, and 0.32 ± 0.04 s−1 for wt and AMPPNP.
DNA Transport Occurs After Hydrolysis of the First ATP.
The rate constant for decatenation by the wt enzyme and ATP suggests that DNA decatenation is occurring after the hydrolysis of at least one ATP. However, the decatenation rate constant is ≈2-fold faster than the rate constant for release of the first hydrolysis products, a step that occurs before the second ATP can be hydrolyzed, suggesting that T segment transport occurs between hydrolysis of the first and second ATP. To directly test this suggestion, single turnover decatenation reactions were performed under two different conditions where topoisomerase II can hydrolyze only one ATP. In the first experiment a heterodimeric mutant/wt topoisomerase II that could bind two ATPs but hydrolyze only one was used. In the second experiment, the wt enzyme was used in the presence of vanadate, an inhibitor that prevents the hydrolysis of the second ATP (13).
Topoisomerase II is a very stable dimer, which does not dissociate or exchange monomers at a detectable rate (17). Therefore, mutant/wt heterodimers cannot be made by simply mixing the separate proteins. To circumvent this problem, E66Q, with one purification tag, was coexpressed in the same yeast cell as the wt enzyme, with a C-terminal truncation that does not affect catalytic activity and a separate purification tag (wtΔC-his). The heterodimers formed by coexpression were separated from the two homodimers by using two affinity chromatography steps, one for each purification tag.
The multiple turnover catalytic activity of the heterodimeric E66Q/wt enzyme was analyzed by steady-state ATPase assays and supercoiled DNA relaxation time courses. The kcat for ATP hydrolysis by the heterodimer (0.6 s−1) was one-tenth of that for the wt homodimer and at least 20-fold faster than for the mutant homodimer. The apparent Km for ATP (130 μM) was unchanged from the wt homodimer. The heterodimer also relaxed supercoiled DNA at about one-tenth the rate of the wt enzyme (not shown). These results are similar to what was previously seen for the analogous gyrase B mutant (23).
Although the steady-state assays showed that the turnover rate for the E66Q/wt heterodimer is about one-tenth that of the wt homodimer, these assays do not indicate which step or steps are slower. Unfortunately, it was not possible to purify enough of the heterodimer to perform presteady-state ATPase assays, so the rate constants for individual steps in its ATPase reaction pathway remain obscure. However, single turnover decatenation experiments with the heterodimer were possible. The results of such an experiment, where decatenation by the wt homodimer is compared with the E66Q/wt heterodimer, are shown in Fig. 3. The decatenation rate constant for the heterodimer is 4.3 ± 0.4 s−1, of the same magnitude as that of the wt homodimer (7.1 ± 1.4 s−1), although reproducibly slower. To determine whether this decreased rate could be caused by a statistical difference in the chances of hydrolyzing two ATP for the homodimer and only one for the heterodimer, the reactions were modeled by computer simulation, as described in Materials and Methods. Assuming an ATP hydrolysis rate constant of 20 s−1, half that of the wt homodimer, the decatenation rate constant for the heterodimer is predicted to be 5.4 s−1. It is presently unclear whether this small difference between the predicted and observed heterodimer decatenation rate constants is significant.
Figure 3.

Hydrolysis of one ATP is sufficient to accelerate DNA transport. Single turnover DNA decatenation time course results are shown for reactions with 1 mM ATP and 25 nM DNA and either 50 nM wt/wtΔC-his (○), 50 nM wt/wtΔC-his plus 100 μM freshly prepared vanadate (□), or 50 nM E66Q/wtΔC-his (▵); rate constants, calculated by fitting data from three separate integrations of the gels, are 6.1 ± 0.5 s−1, 5.9 ± 0.7 s−1, and 4.7 ± 0.7 s−1, respectively.
A very similar result was obtained by including the inhibitor vanadate in a wt homodimer decatenation reaction. Vanadate is a phosphate mimic that binds enzyme-ADP complexes, inhibiting release of the ADP (24). In the presence of vanadate, topoisomerase II hydrolyzes one ATP at the uninhibited rate, but is blocked from sequentially hydrolyzing the second ATP (13). These results have been interpreted to indicate that vanadate binds to the enzyme-ADP-ATP complex after the first Pi is released, preventing the reaction cycle from progressing. When saturating concentrations of vanadate were included in a wt assay, the single turnover decatenation rate was unaffected (Fig. 3). The decatenation rate constant in the presence of vanadate was 6.6 ± 1.3 s−1, within error of the rate constant in the absence of vanadate. Because vanadate does not inhibit DNA transport, these results suggest that DNA transport normally occurs after hydrolysis of the first ATP and before release of the first ADP.
The rate constant for each decatenation experiment, as well as the rate constants for the ATPase reaction up to and including the rate-determining step, are given in Table 1. Taken together, all of these data indicate that topoisomerase II normally catalyzes DNA transport after it hydrolyzes one ATP and before it hydrolyzes the second.
Discussion
Topoisomerase II hydrolyzes one of its two bound ATP ≈5-fold more rapidly than it unlinks single DNA catenanes. If hydrolysis of ATP is blocked, either by using a nonhydrolyzable ATP analog or a hydrolysis-deficient topoisomerase II mutant, the rate of decatenation drops about 20-fold. If the enzyme can hydrolyze only one ATP, the decatenation rate is essentially unchanged from the wt rate in the presence of ATP. Together these data indicate that topoisomerase II normally hydrolyzes one ATP before transporting the T segment of DNA through a transient break in the G segment. Although only ATP binding is absolutely required for DNA transport, hydrolysis of one ATP clearly accelerates the rate of transport.
Studying how enzymes function within a single turnover is crucial to understanding their mechanism. For many ATPases and GTPases, such analysis is most simply done by using nonhydrolyzable analogs of the substrate. Although such analogs are very useful for establishing a general framework for the overall mechanism, one must be careful in interpreting the results. Just because a reaction can occur in the absence of hydrolysis does not mean that hydrolysis does not normally drive the reaction (25). The sequential hydrolysis of ATP by topoisomerase II was missed for many years because it was assumed that reactions occurring in the presence of a nonhydrolyzable ATP analog represented the normal reaction pathway. Because it is doubtful that an enzyme would have a complex, strictly sequential mechanism for no reason, these new results suggest that type II topoisomerases are more complicated than previously predicted. Indeed, it recently was shown that in the presence of ATP these enzymes reduce the steady-state fractions of catenated or knotted DNA molecules 10- to 100-fold below equilibrium values, a finding that could not be explained by a simple reaction mechanism (26).
The structures of several type II DNA topoisomerase fragments have been determined (8, 21, 27, 28), and in each case the dimeric polypeptides show C2 symmetry. Given the symmetric nature of the topoisomerase reaction (the DNA cleavage active sites must simultaneously separate the cleaved G segments to allow transport of the T segment), it is no surprise that the protein is a symmetric dimer. However, at first glance, it is surprising that such an enzyme would use a sequential mechanism for ATP hydrolysis. Although the enzyme quickly binds two ATP with positive cooperativity, it only rapidly hydrolyzes one of those two ATPs (14). The second ATP is not hydrolyzed until after the products of the first hydrolysis, Pi and ADP, are released from the enzyme (13). How can such a sequential mechanism be reconciled with a globally symmetric reaction?
A model that is consistent with this sequential, yet symmetric reaction, and explains the rates of decatenation, is shown in Fig. 4. The overall shapes of the protein domains are taken from four different crystal structures of various type II topoisomerase fragments (8, 21, 27, 28) and their detailed analysis in terms of a predicted reaction mechanism (28). The various domains of the enzyme have been numbered sequentially for clarity, starting at the amino terminus.
Figure 4.

Schematic model of how hydrolysis of one ATP could drive DNA transport by topoisomerase II. The various domains of the topoisomerase II dimer are shown in different colors, with the domains of one monomer numbered starting from the N terminus; the relative orientation of each number and its domain remains constant during the reaction. The C-terminal domain, which is neither conserved nor required for catalytic activity, is not shown. The G and T segments of DNA are shown as green and purple rods, respectively. The exit of the T segment from the topoisomerase is not shown because it is presently unclear at which step it occurs.
Domains 1 and 2 comprise the ATPase region of the protein. Analysis of a wt/ATP binding mutant heterodimer indicated that the same overall conformation exists in both monomers of yeast topoisomerase II whether one or two ATPs are bound (29), hence the protein is shown to have the same structure in Fig. 4 B and C. This is an example of the two monomers in different nucleotide-bound states maintaining overall structural symmetry through allosteric communication. In a similar fashion, the stochastic hydrolysis of one of the two ATPs, or release of the Pi formed, is proposed to trigger a conformational change that includes both monomers (Fig. 4D); the ADP- and ATP-bound monomers once again are predicted to remain globally symmetric. This proposed symmetric conformational change makes sense in terms of the enzyme needing to simultaneously separate the two ends of the cleaved G segment to allow the T segment transport that occurs after hydrolysis of the first and before hydrolysis of the second ATP. This hypothetical enzyme conformation (Fig. 4D) is now incompatible with hydrolysis of the second ATP; the second hydrolysis event would be blocked until the enzyme conformation changed again, returning the ATPase domains to a hydrolysis-competent state. Therefore, a symmetric conformational change that occurs immediately after one ATP is hydrolyzed could enforce a strictly sequential ATPase mechanism.
In this model, altered nucleotide binding states of the 1 domains are transmitted as movements of the 2 domains. These movements drive essentially rigid-body rotations of the 3 domains; amazingly, these domains are actually seen rotated ≈170° relative to each other in two different structures of a yeast topoisomerase II fragment (8, 28). It is the rotation of these domains that is predicted to allow the cleaved G segments of DNA to be separated and the T segment of DNA to be transported through the break. After DNA transport, the T segment is thought to be released through the domain 4 dimer interface. It is presently unclear how this and following steps are correlated with the remainder of the ATPase reaction cycle.
How does this model explain the ability of topoisomerase II to carry out one round of DNA transport in the presence of a nonhydrolyzable ATP analog? The conformational change that occurs between Fig. 4 A and B requires ATP binding. Once that conformation has been stably achieved by binding ATP, or nonhydrolyzable analogs, it is not reversed until after hydrolysis has occurred. If hydrolysis is blocked, the enzyme-DNA complex may slowly sample altered conformations, including that conformation shown in Fig. 4D, without releasing nucleotide or dissociating the amino terminal dimer interface. Once the conformation in Fig. 4D is achieved, it may not readily reverse, and a single round of DNA unlinking can be detected. Another possibility is that ATP binding stabilizes a conformation of the enzyme in which the ends of the G segment are separated. T segment transport then could be driven either rapidly by ATP hydrolysis or more slowly by the free energy of the DNA.
The finding that topoisomerase II binds two ATPs and hydrolyzes them sequentially was unexpected (13). Although several multimeric ATPases, such as kinesin and the F1F0 ATPase, alternate binding and hydrolysis among monomers, the topoisomerase II mechanism is different in that this enzyme first binds both ATP and then sequentially hydrolyzes them. Recently, the β-sliding clamp loader complex of the E. coli DNA polymerase III holoenzyme was shown to have a similar ATPase mechanism (30). It will be interesting to see whether other macromolecular machines are found to use ATP in the same fashion.
Acknowledgments
We are grateful to V. V. Rybenkov for the gift of singly linked DNA catenanes, as well as instructions on producing them, to J. M. Berger for helpful discussions, and to M. M. Hingorani and M. O’Donnell for sharing results before publication. This work was supported by Grant GM51194 from the National Institutes of Health. J.E.L. was supported in part by American Cancer Society Grant JFRA-622. C.L.B. and S.K.M. were supported in part by National Institutes of Health Training Grant 5T32GM08753. S.K.M. also was supported in part by the Huntsman Cancer Institute. T.T.H. was supported in part by National Cancer Institute Training Grant CA09602-06.
Abbreviations
- AMPPNP
adenylyl-β,γ-imidodiphosphate
- wt
wild type
- wtΔC-his
wt topoisomerase II in which the last 95 aa are replaced with a 6× histidine purification tag
- E66Q
topoisomerase II with glutamate 66 mutated to glutamine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Holm C. Cell. 1994;77:955–957. doi: 10.1016/0092-8674(94)90433-2. [DOI] [PubMed] [Google Scholar]
- 2.Warburton P E, Earnshaw W C. BioEssays. 1997;19:97–99. doi: 10.1002/bies.950190203. [DOI] [PubMed] [Google Scholar]
- 3.Berger J M, Wang J C. Curr Opin Struct Biol. 1996;6:84–90. doi: 10.1016/s0959-440x(96)80099-6. [DOI] [PubMed] [Google Scholar]
- 4.Wang J C. Q Rev Biophys. 1998;31:107–144. doi: 10.1017/s0033583598003424. [DOI] [PubMed] [Google Scholar]
- 5.Liu L F, editor. DNA Topoisomerases: Topoisomerase Targeting Drugs. San Diego: Academic; 1994. [Google Scholar]
- 6.Froelich-Ammon S J, Osheroff N. J Biol Chem. 1995;270:21429–21432. doi: 10.1074/jbc.270.37.21429. [DOI] [PubMed] [Google Scholar]
- 7.Maxwell A. Biochem Soc Trans. 1999;27:48–54. doi: 10.1042/bst0270048. [DOI] [PubMed] [Google Scholar]
- 8.Berger J M, Gamblin S J, Harrison S C, Wang J C. Nature (London) 1996;379:225–232. doi: 10.1038/379225a0. [DOI] [PubMed] [Google Scholar]
- 9.Burden D A, Osheroff N. Biochim Biophys Acta. 1998;1400:139–154. doi: 10.1016/s0167-4781(98)00132-8. [DOI] [PubMed] [Google Scholar]
- 10.Sugino A, Higgins N P, Brown P O, Peebles C L, Cozzarelli N R. Proc Natl Acad Sci USA. 1978;75:4838–4842. doi: 10.1073/pnas.75.10.4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osheroff N, Shelton N P, Brutlag D L. J Biol Chem. 1983;258:9536–9543. [PubMed] [Google Scholar]
- 12.Roca J, Wang J C. Cell. 1994;77:609–616. doi: 10.1016/0092-8674(94)90222-4. [DOI] [PubMed] [Google Scholar]
- 13.Harkins T T, Lewis T J, Lindsley J E. Biochemistry. 1998;37:7299–7312. doi: 10.1021/bi9729108. [DOI] [PubMed] [Google Scholar]
- 14.Harkins T T, Lindsley J E. Biochemistry. 1998;37:7292–7298. doi: 10.1021/bi9729099. [DOI] [PubMed] [Google Scholar]
- 15.Giaever G N, Snyder L, Wang J C. Biophys Chem. 1988;29:7–15. doi: 10.1016/0301-4622(88)87020-0. [DOI] [PubMed] [Google Scholar]
- 16.Morris S K, Harkins T T, Tennyson R B, Lindsley J E. J Biol Chem. 1999;274:3446–3452. doi: 10.1074/jbc.274.6.3446. [DOI] [PubMed] [Google Scholar]
- 17.Tennyson R B, Lindsley J E. Biochemistry. 1997;36:6107–6114. doi: 10.1021/bi970152f. [DOI] [PubMed] [Google Scholar]
- 18.Christianson T W, Sikorski R S, Dante M, Shero J H, Hieter P. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- 19.Johnson K A. Methods Enzymol. 1986;134:677–705. doi: 10.1016/0076-6879(86)34129-6. [DOI] [PubMed] [Google Scholar]
- 20.Lindsley J E, Wang J C. J Biol Chem. 1993;268:8096–8104. [PubMed] [Google Scholar]
- 21.Wigley D B, Davies G J, Dodson E J, Maxwell A, Dodson G. Nature (London) 1991;351:624–629. doi: 10.1038/351624a0. [DOI] [PubMed] [Google Scholar]
- 22.Jackson A P, Maxwell A. Proc Natl Acad Sci USA. 1993;90:11232–11236. doi: 10.1073/pnas.90.23.11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kampranis S C, Maxwell A. J Biol Chem. 1998;273:26305–26309. doi: 10.1074/jbc.273.41.26305. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu T, Johnson K A. J Biol Chem. 1983;258:13833–13840. [PubMed] [Google Scholar]
- 25.Rodina M V, Savelsbergh A, Katunin V I, Wintermeyer W. Nature (London) 1997;385:37–41. doi: 10.1038/385037a0. [DOI] [PubMed] [Google Scholar]
- 26.Rybenkov V V, Ullsperger C, Vologodskii A V, Cozzarelli N R. Science. 1997;277:690–693. doi: 10.1126/science.277.5326.690. [DOI] [PubMed] [Google Scholar]
- 27.Cabral J H M, Jackson A P, Smith C V, Shikotra N, Maxwell A, Liddington R C. Nature (London) 1997;388:903–906. doi: 10.1038/42294. [DOI] [PubMed] [Google Scholar]
- 28.Fass D, Bogden C E, Berger J M. Nat Struct Biol. 1999;6:322–326. doi: 10.1038/7556. [DOI] [PubMed] [Google Scholar]
- 29.Lindsley J E, Wang J C. Nature (London) 1993;361:749–750. doi: 10.1038/361749a0. [DOI] [PubMed] [Google Scholar]
- 30.Hingorani M M, Bloom L B, Goodman M F, O’Donnell M. EMBO J. 1999;18:5131–5144. doi: 10.1093/emboj/18.18.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]