

Abstract
Strategies using epitope-based vaccination are being considered for melanoma immunotherapy, in an attempt to overcome failure of other modalities. In the present study, we designed and produced a multiepitope polypeptide for melanoma (MEP-mel), which contains three repeats of four antigenic epitopes (gp100:209–217 (210M); gp100:280–288 (288V); Mart1:26–35 (27L); tyrosinase: 368–376 (370D). The peptides were attached to each other by linkers containing sequences recognized by the proteasome, to improve protein cleavage and antigen presentation. The results show that peptide-specific T cells produced IFN-γ when stimulated with MEP-mel-transfected dendritic cells. The presentation of peptides by MEP-mel-transfected dendritic cells was proteasome-dependent and was more long-lasting than the presentation of exogenously delivered native peptides. When dendritic cells were loaded with MEP-mel protein, weak cross presentation was induced. The production of multiepitope molecules based on several peptides linked by sequences sensitive to proteasomal cleavage represents a promising new tool for the improvement of cancer immunotherapy.
Keywords: melanoma, multiepitope, cytotoxic T cells
Introduction
Melanoma incidence and mortality have risen dramatically during the last century in almost all countries and in fair-skinned populations in particular. In the United States, melanoma rates are increasing faster than those of any other type of cancer, with a twenty-fold increase reported in recent decades; it is the eighth most common type of cancer, causing approximately 2% of all cancer deaths (1). Once it has metastasized, this cancer is associated with only rare durable responses to chemotherapy or any other known treatment (1). Thus, other treatment options are necessary. The potential of harnessing the immune system to induce tumor-specific responses makes immunotherapy a compelling therapeutic alternative. In this context, the use of an antigenic formulation that allows for a wide CD8+ T-cell stimulation remains a major challenge.
The use of protein antigens for vaccination has several potential advantages over the use of peptides. Proteins contain a variety of antigenic epitopes that are not restricted to any given HLA allele. However, protein-based vaccines do not easily prime efficient CD8+ T cell responses (2, 3). It has been shown that in general antigenic epitopes are presented more efficiently by MHC class I to cytotoxic T cells if delivered as peptides, than if delivered as part of an exogenous protein (2). However, in the context of vaccination, the use of peptides was associated with emergence of antigen and/or MHC class I loss variants (4–6). In addition, peptides have the disadvantage of being presented for a relatively short period (usually several hours), due to exogenous loading of the MHC receptors and their recycling (3). On the other hand, peptides derived from proteins or polypeptides are loaded intracellularly on the MHC molecule, and are presented in a more long-lasting manner (3, 7, 8). Thus, one way to optimize an antigenic entity for vaccination would be to produce a polypeptide containing several defined antigenic epitopes. Such a molecule should be designed so it will be efficiently processed, to enable proper presentation of the individual epitopes. Antigen processing of internal proteins involves cleavage by the proteasome, and binding and transport by TAP (Transporter associated with Antigen Processing) to the endoplasmic reticulum. When designing a polypeptide that contains several epitopes, the proteasome cleavage and TAP binding considerations should be taken into account. Collected data of proteasome cleavage products (e.g. (9, 10)) were incorporated into predictive algorithms (11–13). In particular, it has been shown based on an analysis of about 300 naturally processed peptides, that both the C-terminal residue and its flanking residue play a role in determination of proteasome cleavage specificity (10). Furthermore, it was demonstrated that changing the C-terminal flanking residue could enhance immunogenicity of a multiepitope peptide (14). Likewise, there are experimental data by which TAP binding preferences could be determined (15).
In the present study, we have designed and produced a multiepitope polypeptide for melanoma (MEP-mel), which contains four antigenic epitopes. We present results showing that MEP-mel is cleaved by the immunoproteasome of transfected dendritic cells, and the resulting peptides presented to T cells efficiently and lengthily. When MEP-mel is taken up by endocytosis, epitope presentation is less efficient, suggesting that exogenous polypeptides per se do not reach proteasomal cleavage even when privileged epitopes and cleavage signals are inserted.
Materials and Methods
Design of MEP-mel polypeptide
The goal in the combinatorial design of MEP-mel was to optimize the processing of each peptide. We focused on optimizing the proteasomal cleavage and TAP binding signals at the immediate C-terminus flanking region and at the immediate or extended N-terminus flanking region of each peptide. Computation of the proteasome cleavage signals was based on our previous work (10), and on the TAP- binding motifs described by Tampe and colleagues ((15), and personal communication). The flanking regions of each peptide in the MEP-mel were designed according to the following heuristic rules: 1. As both direct and indirect (via amino acid spacer) concatenations of peptides were proved previously to be successful (14, 16), we considered both options. 2. Two types of spacers were searched for: Native protein-derived linkers - derived from the “true” flanking regions of the peptide in its native protein, and artificial linkers –based solely on computational considerations and/or known linkers.
The nucleic acid sequence of MEP-mel encodes T- cell epitopes derived from the following modified melanoma associated antigens: gp100:209–217(210M): IMDQVPFSV; gp100:280–288(288V): YLEPGPVTV; Mart1:27–35(27L): LAGIGILTV; tyrosinase: 368–376(370D): YMDGTMSQV (17–19). All peptides were concatenated via artificial linkers: AAY – a previously proven linker (16); ALL and SSL, which are expected to direct the cleavage to the immediate C-terminus of the preceding peptide and to the immediate N-terminus of the next peptide; RKSYL and RKSY, which are expected to direct the cleavage to the immediate C-terminus of the preceding peptide, but enable a more flexible cleavage at the N-terminus of the next peptide with multiple potential cleavage sites resulting in high TAP-binding scores. A schematic presentation of the polypeptide is shown in Figure 1. Native and modified peptides were synthesized by M. Fridkin, Organic Chemistry, Weizmann Institute of Science, Rehovot, Israel.
Figure 1. Schematic presentation of the MEP-mel molecule.

The four peptides (bottom right) were concatenated via one native (RKSY −/+L) and three artificial linkers (ALL, SSL, AAY) (bottom left) containing sequences recognized by the proteasome. The general scheme (top) shows the linking of the three repeats of the four peptides through the different linkers.
Construction of MEP-mel gene
A set of 12 primers, six on each direction, were designed to produce the gene for MEP- mel. Each primer had 15 bp homology to the flanking primers that serve as a pattern for elongation. The construction of the gene was performed by two PCR reactions. In the first, all 12 primers were mixed, to produce the full gene. This was followed by a second PCR reaction, using primers of both ends of the formed fragment, for propagation of MEP-mel.
The PCR reaction conditions were 1 cycle at 95°C for 5 min; 30 cycles at 94°C for 30 sec, 64°C for 30 sec, and 72°C for 5 min; followed by 1 cycle at 72°C for 10 min. Final products were analyzed by electrophoresis on 0.8% agarose gel stained with ethidium bromide and visualized by UV transillumination. The PCR product was purified with the Concert rapid gel Gibco BRL kit. The amplified fragments were cloned into two plasmids: pcDNA3 (Invitrogen, USA) (5.4 kb, used for transfection of dendritic cells) with KpnI and EcoRI sites, and pQE30 (QIAGEN, USA) (3.4 kb, used for protein production) with BamHI and HindII sites (as previously described (20). DNA sequencing was performed in both directions of the gene (MBC, Rehovot, Israel).
Production and purification of MEP-mel
MEP-mel was expressed in E.coli JM109. The cells were grown to OD 0.5–0.7 on LB medium supplemented with 100 μg/ml of ampicilin. Production of MEP-mel was induced by addition of 1mM isopropyl β-D-thiogalactoside (IPTG) to the medium, and growing the bacteria for 3 hours at 37°C. The cells were centrifuged (5,000g for 30 min) and the pellet was dissolved in PBS buffer and lysed by sonication on ice. The disrupted bacteria were centrifuged (30 min, 4°C, 17,000g) to separate soluble proteins from inclusion bodies (IB). IB were partially purified by shaking in wash buffer (2% triton,0.01M Tris HCl, 300Mm NaCl ) and centrifugation (30 min, 4°C, 17000g) followed by a second wash with 25 mM sodium phosphate buffer. This step was repeated, and then the pellet was dissolved overnight in solubilization buffer 6M GuHCl and centrifuged (30 min, 4°C, 17000g). The supernatant was loaded on a Ni-NTA affinity chromatography column (QIAGEN). Following a wash step (250mM imidizole and 8M urea in 25 mM sodium phosphate buffer) MEP-mel was eluted with 0.5M NaOH. The eluted protein was dialyzed three times against PBS. Identification and determination of purity of MEP-mel were by SDS-PAGE followed by Coomasie staining or immunoblot using monoclonal anti polyhistidine antibodies (Sigma Aldrich). Concentration of the purified MEP-mel was calculated following determination of total protein by Bradford test (BioRad, Hercules, CA, USA).
Generation of dendritic cells (DC)
Peripheral blood mononuclear cells (PBMC) were obtained from healthy volunteers and from patients undergoing treatment for melanoma, as part of their Institutional Review Board approved protocol. PBMC-derived DC were generated from adherent PBMC, cultured in RPMI 1640 with 10% heat-inactivated human AB serum, supplemented with 1000 IU/ml IL-4 (R&D Systems, Minneapolis, MN) and 1000 IU/ml granulocyte-macrophage colony-stimulating factor (GM-CSF; Amgen-Immunex, Seattle, WA) on days 1 and 4 or 5. On day 5 or 6 DC were induced to mature by the addition of soluble trimeric CD40L (Immunex, 1μg/ml) and LPS (Sigma-Aldrich, 5μg/ml), or tumor necrosis factor-α (TNF, R&D Systems, 1000U/ml) and prostaglandin E2 (Sigma, 1μM). DC were used as immature (day 5 or 6) or mature (day 6 or 7) cells.
DNA electroporation
DNA electroporation was performed on immature DC (day 6) with the AMAXA electroprator, using the Human Dendritic Cell Nucleofector™ Kit I (AMAXA biosystems, Cologne, Germany). Briefly, 2.5×106 dendritic cells were resuspended in 100 μl nucleofector™ solution and electroporated in a cuvette with an electrode gap of 2mm. DNA was added immediately prior to electroporation at 5μg/sample. DC were recovered in pre-warmed medium plus GM-CSF and IL-4 to a final concentration of 1 × 106 cells/ml. Viability of cells 24 hours post transfection ranged from 45–70%, as detected by propidium iodide staining.
Transfected cells were characterized by flow cytometry or immunohistochemical staining 24 hours post-transfection.
Production of peptide-specific cytotoxic T-cell lines
T-lines against the modified melanoma peptides gp100 209–217 (210M), gp100 280–288 (288V), Mart126–35 (27L), and the MUC1 63–71 peptide were prepared as described by Riley et al (21). Recognition of peptides by individual T cell microcultures was determined by measurement of IFN-γrelease as described (21). Briefly, PBMC were incubated with each peptide for 7 days, with the addition of 300 IU IL-2 (Chiron) after the second day. The cells were restimulated twice with peptide, at weekly intervals, in the presence of irradiated allogeneic feeder cells. IFN-γ secretion of individual wells was measured in the presence of peptide-loaded T2 cells. Positive cells were restimulated once more with peptide and feeder cells, and IFN-γ was determined after one week with peptide-loaded T2 cells. Positive wells were then stimulated with OKT3 (Orthoclone, Janssen-Cilag) in the presence of allogeneic feeder cells for 10–13 days, and their activity was evaluated against peptide-loaded T2 cells and against melanoma targets. With this protocol, the percentage of CD3+CD8+ within a population ranged from 50–90%. In many cases, the lines with a higher percentage of CD4+ cells were purified by positive sorting (using CD8-coated BD Imag™ particles) to >90% CD8+cells. However, no difference in response to T2 cells loaded with the specific peptide was detected between sorted and unsorted populations (not shown).
Cytokine release assay
For functional detection of antigen presentation on DC, antigen-specific CD8+ T cell lines were co-incubated with (a) MEP-mel transfected immature DC expressing HLA-A*0201 or (b) immature DC or a lymphoblastoid B cell line expressing HLA-A*0201 (LCL721.221 cells (22)) loaded with MEP-mel protein. (a) Following transfection with plasmid DNA, modified DC were distributed into 96-well flat bottom plates, 105 cells per 100μl per well in RPMI 1640 with 10% heat-inactivated human AB serum, supplemented with GM-CSF and IL-4, and let to recover overnight.. Then, responder T cells were added, 100 μl/well (t = 0) at a 1:1 ratio. In the longitudinal study, T cells were gradually added at later time points. Supernatants were collected 16 hours after the addition of responder T cells. IFN-γ secretion was measured in supernatants by ELISA, according to the manufacturer’s instructions (Pierce-Endogen, Cambridge, MA). (b) Immature DC or LCL721.221 cells were incubated for 4 hours with MEP-mel, 10–30 μg/ml, in the absence of serum, and then for further 40 hours after the addition of 10% heat-inactivated human AB serum and the maturation factors TNF and prostaglandin E2. Addition of responder T cells and measurement of IFN-γ secretion was performed as above. As positive controls for T cell function, T2 cells were incubated with 1 μM of the appropriate peptide for 1–3 h at 37°C, and added to T cells as above.
Proteasomal blocking assay
Lactacystin is an irreversible proteasome blocker that works by inhibiting trypsin-like and chymotrypsin-like activities of the 20S subunit (23). DC were incubated for 1 hour with 25μM lactacystin (Calbiochem), within 2 hours post transfection, followed by two washes and overnight recovery in medium with 10% human AB serum supplemented with IL-4 and GM-CSF. Reactive T cell clones were added, and supernatants collected after overnight co-incubation, for measurement of IFN-γ secretion by the T cells.
To exclude a direct effect of lactacystin on DC, its effect on exogenous presentation of peptides was assessed. DC were pre-treated with lactacystin for one hour, washed twice, incubated for two hours with peptides, and after two more washes co-incubated overnight with T cells.
Statistical evaluation
The two-tailed Student’s t-test was used. Each experiment was performed at least twice. Error bars indicate standard error of the mean.
Results
1. MEP-mel production
pcDNA30-MEP-mel plasmid was propagated in JM109 E. coli and purified. The plasmid was obtained in the desired size (6 KB), purified and used for DC transfection. MEP-mel protein was produced in E. coli JM109 at levels of 25 mg/liter and over 90% purity (Figure 2). The two high molecular weight bands seen in the gel are irrelevant to MEP-mel, as demonstrated by the fact that they do not label with monoclonal anti polyhistidine antibodies (not shown).
Figure 2. MEP-mel following purification, run on SDS-PAGE and stained with Coomassie blue.
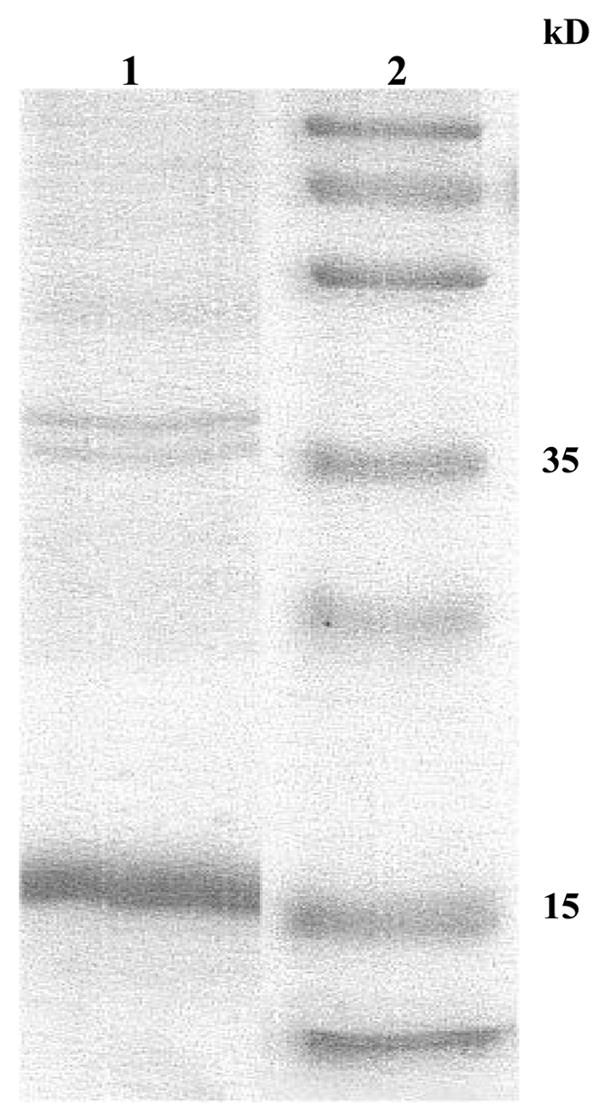
Lane 1 MEP-mel (expected MW: 17kDa); lane 2-molecular size marker
2. Activation of peptide-specific T cells by MEP-mel cDNA-electroporated DC
DC were electroporated with pcDNA3-MEP-mel. In a control group of DC electroporated with Green Fluorescent Protein (GFP) DNA, 19–38% of the total number of cells produced fluorescent protein, and cell viability ranged from 40–75%.
To determine if DC transfected with MEP-mel cDNA present MHC class I-restricted tumor associated epitopes to peptide-specific cytotoxic T-cells, IFN-γ secretion by T cell lines specific for gp100:209–217, gp100:280–288, and Mart1:26–35 was measured. Since these T lines were produced and identified based on their MHC class I peptide specificity, DC loaded with the appropriate native peptide served as a control for T-cell activity and specificity, as well as a control for MHC class I restriction,. The results showed that both MEP-mel-transfected DC and exogenously peptide-loaded DC induced significant IFN-γ secretion by reactive T cells when coincubated for the standard 16-hour overnight period (t = 0). However, presentation of the native peptide markedly decreased in subsequent time points, whereas MEP-mel transfected DC effectively stimulated T cells for as long as 72 hours post transfection. Similar results were obtained for gp100–209-specific T cells (Figure 3, top) and for MART-1-specific T cells (Figure 3, bottom).
Figure 3. Stimulation of peptide-specific CD8+ T cells following incubation with MEP-mel transfected DC.
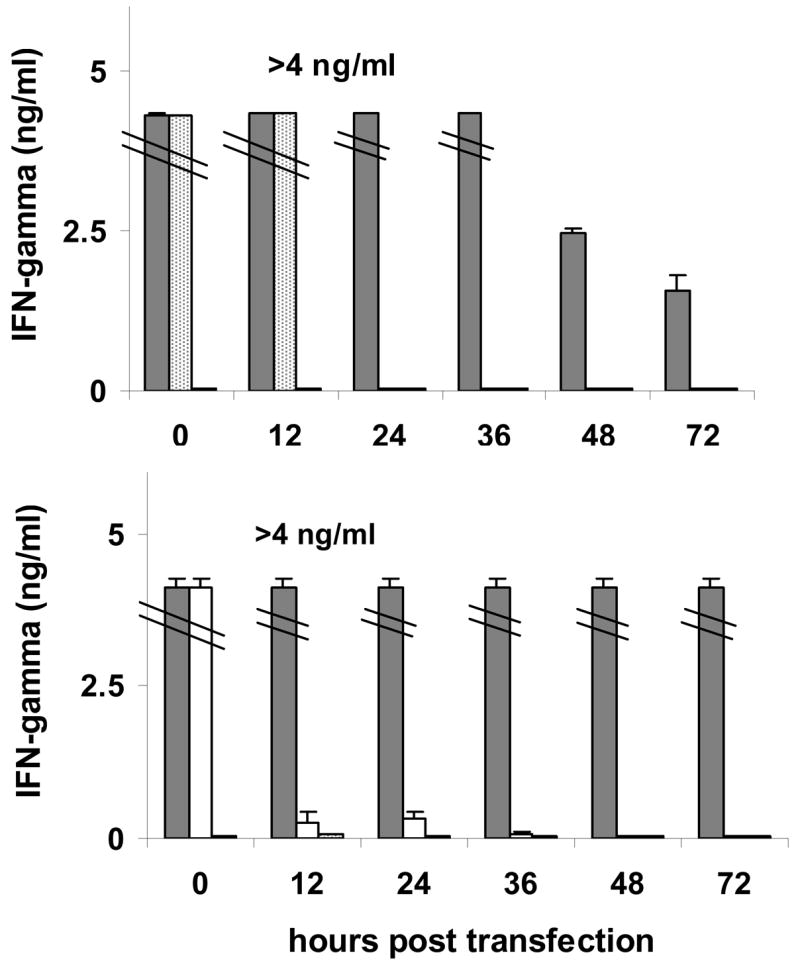
Immature DC transfected with MEP-mel (grey bars) and mature DC loaded with gp100–209 (dotted bars) or with mart1–27 (white bars) were used to stimulate a gp100–209 (top) or a mart1–27 (bottom) specific T-cell line. Each time point included two negative controls: GFP (Green Fluorescent Protein)-transfected DC (0–500 pg/ml) and Hepatitis B virus (HBVc:18–26 (23Y)-loaded DC (0–20 pg/ml). Bars represent standard error of the mean of two independent experiments.
3. Proteasome cleavage is required for processing and presentation of cDNA-encoded MEP-mel
In order to verify that proper presentation of the epitopes that compose MEP-mel is dependent on proteasome degradation, a T-cell stimulation assay was performed in the presence of lactacystin. Incubation with this proteasome inhibitor decreased the stimulation of four T cell lines specific for the peptides included in MEP-mel in the range of 60–92% (Figure 4). Background IFN-γ secretion of T cells only was in the range of 40–125 pg/ml, measured with DC transfected with GFP, and was not affected by lactacystin (62–96 pg/ml). In another control group, lactacystin-treated DC were loaded exogenously with peptide. In this group, IFN-γ secretion was above 500 pg/ml, without a difference between lactacystin-treated and –untreated cells. These results rule out the possibility that the decrease in activity in lactacystin-treated transfected cells was due to direct cell damage by the proteasome blocker. Taken together, the results indicate that peptide presentation by MEP-mel-transfected DC is proteasome-dependent.
Figure 4. Antigen presentation by MEP-mel transfected DC is proteasome-dependent.
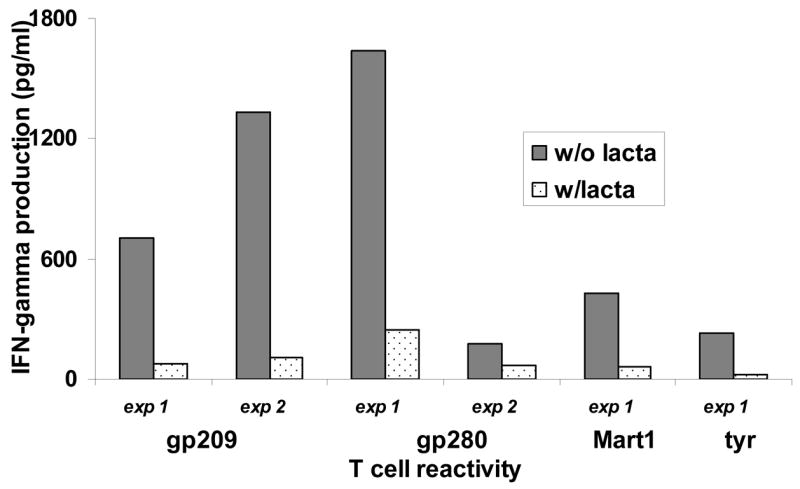
MEP-mel-transfected DC incubated with the proteasome blocker lactacystin inhibited stimulation of CD8+ T-cell lines specific for the four peptides contained in MEP-mel. Individual results from two independent experiments (exp1 and exp2) are presented. w/lacta – with lactacystin; w/o lacta – without lactacystin.
4. Exogenous MEP-mel protein presentation (cross presentation)
MEP-mel protein was produced in E. coli and purified. Due to the hydrophobic nature of the protein, solubility was only achieved in 8M urea or 6M guanidine. Immature DC or LCL721.221 cells were loaded with MEP-mel protein, and maturation of DC was induced for 48 hours in the presence of TNF-α and prostaglandin E2 (24). The antigen presenting cells were co-incubated with HLA-A*0201 lymphocyte lines specific for the peptides present in the MEP-mel construct, and a control unrelated T-cell line against a MUC1 peptide (LLLTVLTVV)(25). Table 1 shows that MEP-mel-loaded DC induced IFN-γ production. However, upon comparison of the amounts of IFN-γ produced by protein loading (up to 250 pg/ml) and by DC transfection (>4,000 pg/ml, Figure 3), DC protein loading was clearly less effective Using FITC-labeled MEP-mel, we confirmed by confocal microscopy that MEP-mel was taken up very efficiently by DC (not shown).
Table 1. IFN-γ secretion by peptide-specific T cell lines in response to stimulation by antigen presenting cells loaded with MEP-mel protein.
DC or antigen presenting LCL721.221cells were loaded with MEP-mel protein or with peptides, and coincubated overnight with peptide-specific T cells. MUC1 and MUC1-specific T cells served as negative control. IFN-γ levels secreted into the supernatant were determined.
| APC | Loading | T-cell specificity (IFN-γ secretion, pg/ml) | ||
|---|---|---|---|---|
| DC | Gp209 | Mart1 | MUC1 | |
| None | 66 | 68 | 49 | |
| MEP-mel | 235 | 135 | 59 | |
| Specific peptide | 5840 | 7220 | 16460 | |
| Control peptide | 140 | 53 | 43 | |
| LCL | None | 41 | 46 | 297 |
| MEP-mel | 83 | 125 | 69 | |
| Specific peptide | 5793 | 5253 | 14360 | |
| Control peptide | 81 | 64 | 406 | |
Discussion
T lymphocytes that recognize antigens expressed on tumor cells are essential effector cells that mediate tumor regression. Active immunization with tumor vaccines aims at the generation of tumor-specific T cells. Strategies used for vaccination have included delivery of antigenic peptides in combination with adjuvants, or loaded on antigen presenting cells (26, 27). However, loss of immunogenic epitopes by tumors has urged the development of vaccines against multiple epitopes. Combinations of peptides have been delivered as a mixture in a single emulsion (28–30). However, the presence of an immunodominant epitope may jeopardize an unbiased immune response to the mixture (16, 31). A different multiepitope approach has been the use of a polypeptide composed of a string of ten epitopes (32). Studies in mice with multiepitope DNA-based vaccines containing melanoma sequences have focused attention to the importance of linkers as part of the recombinant complex (16). Another polyepitope, produced in poxvirus and containing ten HLA-A2-restricted epitopes derived from five melanoma antigens, elicited a CTL response in vitro (33). The advantage of using antigens with point mutations to improve peptide-MHC binding has also been demonstrated (34).
The goal of this study was to design a molecule consisting of several repeats of four immunogenic HLA*0201 peptides, chosen for their ability to elicit specific CD8+ T cells when used for vaccination (17–19). Since the direct concatenations or the concatenation by native protein-derived linkers of the four epitopes yielded relatively poor cleavage scores and poor TAP-binding scores, we designed a multiepitope construct in which the melanoma epitopes are attached to each other by linkers containing sequences recognized by the proteasome, with the aim of improving protein cleavage and antigen presentation. By improving cleavage rates the availability of low cleavage epitopes could theoretically compensate for lower peptide-MHC binding and augment peptide density on the cell surface. Since proteasomal cleavage efficacy of the various linkers was not known, MEP-mel was designed in such a way that each peptide was attached to several linkers. For this purpose, the sequence of each peptide was introduced three times into MEP-mel. Nevertheless, competition between the epitopes for the available HLA-A2 molecules remains a theoretical possibility.
The results presented in this study show that peptide-specific T cells produced IFN-γ when stimulated with MEP-mel-transfected DC. The multiepitope protein was produced intracellularly, and degraded to the appropriate peptides, which were presented on DC to peptide-specific cytotoxic T cells. The presentation of peptides by MEP-mel-transfected DC was proteasome-dependent and was more long-lasting than the presentation of exogenously delivered native peptides.
The ultimate goal of cell immunotherapy is to use an antigenic formulation allowing a wide tumor associated peptide repertoire presentation. Using DNA electroporation of human DC we induced stable and efficient antigen presentation. However, genetic modifications raise safety issues such as insertional mutagenesis and promotor’s effects (35). To attempt to bypass these disadvantages, we delivered the multiepitope polypeptide protein in addition to the MEP-mel-encoding DNA gene. Under the experimental conditions used in this study, MEP-mel protein was taken up by DC, but the extent of cross priming was not very efficient. Thus, our data show that cDNA MEP-mel, rather than MEP-mel protein, is a powerful source of prolonged antigen presentation.
Acknowledgments
This work was supported by NIH grant 1 R21 CA114160-01A1 (to S.F.), by the Israel Ministry of Commerce, and by the Chief Scientist of the Israel Ministry of Health. Several of the T cell lines used were kindly provided by the Surgery Branch, NCI, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Langley RGBBR, Mihm MC, Fitzpatrick TB, Sober AJ. Neoplasms: Cutaneous Melanoma. 2003 [Google Scholar]
- 2.van der Bruggen P, Van den Eynde BJ. Curr Opin Immunol. 2006;18:98–104. doi: 10.1016/j.coi.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 3.Schnurr M, Chen Q, Shin A, Chen W, Toy T, Jenderek C, Green S, Miloradovic L, Drane D, Davis ID, Villadangos J, Shortman K, Maraskovsky E, Cebon J. Blood. 2005;105:2465–72. doi: 10.1182/blood-2004-08-3105. [DOI] [PubMed] [Google Scholar]
- 4.Riker A, Cormier J, Panelli M, Kammula U, Wang E, Abati A, Fetsch P, Lee KH, Steinberg S, Rosenberg S, Marincola F. Surgery. 1999;126:112–20. [PubMed] [Google Scholar]
- 5.Jager E, Ringhoffer M, Karbach J, Arand M, Oesch F, Knuth A. Int J Cancer. 1996;66:470–6. doi: 10.1002/(SICI)1097-0215(19960516)66:4<470::AID-IJC10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo NP, Hughes MS, Schwartzentruber D, Berman DM, Schwarz SL, Ngo LT, Mavroukakis SA, White DE, Steinberg SM. J Immunol. 2005;175:6169–76. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 7.Amoscato AA, Prenovitz DA, Lotze MT. J Immunol. 1998;161:4023–32. [PubMed] [Google Scholar]
- 8.Ludewig B, McCoy K, Pericin M, Ochsenbein AF, Dumrese T, Odermatt B, Toes RE, Melief CJ, Hengartner H, Zinkernagel RM. J Immunol. 2001;166:3678–87. doi: 10.4049/jimmunol.166.6.3678. [DOI] [PubMed] [Google Scholar]
- 9.Nussbaum AK, Dick TP, Keilholz W, Schirle M, Stevanovic S, Dietz K, Heinemeyer W, Groll M, Wolf DH, Huber R, Rammensee HG, Schild H. Proc Natl Acad Sci U S A. 1998;95:12504–9. doi: 10.1073/pnas.95.21.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altuvia Y, Margalit H. J Mol Biol. 2000;295:879–90. doi: 10.1006/jmbi.1999.3392. [DOI] [PubMed] [Google Scholar]
- 11.Kuttler C, Nussbaum AK, Dick TP, Rammensee HG, Schild H, Hadeler KP. J Mol Biol. 2000;298:417–29. doi: 10.1006/jmbi.2000.3683. [DOI] [PubMed] [Google Scholar]
- 12.Nussbaum AK, Kuttler C, Hadeler KP, Rammensee HG, Schild H. Immunogenetics. 2001;53:87–94. doi: 10.1007/s002510100300. [DOI] [PubMed] [Google Scholar]
- 13.Kesmir C, Nussbaum AK, Schild H, Detours V, Brunak S. Protein Eng. 2002;15:287–96. doi: 10.1093/protein/15.4.287. [DOI] [PubMed] [Google Scholar]
- 14.Livingston BD, Newman M, Crimi C, McKinney D, Chesnut R, Sette A. Vaccine. 2001;19:4652–60. doi: 10.1016/s0264-410x(01)00233-x. [DOI] [PubMed] [Google Scholar]
- 15.Uebel S, Kraas W, Kienle S, Wiesmuller KH, Jung G, Tampe R. Proc Natl Acad Sci U S A. 1997;94:8976–81. doi: 10.1073/pnas.94.17.8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Velders MP, Weijzen S, Eiben GL, Elmishad AG, Kloetzel PM, Higgins T, Ciccarelli RB, Evans M, Man S, Smith L, Kast WM. J Immunol. 2001;166:5366–73. doi: 10.4049/jimmunol.166.9.5366. [DOI] [PubMed] [Google Scholar]
- 17.Nagorsen D, Servis C, Levy N, Provenzano M, Dudley ME, Marincola FM, Levy F. Cancer Immunol Immunother. 2004;53:817–24. doi: 10.1007/s00262-004-0532-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parkhurst MR, Salgaller ML, Southwood S, Robbins PF, Sette A, Rosenberg SA, Kawakami Y. J Immunol. 1996;157:2539–48. [PubMed] [Google Scholar]
- 19.Skipper JC, Hendrickson RC, Gulden PH, Brichard V, Van Pel A, Chen Y, Shabanowitz J, Wolfel T, Slingluff CL, Jr, Boon T, Hunt DF, Engelhard VH. J Exp Med. 1996;183:527–34. doi: 10.1084/jem.183.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gelbart Y, Frankenburg S, Pinchasov Y, Krispel S, Eliahu D, Drize O, Morag E, Bartfeld D, Lotem M, Peretz T, Pitcovski J. Protein Expr Purif. 2004;34:183–9. doi: 10.1016/j.pep.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 21.Riley JP, Rosenberg SA, Parkhurst MR. J Immunol Methods. 2003;276:103–19. doi: 10.1016/s0022-1759(03)00078-4. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu Y, DeMars R. J Immunol. 1989;142:3320–8. [PubMed] [Google Scholar]
- 23.Oda K, Ikehara Y, Omura S. Biochem Biophys Res Commun. 1996;219:800–5. doi: 10.1006/bbrc.1996.0314. [DOI] [PubMed] [Google Scholar]
- 24.Frankenburg S, Elias O, Gelbart Y, Drize O, Lotem M, Ingber A, Peretz T, Pitcovski J. Immunol Lett. 2004;94:253–9. doi: 10.1016/j.imlet.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 25.Carmon L, El-Shami KM, Paz A, Pascolo S, Tzehoval E, Tirosh B, Koren R, Feldman M, Fridkin M, Lemonnier FA, Eisenbach L. Int J Cancer. 2000;85:391–7. [PubMed] [Google Scholar]
- 26.Davis ID, Jefford M, Parente P, Cebon J. J Leukoc Biol. 2003;73:3–29. doi: 10.1189/jlb.0502261. [DOI] [PubMed] [Google Scholar]
- 27.Jaeger EJD, Knuth A. Current Opinion in Immunology. 2002;14:178–182. doi: 10.1016/s0952-7915(02)00318-7. [DOI] [PubMed] [Google Scholar]
- 28.Valmori D, Dutoit V, Ayyoub M, Rimoldi D, Guillaume P, Lienard D, Lejeune F, Cerottini JC, Romero P, Speiser DE. Cancer Immun. 2003;3:15. [PubMed] [Google Scholar]
- 29.Slingluff CL, Jr, Petroni GR, Yamshchikov GV, Hibbitts S, Grosh WW, Chianese-Bullock KA, Bissonette EA, Barnd DL, Deacon DH, Patterson JW, Parekh J, Neese PY, Woodson EM, Wiernasz CJ, Merrill P. J Clin Oncol. 2004;22:4474–85. doi: 10.1200/JCO.2004.10.212. [DOI] [PubMed] [Google Scholar]
- 30.Rosenberg SA, Sherry RM, Morton KE, Yang JC, Topalian SL, Royal RE, Kammula US, Restifo NP, Hughes MS, Schwarz SL, Ngo LT, Mavroukakis SA, White DE. J Immunother. 2006;29:224–31. doi: 10.1097/01.cji.0000190399.98802.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palmowski MJ, Choi EM, Hermans IF, Gilbert SC, Chen JL, Gileadi U, Salio M, Van Pel A, Man S, Bonin E, Liljestrom P, Dunbar PR, Cerundolo V. J Immunol. 2002;168:4391–8. doi: 10.4049/jimmunol.168.9.4391. [DOI] [PubMed] [Google Scholar]
- 32.Alexander J, Oseroff C, Dahlberg C, Qin M, Ishioka G, Beebe M, Fikes J, Newman M, Chesnut RW, Morton PA, Fok K, Appella E, Sette A. J Immunol. 2002;168:6189–98. doi: 10.4049/jimmunol.168.12.6189. [DOI] [PubMed] [Google Scholar]
- 33.Mateo L, Gardner J, Chen Q, Schmidt C, Down M, Elliott SL, Pye SJ, Firat H, Lemonnier FA, Cebon J, Suhrbier A. J Immunol. 1999;163:4058–63. [PubMed] [Google Scholar]
- 34.Tine JA, Firat H, Payne A, Russo G, Davis SW, Tartaglia J, Lemonnier FA, Demoyen PL, Moingeon P. Vaccine. 2005;23:1085–91. doi: 10.1016/j.vaccine.2003.01.001. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Troilo PJ, Wang X, Griffiths TG, Pacchione SJ, Barnum AB, Harper LB, Pauley CJ, Niu Z, Denisova L, Follmer TT, Rizzuto G, Ciliberto G, Fattori E, Monica NL, Manam S, Ledwith BJ. Gene Ther. 2004;11:711–21. doi: 10.1038/sj.gt.3302213. [DOI] [PubMed] [Google Scholar]