

Abstract
Dendritic cells (DCs) produce cytokines and are susceptible to cytokine-mediated activation. Thus, interaction of resting immature DCs with TLR ligands, for example nucleic acids, or with microbes leads to a cascade of pro-inflammatory cytokines and skewing of T cell responses. Conversely, several cytokines are able to trigger DC activation (maturation) via autocrine, for example TNF and plasmacytoid DCs, and paracrine, for example type I IFN and myeloid DCs, pathways. By controlling DC activation, cytokines regulate immune homeostasis and the balance between tolerance and immunity. The increased production and/or bioavailability of cytokines and associated alterations in DC homeostasis have been implicated in various human inflammatory and autoimmune diseases. Targeting these cytokines with biological agents as already is the case with TNF and IL-1 represents a success of immunology and the coming years will expand the range of cytokines as therapeutic targets in autoinflammatory and autoimmune pathology.
Keywords: Dendritic cells, IL-1, IL-12, IL-23, TNF-α, IFN-α
1. Introduction
The immune system is composed of a non-antigen-specific innate limb and an antigen-specific adaptive limb [1]. Innate immunity, borne by cells such as granulocytes and macrophages and proteins such as complement and cytokines, includes a variety of prompt reactions in response to infectious agents and other challenges. An excessive response results in inflammatory processes. The adaptive immunity, borne by lymphocytes, is acquired in weeks or months. It is characterized by an exquisite specificity for the eliciting antigen as well as memory, which allows a faster and stronger response upon re-exposure to the antigen. Adaptive responses can be immunogenic, leading to resistance to infections and possibly cancer, or tolerogenic avoiding response against self.
Indeed, to efficiently protect us from invading microorganisms, the adaptive immune system must distinguish self from non-self as immune responses against self can create a wide repertoire of autoimmune diseases. Anti-self immune responses are prevented through a variety of mechanisms occurring at various levels during the development of the immune system [2]. Autoreactive lymphocytes can be deleted, rendered anergic or rendered suppressive [3–5]. Suppressor T cells, also called regulatory T cells, suppress autoreactive responses both in an antigen-specific and a non-antigen-specific fashion. These immunological events happen either in the primary lymphoid organs (bone marrow and thymus) and are thus collectively called “central tolerance” or in the periphery and are then called “peripheral tolerance”. Clinical autoimmunity arises as a result of an altered balance between the autoreactive cells and the regulatory mechanisms designed to counterbalance them.
DCs are specialized to capture and process antigens to present their peptides to lymphocytes. They are found in all tissues including blood and lymphoid organs [6–12]. In peripheral tissues, DCs are found in an immature stage specialized in the capture of antigens. In response to microbes, DCs undergo a complex process of maturation into antigen-presenting cells. This happens while the DCs migrate from the periphery into the draining lymph node through the lymphatics. In the steady state, DCs also migrate at a low rate without undergoing activation. Then they present self-antigens to lymphocytes in the absence of costimulation thereby leading to peripheral tolerance. Various mouse models have demonstrated that DCs bearing self-antigens are able to induce autoimmune diseases [13–15]. Furthermore alterations of DC homeostasis have been directly implicated in various human autoimmune diseases including type I diabetes, multiple sclerosis, and systemic lupus erythematosus (SLE) [16,17].
Here we review our current understanding of dendritic cell function in tolerance and how cytokines interfere with these processes to generate autoimmunity.
2. Dendritic cells
2.1. Dendritic cell maturation
Dendritic cells (DCs) are a heterogeneous family of cells of haematopoietic origin that are specialized in the handling of antigens, i.e. those from infectious agents and self, and their presentation to lymphocytes. Though most of the current knowledge relates to the presentation of peptides to T cells in the context of MHC classes I and II molecules, DCs can present glycolipids and glycopeptides to T cells and NKT cells as well as polypeptides to B cells. DCs undergo a complex maturation process from antigen-capturing cells into antigen-presenting cells. Numerous agents activate DCs including: microbes, dying cells, cells of the innate immune system and cells of the adaptive immune system. pathogen-associated molecular patterns (PAMPs) from microbes [18] signal DCs and other cell types through a variety of pattern-recognition receptors (PRR) including toll-like receptors (TLRs) [19,20]; cell surface C-type lectins receptors (CLRs) [21,22] and intracytoplasmic NOD-like receptors (NLRs) [23,24]. TLRs have been given the most attention until now and appear to be particularly important in the context of autoimmunity and most specifically SLE. Distinct DC subsets display different TLRs as will be discussed hereunder. Lysates of dying cells induce the maturation of DCs [25], and some components involved in dying cells enhance antigen presentation by DCs leading to T cell immunity [25,26]. These endogenous activating molecules are collectively called damage-associated molecular pattern molecules (DAMPs) [27]. They include heat shock proteins (HSPs) [28], high mobility group box 1 protein (HMGB1) [29], β-defensin [30] and uric acid [31].
DCs can secrete a diversified panel of chemokines that attract different cell types at different times of the immune response [32]. They also express a unique set of costimulatory molecules which permit the activation of naïve T cells and thus allow the launching of primary immune response. Through the cytokines they secrete, e.g.: IL-12, IL-23 or IL-10 as well as the surface molecules they express, e.g.: OX40-L [33] or ICOS-l [34] DCs can polarize naïve T cells into Th1, Th2, Treg or Th17.
2.2. Dendritic cell subsets
There are two main pathways of DC ontogeny from hematopoietic progenitor cells (HPCs). One pathway generates myeloid DCs (mDCs); while another generates plasmacytoid DCs (pDCs), a subset capable of secreting large amounts of type I IFN in response to viral stimulation [35,36] (Fig. 1). At least six DC subsets have been described in mouse spleen and lymph nodes, including conventional DCs (formerly designated myeloid DCs and lymphoid DCs) and plasmacytoid DCs [11,37]. They are distinguished according to surface markers such as CD11b, CD8a and CD11c, as well as by their functions [38–40]. Myeloid DCs are found in three compartments: (1) peripheral tissue, (2) secondary lymphoid organ and (3) blood. In the skin, two distinct types of mDCs are found in two distinct layers. Langerhans cells (LCs), which express CD1a and Langerin reside in the epidermis, while interstitial DCs (intDCs), which express DC-SIGN and CD14 reside in the dermis [41]. Plasmacytoid DCs, circulating in the blood and secondary lymphoid organs by crossing high endothelial venules, express BDCA2, ILT-7 and CD123, and secrete large amounts of type I interferons (IFNs) in response to viruses and/or TLR7−9 ligands [9]. Blood plasmacytoid DCs express TLR1, 6, 7, 9 and 10, but nor TLR4, while blood myeloid DCs express TLR1, 2, 3, 4, 5, 6, 7, 8 and 10, but not TLR9 [42,43]. Epidermal Langerhans cells isolated from skin lack the expression of TLR4 and TLR5, while dermal interstitial DCs express many TLRs including TLR2, 4 and 5 [44]. In the human, CLRs permit to distinguish DC subsets with BDCA2 specifically expressed on plasmacytoid DCs [45], Langerin expressed on Langerhans cells [46], and DC-SIGN expressed on interstitial DCs [47]. Many other C-type lectins are more promiscuous, and are, as is the case with TLRs, expressed on various cell types including endothelial cells and neutrophils. C-type lectins expressed on DCs act as anchors for a large number of microbes including viruses, bacteria, parasites and fungi, and allow their internalization, but they also act as adhesion molecules between DCs and other cell types including endothelial cells, T cells and neutrophils. Abnormalities in dendritic cell homeostasis have been implicated in various human diseases, including cancer, autoimmune diseases, allergy and infections.
Fig. 1.
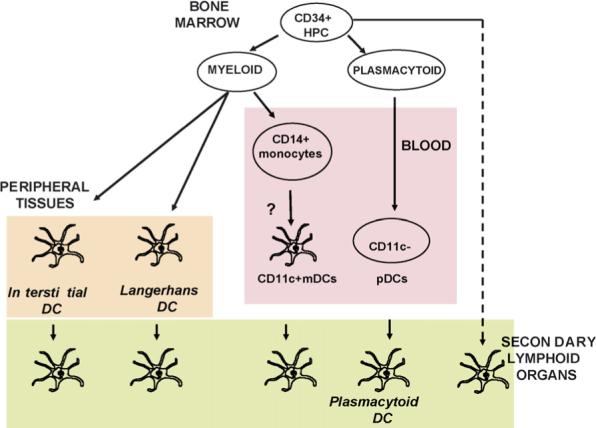
Dendritic cells are composed of subsets. DC progenitors originate from bone marrow CD34+FLT3+ hematopoietic progenitor cells (HPCs). A myeloid pathway generates both Langerhans cells (LCs), found in stratified epithelia such as the skin, and interstitial (int)DCs, found in all other tissues. It also generates mDCs circulating in the blood. Upon inflammation monocytes can yield mDCs. Another pathway generates plasmacytoid DCs (pDCs), which secrete large amounts of IFN-α/β after viral infection. Activated (mature) mDCs and pDCs traffic to secondary lymphoid organs either via afferent lymphatics (mDCs) or blood (pDCs). Langerhans DCs home to cell zones while interstitial DCs home to follicles consistent with their functional specialization, i.e. generation of cellular (Langerhans DCs) and humoral (interstitial DCs) immunity, respectively. The origin of resident lymph node DCs remains to be determined.
2.3. Dendritic cells and immune tolerance
Central tolerance, induced in the thymus or bone marrow, plays a pivotal role in the prevention of undesired attacks against self. Anti-self immune responses are prevented through a variety of mechanisms occurring at various levels of the immune system development [2]. Autoreactive lymphocytes can be either deleted, or rendered anergic or rendered suppressive [3–5]. Suppressor T cells also called regulatory T cells are also generated which suppress autoreactive responses both in an antigen-specific fashion and a non-antigen-specific fashion. Many peripheral auto-antigens through their expression in thymic medullary epithelial cells (a process regulated by the autoimmune regulator AIRE) are known to be responsible for the so-called negative selection [48]. Moreover, cytokines like thymic stromal lymphopoietin (TSLP) produced by epithelial cells of thymic Hassall's corpuscles promote the conversion of CD4+CD25-thymocytes into CD4+CD25+Foxp3+ T regulatory cells (Tregs) [49]. Dendritic cells in the thymus are also involved in the process of central tolerance [50].
It is clear, however, that negative selection in the thymus does not eliminate all autoreactive cells. Thus, tolerance induced in the periphery becomes a very important mechanism to maintain control of emerging autoreactivity. The mechanisms involved in peripheral tolerance are not entirely understood, but there is evidence that “resting” immature DCs that capture self-antigens in the steady state play an important role in this process. Indeed, under these conditions, DCs capture apoptotic bodies and/or cellular debris arising from normal cell turnover, migrate to draining lymph nodes and silence T cells reacting to these antigens [51]. The myeloid DC subset appears to be the most potent cell able to capture self-apoptotic bodies.
This phenomenon needs to be tightly regulated, as an unusual load of apoptotic bodies can induce systemic autoimmune disease [52]. Indeed, dead cells may also contribute to DC maturation, as it has been shown for necrotic or late apoptotic cells, but no early apoptotic cells. As discussed above, endogenous activating molecules are collectively called damage-associated molecular pattern molecules [27]. There is evidence that plasmacytoid DCs in their resting state are involved in tolerance induction [53–55]. pDCs stimulated via CD40 induce IL-10-secreting regulatory CD4+ T cells [34] as well as suppressor CD8+ T cells [56].
Recently, the concept that “immature DCs are tolerogenic whereas mature DCs are immunogenic” has been challenged by several studies showing that fully mature DCs can induce tolerance and differentiation of regulatory T cells [57–59]. In fact, the integration of different signals by the DCs, including Ag dose, cytokine milieu at sites of inflammation, encountered pathogen etc., will determine whether DCs will become tolerogenic vs. immunogenic. Possibly, peripheral tolerance is actively maintained by “tolerogenic” DCs [60]. In addition to deleting T cells, tolerogenic DCs induce the differentiation and proliferation of T cells with regulatory/suppressor functions [3,61]. Some pathogens have a capacity to actively render DCs tolerogenic [62]. Although the specific markers of tolerogenic DCs are yet to be determined, expression of inhibitory immunoglobulin like transcript (ILT) receptors might be their feature [63]. In vitro-generated DCs exposed to IL-10 express ILT-3, which is associated with their tolerogenic functions [64]. Studies in mice suggest that DCs might be used in the treatment of autoimmunity through their ability to induce regulatory T cells. Thus, repetitive injections of “semi-mature” DCs induce antigen-specific protection of mice from experimental autoimmune encephalomyelitis and thyroiditis [57,58]. In NOD mice which spontaneously develop diabetes, DCs can induce the generation of Tregs in vitro which provide a therapeutic benefit even after onset of disease [59]. Indeed, Tregs appear to suppress DCs that induce autoimmunity by presenting autoantigens [59,65].In keeping with this, animals which are depleted of Tregs show autoimmunity that is associated to expansion of activated DCs [66,67]. While immunogenic DCs have been used in clinical trials to treat mainly patients with cancer, understanding the mechanisms underlying the tolerogenic functions of DCs, such as those generated with IL-10 [68,69] or those infected with RSV [70], opens new avenues for the treatment of autoimmunity or the induction of specific tolerance in organ transplants.
Cytokines secreted to induce a specific immune response against an invading pathogen might interfere with DC homeostasis and induce an autoimmune response that can be responsible for tissue pathology. Indeed, clinical and epidemiological studies have suggested a link between infectious agents and chronic inflammatory disorders, including autoimmune diseases [71].
3. Cytokines, inflammation and autoimmunity
Cytokines represent critical mediators of the autoimmune process. They may represent products of DCs and/or induce the differentiation of immature DCs into mature DCs that can select autoreactive lymphocytes (Fig. 2).
Fig. 2.
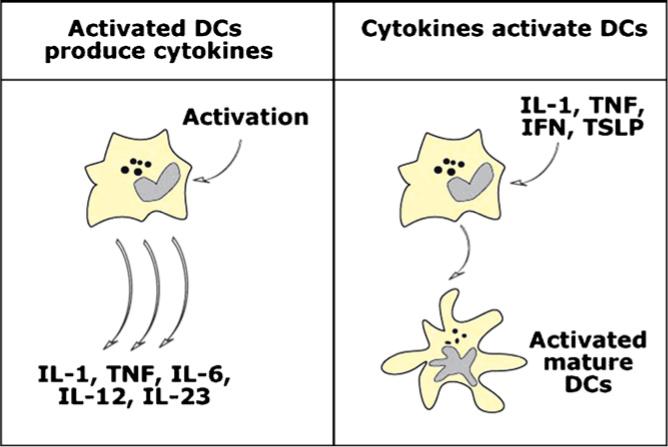
Cytokines and dendritic cell activation. DCs both produce cytokines and are susceptible to cytokine-mediated activation. Thus, exposure to DC activators, for example TLR ligands or microbes, triggers secretion of pro-inflammatory cytokines including type I interferons (IFN), acute phase cytokines such as TNF and IL-6, IL-1 as well as IL-12 family (left panel). Several cytokines are able to trigger DCs activation (maturation) either in autocrine or paracrine fashion including IL-1, TNF, type I IFNs and TSLP (right panel).
3.1. IL-1 and its family
The IL-1 family plays an important role in inflammation and host defense. Up to 11 members of this family have been identified to date [72,73]. Of those, only five have been thoroughly studied: IL-1α, IL-1b, IL-18, IL-1RA and the recently reported IL-33. The remaining six (IL-1F5; IL-1F6; IL-1F7; IL-1F8; IL-1F9; IL-1F10) have been shown to be expressed in various cell types or tissues but their functions remain to be determined.
IL-1α and IL-1β are pro-inflammatory cytokines. Both are synthesized as precursor molecules (pro-IL-1α and pro-IL-1β) by many different cell types. Pro-IL-1α is biologically active and needs to be cleaved by calpain to generate the smaller mature protein. By contrast, pro-IL-1β is biologically inactive and requires enzymatic cleavage by caspase-1 in order to become active. IL-1α is primarily bound to the membrane whereas IL-1β is secreted and thus represents the predominant extracellular form of IL-1 (reviewed in Ref. [74,75]). IL-33 is a new member of the IL-1 family that is produced as a propeptide requiring cleavage by caspase-1. It binds to IL-1R4 (ST2) and stimulates T helper 2 (Th2) responses [73]. IL-1 is an activator of DCs, though it is not yet clear whether such IL-1-activated DCs display unique biological functions [73].
Interleukin-1 is involved in the pathogenesis of numerous diseases with an inflammatory component [76]. This is best demonstrated by the therapeutic benefits of treatment of patients with IL-1 antagonists such as IL-1-RA. These diseases include Systemic onset Juvenile Idiopathic Arthritis (SoJIA) [77], which represents up to 20% of chronic inflammatory arthritis in childhood. IL-1RA has also shown therapeutic efficacy in gout [78], type II diabetes [79] as well as a series of hereditary diseases causing periodic inflammatory symptoms and grouped under the term “familial autoinflammatory syndromes” [80]. Whether the beneficial effects are due to the inhibition of DC activation is not demonstrated.
3.2. IL-6 and its family
The IL-6 family is composed of IL-6, IL-11, leukaemia inhibitory factor (LIF), oncostatin M (OSM), ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-1) and cardiotrophin-like cytokine (CLC). Members of this family display pro- but also anti-inflammatory effects and play a central role in hemopoiesis as well as in innate and adaptive immune responses.
Activation of IL-6 signalling is mediated through the IL-6/sIL-6R complex, a process known as “trans-signalization” and a unique example of a soluble cytokine receptor displaying agonistic effects. IL-6 is secreted by many cell types, including B and T lymphocytes, monocytes, fibroblasts, keratinocytes, endothelial cells, mesenchymal cells and certain types of tumor cells. IL-6 induces the differentiation of B lymphocytes into plasma cells as well as the proliferation of T lymphocytes, differentiation of cytotoxic T cells and IL-2 production. IL-6 also induces the differentiation of macrophages and megakaryocytes [81].
Recently, IL-6 has been described to participate in the differentiation of a novel T cell subset, Th17, which displays pro-inflammatory functions [82,83]. IL-23 is responsible for the expansion of Th17 previously differentiated, while IL-6 and TGF-β are responsible for the differentiation of Th17 from their naïve precursors. TGF-β induces Foxp3 which leads to the formation of Tregs, while IL-6 inhibits Foxp3 expression induced by TGF-β, and favors the formation of Th17 together with TGF-β [84–86].
IL-6 is likely to be involved in the pathogenesis of inflammatory and autoimmune diseases. It plays an important role in bone biology by inducing the differentiation and activation of osteoclasts and it mediates periarticular destruction of bone and cartilage in experimental models of arthritis [87]. IL-6 levels are increased in the serum of children with Systemic onset Juvenile Arthritis in a disease activity-dependent manner [88], and blocking its receptor is emerging as an effective therapy both in SoJIA [89] as well as in adult RA [90].
3.3. IL-12 and its family
IL-12, a heterodimeric cytokine produced mainly by activated myeloid DCs, plays a pivotal role in the differentiation and expansion of Th1 cells [91–95].The recent discovery of IL-23 has led to a re-evaluation of interleukin-12 biology, as they share a common p40 subunit. In animal models, predisposition to autoimmunity can be explained by abnormal levels of IL-12 secreted by APCs [96]. Furthermore, IL-12 administration has been shown to switch tolerance mediated by intravenous or orally administered antigens into immunity [97]. In addition blocking IL-12 in patients with active Crohn's disease using a specific antibody can induce stable remission [98].
IL-23 is a cytokine that drives autoimmune diseases, including psoriasis and inflammatory bowel diseases [99,100]. IL-23 is secreted by human DCs exposed to gram-negative bacteria [101]. As mentioned above, IL-23 promotes the development and expansion of activated CD4+ T cells that produce IL-17, IL-17F, IL-6 and TNF and are called Th17. Their differentiation is inhibited by IFN-gamma, IL-4 and IL-2 [102]. Genetic analysis of these Th17 cells identified a unique expression pattern of pro-inflammatory cytokines and revealed a unique role in different mouse models of autoimmune inflammation [103]. Given that the levels of IL-23 p19 and IL-17 are elevated in human diseases including multiple sclerosis, psoriasis and Crohn's disease, it is possible that these cytokines mediate human diseases [104–106]. Indeed, the therapeutic efficacy of an IL-12/23 p40 monoclonal antibody in psoriasis has recently been established [107]. The Th17 pathway has also been implicated in multiple sclerosis (MS) [108]. DCs, i.e. monocyte-derived DCs from MS patients, secrete more IL-23 but equivalent amounts of IL-12 compared to healthy controls [104]. Patients with MS also appear to have increased numbers of IL-17-expressing cells [109]. Finally, a subset of infiltrating T cells express IL-17 in RA synovium [110].
The implication of IL-27 in autoimmunity is less clear as this cytokine can have pro- and anti-inflammatory properties [111,112].
3.4. TNF-α
TNF-α was among the first cytokines whose dysregulation was proposed to contribute to the pathogenesis of various autoimmune disorders. More importantly, TNF blockers have been extensively used and validated as an efficacious treatment for RA, Crohn's disease and psoriasis [113,114]. This clearly represents one of the greatest successes of immunology though the mechanisms of action remain unclear. Inasmuch as TNF induces many cell types, including DCs, to secrete pro-inflammatory cytokines, it is likely that TNF blocking results in their decreased secretion. Alternatively, anti-TNF-α therapy might generate a newly differentiated population of Treg cells distinct from natural Tregs, which seem to be defective in RA patients [115].
However, TNF antagonists are not without adverse effects, including reactivation of tuberculosis and induction of reversible systemic autoimmunity like SLE. In fact TNF blockers enhance the production of type I IFNs by pDCs exposed to viruses whereas TNF inhibits it. Type I IFNs, as described below, have been implicated as important mediators of autoimmune diseases in humans. Interestingly, transcription of type I IFN-inducible genes is observed in juvenile arthritis patients treated with TNF blockers. These data suggests that TNF represent an endogenous mechanism to control IFN production by pDCs [116]. Indeed, TNF produced by pDCs in response to viral activation acts as an autocrine maturation factor for these cells. Once they mature, pDCs are unable to secrete type I IFNs. It is therefore conceivable that blocking TNF would keep pDCs at an immature stage where they can continue to produce type I IFNs. Thus, based on these observations, immunity can be viewed as a dynamic system driven by opposite vectors, i.e. TNF-type I IFNs. The sum of the vectors yields an equilibrium point which allows protective immunity when vectors are equal. This dynamic system can accommodate the prevalence of either vector to a certain extent. However, when one of the vectors prevails beyond a certain threshold, the equilibrium point moves into a zone of immunopathology, including arthritis when the TNF vector prevails and SLE and others when type I IFN production prevails (Fig. 3) [16].
Fig. 3.

Cross-regulation of TNF and IFN-α in autoimmune diseases. TNF and IFN-α represent opposite vectors (paths) of immune responses. The sum of the vectors yields an equilibrium point, which allows protective immunity when vectors are equal. When one of the vectors prevails beyond a certain threshold, the equilibrium point moves into a zone of autoimmunity: an excess of IFN-α/β is pathogenic in SLE, Sjogren's, dermatomyositis and early stages of psoriasis while excess of TNF is pathogenic in rheumatoid arthritis, inflammatory bowel disease (IBD), Crohn's disease and psoriasis.
3.5. Type I interferons (IFNs)
Type I IFNs (IFN-α/β), major controllers of viral infections, play a role in several human autoimmune diseases, most particularly SLE. Indeed, SLE is the first autoimmune disorder where alterations in the type I IFN system were reported. In 1979, Notkins and colleagues described the presence of IFN activity in the serum of patients with SLE [117]. More importantly, induction of autoimmunity, including appearance of anti-nuclear antibodies and occasionally clinical symptoms of SLE, were reported during repeated administration of recombinant IFN-α to patients with various malignancies or chronic viral infection [118]. Recent studies have identified an “IFN signature” in the majority of patients with active SLE [119-121]. IFN-α in SLE patients is mainly secreted by pDCs and understanding what drives its unabated secretion in SLE patients remains an area of intense investigation. Type I IFNs can contribute to the breaking of tolerance through different mechanisms including direct effect on APCs, T cells and B cells.
3.5.1. IFN-α and DC alteration
We have shown that SLE blood constitutes a DC-inducing environment, as it promotes the differentiation of healthy monocytes into mDCs. The DC-inducing property of SLE sera is mainly mediated through IFN-α [122]. Indeed, blood SLE monocytes display DC-like functions as they capture antigens and autoantigens and present them to CD4+ and CD8+ T cells. Thus, type I IFN-induced unabated DC activation could promote the expansion of autoreactive T cells. SLE DCs are characterized by their unique in vitro ability to promote the differentiation of CD8+ T lymphocytes in CTLs able to generate nucleosomes and granzyme B-dependent autoantigens. Interestingly, terminally differentiated effector CD8+ T lymphocytes (CCR7− , CD45RA+) are expanded in the blood of SLE patients and this expansion correlates with disease activity as assessed by the SLE Disease Activity Index (SLEDAI) [123]. These cells can induce direct tissue damage as they represent the main cell subset infiltrating the kidney in lupus nephritis, where they adopt a periglomerular localization. A direct correlation is found between the lupus nephritis activity score and the number of periglomerular infiltrating CD8+ T lymphocytes. No correlation was found, however, with the chronicity score. In a similar way, autoreactive CD8+ T lymphocytes directed against myelin epitopes are expanded in patients with CNS lupus involvement. None of those autoreactive cells were found in a control population without neurologic involvement or in SLE patients in whom thrombosis was responsible for the neurologic symptoms. IFN also appears to be associated to other autoimmune diseases including myositis [124,125], Sjogren's syndrome [126,127] and the initial phase of psoriasis [128].
3.5.2. IFN-α and B cell activation
The key role of B lymphocytes in SLE has been known for a long time and it has recently been reinforced by the observation that treatment of patients with the B cell depleting CD20 antibody leads to disease improvement [129]. Through their direct effect on B cells, type I IFNs enhance primary antibody responses to soluble proteins and induce the production of all subclasses of IgG in mice [130]. IFN-α up-regulates CD38, a germinal center B cell and plasma cell marker, on B lymphocytes and BAFF (B cell activating factor) on monocytes and mDCs. BAFF in turn contributes to the survival of autoreactive B lymphocytes [131]. In addition, IFN-α promotes the differentiation of activated B lymphocytes into plasmablasts. pDCs activated with viruses secrete IFN-α and IL-6, which permits plasmablasts to become antibody-secreting plasma cells [132]. The same effect is observed when pDCs are activated with SLE immune complexes containing nucleic acids that bind TLRs [133,134]. This could contribute to amplify the production of type I IFNs and subsequently the differentiation of autoreactive plasma cells that would further secrete autoantibodies, thus perpetuating this pathogenic loop.
4. Toll-like receptor (TLR) ligands and autoimmunity
Infections frequently precede the occurrence of either organ-specific or systemic autoimmune diseases. Molecular mimicry, however, cannot account for all the autoimmune responses that have been linked to infectious diseases. TLRs are key components of the innate immune system. These receptors activate multiple pathways of inflammation that eventually permit to eradicate invading pathogens [135]. Microbial-derived TLR ligands include a wide range of molecules with strong adjuvant activity that can activate DCs, macrophages and other APCs [136]. TLRs are involved in the pathogenesis of autoimmune disorders [14], and endogenous ligands also activate these receptors [137–139]. Exposure to TLR3 or TLR7 ligands is required, for example, to induce autoimmune diabetes in transgenic mice that harbour large numbers of pancreatic islet-reactive cytotoxic T cells. In this model, TLR-induced local production of IFN-α triggers the recruitment of autoreactive T cells into the pancreatic islets [140]. TLR3 and TLR9 ligation are also key events in the development of autoimmune myocarditis by inducing the maturation of DCs pulsed with heart-specific self-peptide [14]. In humans, TLR activation has been reported as a pathogenic event mainly in the context of systemic autoimmune diseases such as SLE. Early studies demonstrated that immune complexes were potent stimuli for IFN-α secretion by pDCs in an Fc receptor (CD32)-dependent manner [141,142]. Indeed, chromatin and/or ribonucleoprotein-containing immune complexes are internalized by pDCs via FcgRIIa, reach the endosomal compartment and activate IFN-α secretion through TLR9 and/or 7-dependent pathways [143,144]. Sera from SLE patients can also induce IFN-α secretion in a TLR7/8-dependent manner [133]. Accordingly, there is a correlation between the presence of an IFN gene signature in blood leukocytes and the detection of autoantibodies directed against ribonucleoproteins in the sera of SLE patients [145]. Since INF-α induces the transcription of TLR7 itself, a self-amplifying loop could take place at this stage as well, thus explaining the above-described correlation.
The contribution of immune complexes and TLR signalling to the generation of autoantibodies characteristic of SLE has been the purpose of several studies in murine SLE models. Chromatin-containing IC activate transgenic autoreactive B cells via sequential engagement of the B cell antigen receptor (BCR) and TLR9. In vivo, TLR9 contributes to the development of anti-dsDNA antibodies, as lupus-prone (Fas-deficient) mice that lack TLR9 on the mixed MRL-B6−129 background fail to generate these antibodies [146]. Unexpectedly, in another mouse model (MRL/lpr), TLR9 deficiency leads to an increased production of autoantibodies and a more severe lupus-like disease [147]. Thus, depending on the genetic background, TLR9 seems to deliver a pathogenic or a protective signal. TLR7 signalling contributes in turn to induce an SLE-like syndrome in most of the murine models so far studied. Indeed, as shown for pDCs, immune complexes that contain TLR7 ligands (RNA and RNA-associated autoantigens) activate autoreactive B cells in vitro [148]. In vivo, FcRIIb−/− mice develop enhanced autoimmunity when crossed to the Y-linked autoimmune accelerator (Yaa) locus which harbours a duplication in the TLR7 gene [149]. Thus, naturally occurring differences in expression of the TLR7 gene as well as environmental factors that induce TLR7 expression (CD40-L and/or IFN-α) could contribute to SLE pathogenesis.
5. High mobility group box 1 protein and SLE
High mobility group box 1 protein, an abundant nuclear protein displaying potent pro-inflammatory effects when released extracellularly, can mediate the activation of TLR9 by DNA-containing immune complexes through a mechanism involving the immunoglobulin superfamily member RAGE, which is the best-characterized receptor for HMGB1. Necrosis or tissue injury causes HMGB1 to be released from cells; it then binds to DNA-containing immune complexes in serum and then the resultant complexes regulate the expansion of autoreactive B cells and the production of IFNα by pDCs. RAGE is involved in the recognition of HMGB1- and DNA-containing immune complexes [150], and the DNA-dependent association of TLR9 and RAGE, along with the endosomal localization of RAGE, raise the possibility that RAGE determines the subcellular localization and/or retention of DNA-TLR9 complexes in the endosome (Fig. 4). These effects may be important in perpetuating inflammatory amplification loops in SLE.
Fig. 4.
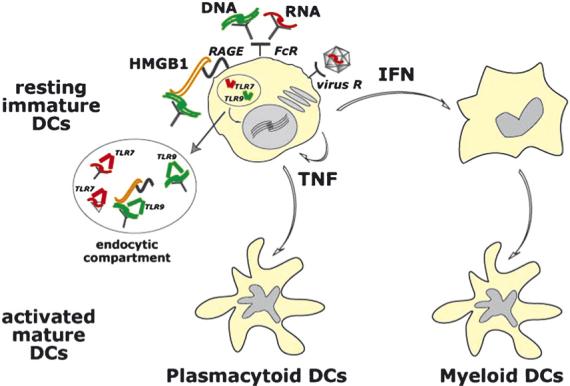
Nucleic acids regulate activation of DC subsets. Interaction of resting immature pDCs with nucleic acids leads to a cascade of cytokine secretion including high amounts of type I IFN as well as TNF. This leads to generation of activated (mature) DCs derived from pDCs, via autocrine TNF, and from immature mDCs, via paracrine type I IFN, both of which drive T and B cell responses. Nucleic acids, abundant in autoimmune diseases such as SLE, can be presented to pDCs via several pathways such as i) immune complexes containing double stranded DNA (green) or single stranded RNA (red) which are taken up via Fc receptors; ii) viruses (for example RNA viruses) taken up via surface receptors; and iii) complexes of nuclear protein HMGB1 bound to DNA-containing immune complexes which are taken up by pDCs via the interaction of HMGB1 with surface protein of the immunoglobulin superfamily RAGE. Ultimately, captured nucleic acids are targeted to endocytic compartments where they bind TLR7 (RNA) or TLR9 (DNA) resulting in activation of signalling pathways that trigger transcription of inflammatory cytokines.
6. Conclusions
Much progress has occurred in the understanding of the biological basis of autoimmunity in the past decade leading to identification of cytokines as the major regulators of immune homeostasis and the balance between tolerance and immunity. This permitted generation of new treatments with TNF and IL-1 antagonists on top (Table 1). TNF blockade is clinically useful in several autoimmune diseases. Blocking IL-1 effectively treats patients with juvenile arthritis, familial periodic fever syndromes and type II diabetes. New cytokine targets are being identified including: IL-12/23 blockade in Crohn's disease, multiple sclerosis and psoriasis and type I IFNs in SLE, Sjogren's syndrome, autoimmune myositis and perhaps early stages of psoriasis (Table 1).
Table 1.
Cytokine targets in human inflammatory and autoimmune diseases
| Cytokine | Disease | Therapeutic effect | References |
|---|---|---|---|
| IL-1 | NOMID | Yes | [151] |
| FCA | Yes | [152] | |
| Muckle-Wells | Yes | [153] | |
| SoJIA | Yes | [77] | |
| Gout | Yes | [78] | |
| Type II diabetes | [79] | ||
| IL-6 | SoJIA | Yes | [89] |
| RA | Yes | [90] | |
| IL-12/23 | Crohn's | Yes | [98] |
| MS | Yes | [154] | |
| Psoriasis | Yes | [107,155] | |
| IL-17 | RA | Not tested | |
| MS | Not tested | ||
| TNF-α | RA | Yes | [113] |
| JIA | Yes | [156] | |
| Crohn's | Yes | [157] | |
| Psoriasis | Yes | [114] | |
| IFN-α | SLE | Not tested | |
| Myositis | Not tested | ||
| Psoriasis | Not tested | ||
| Sjogren's | Not tested |
Yet, the causative links between cytokine imbalance and alterations in DC homeostasis remain to be determined for many of the cytokines identified. Likewise, the precise mechanisms of action of cytokine blockade remain unclear. Are we targeting a cytokine that represents a product of activated DCs and which triggers the cascade of inflammation by acting on other cells? Or are we targeting a cytokine that drives uncontrolled DC activation leading to break in tolerance? These scientific challenges will keep us busy for a while!
Acknowledgments
Supported by Baylor Health Care System Foundation, the Alliance for Lupus Research (VP), the Dana Foundation, Defense Advanced Research Planning Agency (JB), The National Institutes of Health (U19 AIO57234-02, P01 CA084512, R01 CA078846 to JB, R0-1 AR050770-01 and P50 ARO54083 to VP). JB holds the W.W. Caruth, Jr. Chair in Organ Transplantation Immunology. AKP holds the Michael A. Ramsay Chair for Cancer Immunology Research. We thank Dr. Michael Ramsay and Dr. William Duncan for their continuous support.
Biography
 Jacques Banchereau, PhD is the director of Baylor Institute for Immunology Research in Dallas and holds the W.W. Caruth, Jr. Chair in Organ Transplantation Immunology. He received his PhD in biochemistry from the University of Paris in 1980 and later served as director of the Schering Plough Laboratory for Immunological Research near Lyon, France, where he was among the first to discover how to grow human dendritic cells. Dr. Banchereau came to Baylor in 1996 to develop the Baylor Institute for Immunology Research. He is an adjunct professor of Microbiology and a member of the Cancer Immunobiology Center at The University of Texas Southwestern Medical Center. Dr. Banchereau also holds an adjunct professorship in biomedical studies at Baylor University Medical Center in Waco, TX. He has served on the National Institutes of Health's Experimental Immunology Study Section, Center for Scientific Review, in the area of Experimental Immunology. He has published more than 260 papers and 160 book chapters and reviews in major international journals. His research interests center around various areas of immunology and cancer including dendritic cells, novel cytokines and antibody-producing B lymphocytes.
Jacques Banchereau, PhD is the director of Baylor Institute for Immunology Research in Dallas and holds the W.W. Caruth, Jr. Chair in Organ Transplantation Immunology. He received his PhD in biochemistry from the University of Paris in 1980 and later served as director of the Schering Plough Laboratory for Immunological Research near Lyon, France, where he was among the first to discover how to grow human dendritic cells. Dr. Banchereau came to Baylor in 1996 to develop the Baylor Institute for Immunology Research. He is an adjunct professor of Microbiology and a member of the Cancer Immunobiology Center at The University of Texas Southwestern Medical Center. Dr. Banchereau also holds an adjunct professorship in biomedical studies at Baylor University Medical Center in Waco, TX. He has served on the National Institutes of Health's Experimental Immunology Study Section, Center for Scientific Review, in the area of Experimental Immunology. He has published more than 260 papers and 160 book chapters and reviews in major international journals. His research interests center around various areas of immunology and cancer including dendritic cells, novel cytokines and antibody-producing B lymphocytes.
 Patrick Blanco, MD, PhD is currently an assistant professor in the Department of Immunology at the University of Bordeaux 2 and at the University Hospital of Bordeaux. He obtained his degree in medicine (1998) and in internal medicine (2001) at the University of Bordeaux “Victor Segalen”. He has worked in the laboratory of Professor Jacques Banchereau as a postdoctoral Fellow from 1999 to 2001 (Baylor Institute for Immunology Research, Dallas, TX, USA). His main field of interest is the study of the pathogenesis and treatment of autoimmune diseases. In particular, he was among the first to delineate the implication of dendritic cells and CD8+ T lymphocyte in the generation of the autoimmune response and tissue lesions in systemic lupus erythematosus.
Patrick Blanco, MD, PhD is currently an assistant professor in the Department of Immunology at the University of Bordeaux 2 and at the University Hospital of Bordeaux. He obtained his degree in medicine (1998) and in internal medicine (2001) at the University of Bordeaux “Victor Segalen”. He has worked in the laboratory of Professor Jacques Banchereau as a postdoctoral Fellow from 1999 to 2001 (Baylor Institute for Immunology Research, Dallas, TX, USA). His main field of interest is the study of the pathogenesis and treatment of autoimmune diseases. In particular, he was among the first to delineate the implication of dendritic cells and CD8+ T lymphocyte in the generation of the autoimmune response and tissue lesions in systemic lupus erythematosus.
 Karolina Palucka, MD, PhD earned her MD in 1988 from Warsaw Medical Academy in Poland. She went on to complete her PhD in hematology and immunology in 1993 at the Karolinska Institute in Stockholm, Sweden. Dr. Palucka is an investigator at the Baylor Institute for Immunology Research in Dallas, where she began in 1998 as a senior research associate. She holds the Michael A.E. Ramsay Chair for Cancer Immunology Research. She also oversees the Flow Cytometry Core and the GMP Cell Core. In August 2005, she was appointed to an adjunct professorship in biomedical studies at Baylor, Waco. Dr. Palucka and her team focus on understanding how the human immune system works and how it may be manipulated to fight cancer. She also leads a project to develop a mouse model of the human immune system, which is being used to study human tumors and how they influence dendritic cell function. These ‘humanized’ mice are also being used to develop improved vaccine strategies.
Karolina Palucka, MD, PhD earned her MD in 1988 from Warsaw Medical Academy in Poland. She went on to complete her PhD in hematology and immunology in 1993 at the Karolinska Institute in Stockholm, Sweden. Dr. Palucka is an investigator at the Baylor Institute for Immunology Research in Dallas, where she began in 1998 as a senior research associate. She holds the Michael A.E. Ramsay Chair for Cancer Immunology Research. She also oversees the Flow Cytometry Core and the GMP Cell Core. In August 2005, she was appointed to an adjunct professorship in biomedical studies at Baylor, Waco. Dr. Palucka and her team focus on understanding how the human immune system works and how it may be manipulated to fight cancer. She also leads a project to develop a mouse model of the human immune system, which is being used to study human tumors and how they influence dendritic cell function. These ‘humanized’ mice are also being used to develop improved vaccine strategies.
 Virginia Pascual, MD received her MD from Facultad de Medicina, Universidad Complutense in Madrid. Dr. Pascual joined the faculty at Baylor Institute for Immunology Research in 1999 as an Investigator. She has been an adjunct professor of biomedical studies at Baylor, Waco since August 2005. In her clinical practice, she specializes in pediatric rheumatology and her research focuses on understanding autoimmune diseases in children. Dr. Pascual's group discovered that interleukin-1 is a major cause of the joint inflammation in a type of juvenile arthritis and that blocking this cytokine alleviated symptoms. They have also elucidated the role of interferon-α in systemic lupus erythematosus (SLE). She is the principal investigator on an NIH P50 Center of Research Translation award that established a Center for Lupus Research at BIIR.
Virginia Pascual, MD received her MD from Facultad de Medicina, Universidad Complutense in Madrid. Dr. Pascual joined the faculty at Baylor Institute for Immunology Research in 1999 as an Investigator. She has been an adjunct professor of biomedical studies at Baylor, Waco since August 2005. In her clinical practice, she specializes in pediatric rheumatology and her research focuses on understanding autoimmune diseases in children. Dr. Pascual's group discovered that interleukin-1 is a major cause of the joint inflammation in a type of juvenile arthritis and that blocking this cytokine alleviated symptoms. They have also elucidated the role of interferon-α in systemic lupus erythematosus (SLE). She is the principal investigator on an NIH P50 Center of Research Translation award that established a Center for Lupus Research at BIIR.
References
- 1.Germain RN. An innately interesting decade of research in immunology. Nat Med. 2004;10:1307–20. doi: 10.1038/nm1159. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 3.Sakaguchi S. Naturally arising Foxp3-expressing CD25 + CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–52. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 4.Shevach EM. Regulatory/suppressor T cells in health and disease. Arthritis Rheum. 2004;50:2721–4. doi: 10.1002/art.20500. [DOI] [PubMed] [Google Scholar]
- 5.Tang Q, Bluestone JA. Regulatory T-cell physiology and application to treat autoimmunity. Immunol Rev. 2006;212:217–37. doi: 10.1111/j.0105-2896.2006.00421.x. [DOI] [PubMed] [Google Scholar]
- 6.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 7.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 8.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–67. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 9.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 10.Mellman I. Antigen processing and presentation by dendritic cells: cell biological mechanisms. Adv Exp Med Biol. 2005;560:63–7. doi: 10.1007/0-387-24180-9_9. [DOI] [PubMed] [Google Scholar]
- 11.Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 12.Ueno H, Klechevsky E, Morita R, Aspord C, Cao T, Matsui T, et al. Dendritic cell subsets in health and disease. Immunol Rev. 2007;219:118–42. doi: 10.1111/j.1600-065X.2007.00551.x. [DOI] [PubMed] [Google Scholar]
- 13.Dittel BN, Visintin I, Merchant RM, Janeway CA., Jr Presentation of the self antigen myelin basic protein by dendritic cells leads to experimental autoimmune encephalomyelitis. J Immunol. 1999;163:32–9. [PubMed] [Google Scholar]
- 14.Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med. 2003;9:1484–90. doi: 10.1038/nm960. [DOI] [PubMed] [Google Scholar]
- 15.Bondanza A, Zimmermann VS, Dell'Antonio G, Dal Cin E, Capobianco A, Sabbadini MG, et al. Cutting edge: dissociation between autoimmune response and clinical disease after vaccination with dendritic cells. J Immunol. 2003;170:24–7. doi: 10.4049/jimmunol.170.1.24. [DOI] [PubMed] [Google Scholar]
- 16.Banchereau J, Pascual V, Palucka AK. Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 2004;20:539–50. doi: 10.1016/s1074-7613(04)00108-6. [DOI] [PubMed] [Google Scholar]
- 17.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–92. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Janeway CA, Jr, Yagi J, Conrad PJ, Katz ME, Jones B, Vroegop S, et al. T-cell responses to Mls and to bacterial proteins that mimic its behavior. Immunol Rev. 1989;107:61–88. doi: 10.1111/j.1600-065x.1989.tb00003.x. [DOI] [PubMed] [Google Scholar]
- 19.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 20.Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 21.Figdor CG, van Kooyk Y, Adema GJ. C-type lectin receptors on dendritic cells and Langerhans cells. Nat Rev Immunol. 2002;2:77–84. doi: 10.1038/nri723. [DOI] [PubMed] [Google Scholar]
- 22.Geijtenbeek TB, van Vliet SJ, Engering A, 't Hart BA, van Kooyk Y. Self- and nonself-recognition by C-type lectins on dendritic cells. Annu Rev Immunol. 2004;22:33–54. doi: 10.1146/annurev.immunol.22.012703.104558. [DOI] [PubMed] [Google Scholar]
- 23.Martinon F, Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 2005;26:447–54. doi: 10.1016/j.it.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Ting JP, Davis BK. CATERPILLER: a novel gene family important in immunity, cell death, and diseases. Annu Rev Immunol. 2005;23:387–414. doi: 10.1146/annurev.immunol.23.021704.115616. [DOI] [PubMed] [Google Scholar]
- 25.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–55. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 26.Sauter B, Albert ML, Francisco L, Larsson M, Somersan S, Bhardwaj N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–34. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–78. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 28.Srivastava PK, Maki RG. Stress-induced proteins in immune response to cancer. Curr Top Microbiol Immunol. 1991;167:109–23. doi: 10.1007/978-3-642-75875-1_7. [DOI] [PubMed] [Google Scholar]
- 29.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–42. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 30.Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–9. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- 31.Rock KL, Hearn A, Chen CJ, Shi Y. Natural endogenous adjuvants. Springer Semin Immunopathol. 2005;26:231–46. doi: 10.1007/s00281-004-0173-3. [DOI] [PubMed] [Google Scholar]
- 32.Piqueras B, Connolly J, Freitas H, Palucka AK, Banchereau J. Upon viral exposure, myeloid and plasmacytoid dendritic cells produce 3 waves of distinct chemokines to recruit immune effectors. Blood. 2006;107:2613–8. doi: 10.1182/blood-2005-07-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flynn S, Toellner KM, Raykundalia C, Goodall M, Lane P. CD4 T cell cytokine differentiation: the B cell activation molecule, OX40 ligand, instructs CD4 T cells to express interleukin 4 and upregulates expression of the chemokine receptor, Blr-1. J Exp Med. 1998;188:297–304. doi: 10.1084/jem.188.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ito T, Yang M, Wang YH, Lande R, Gregorio J, Perng OA, et al. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J Exp Med. 2007;204:105–15. doi: 10.1084/jem.20061660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med. 1997;185:1101–11. doi: 10.1084/jem.185.6.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–7. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 37.Shortman K, Liu Y-J. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–61. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 38.Pulendran B, Smith JL, Jenkins M, Schoenborn M, Maraskovsky E, Maliszewski CR. Prevention of peripheral tolerance by a dendritic cell growth factor: Flt3 ligand as an adjuvant. J Exp Med. 1998;188:2075–82. doi: 10.1084/jem.188.11.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz V, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–11. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 40.Soares H, Waechter H, Glaichenhaus N, Mougneau E, Yagita H, Mizenina O, et al. DEC-205/CD205+ dendritic cells induce CD4+ T cells to produce IFN-γ by a CD70 dependent, IL-12 independent mechanism. J Exp Med. 2007;204:1095–106. doi: 10.1084/jem.20070176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valladeau J, Saeland S. Cutaneous dendritic cells. Semin Immunol. 2005;17:273–83. doi: 10.1016/j.smim.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 42.Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–9. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur J Immunol. 2001;31:3388–93. doi: 10.1002/1521-4141(200111)31:11<3388::aid-immu3388>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 44.van der Aar AM, Sylva-Steenland RM, Bos JD, Kapsenberg ML, de Jong EC, Teunissen MB. Loss of TLR2, TLR4, and TLR5 on Langerhans cells abolishes bacterial recognition. J Immunol. 2007;178:1986–90. doi: 10.4049/jimmunol.178.4.1986. [DOI] [PubMed] [Google Scholar]
- 45.Dzionek A, Sohma Y, Nagafune J, Cella M, Colonna M, Facchetti F, et al. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J Exp Med. 2001;194:1823–34. doi: 10.1084/jem.194.12.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Valladeau J, Ravel O, Dezutter-Dambuyant C, Moore K, Kleijmeer M, Liu Y, et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 2000;12:71–81. doi: 10.1016/s1074-7613(00)80160-0. [DOI] [PubMed] [Google Scholar]
- 47.Geijtenbeek TB, Torensma R, van Vliet SJ, van Duijnhoven GC, Adema GJ, van Kooyk Y, et al. Identification of DC-SIGN, a novel dendritic cell-specific ICAM-3 receptor that supports primary immune responses. Cell. 2000;100:575–85. doi: 10.1016/s0092-8674(00)80693-5. [DOI] [PubMed] [Google Scholar]
- 48.Goldrath AW, Hedrick SM. Central tolerance matters. Immunity. 2005;23:113–4. doi: 10.1016/j.immuni.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe N, Wang YH, Lee HK, Ito T, Cao W, Liu YJ. Hassall's corpuscles instruct dendritic cells to induce CD4+ CD25+ regulatory T cells in human thymus. Nature. 2005;436:1181–5. doi: 10.1038/nature03886. [DOI] [PubMed] [Google Scholar]
- 50.Hengartner H, Odermatt B, Schneider R, Schreyer M, Walle G, MacDonald HR, et al. Delection of self-reative T cells before entry into the thymus medulla. Nature. 1988;336:388–90. doi: 10.1038/336388a0. [DOI] [PubMed] [Google Scholar]
- 51.Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci USA. 2002;99:351–8. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rovere P, Sabbadini MG, Fazzini F, Bondanza A, Zimmermann VS, Rugarli C, et al. Remnants of suicidal cells fostering systemic autoaggression. Apoptosis in the origin and maintenance of autoimmunity. Arthritis Rheum. 2000;43:1663–72. doi: 10.1002/1529-0131(200008)43:8<1663::AID-ANR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 53.de Heer HJ, Hammad H, Soullie T, Hijdra D, Vos N, Willart MA, et al. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med. 2004;200:89–98. doi: 10.1084/jem.20040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat Immunol. 2006;7:652–62. doi: 10.1038/ni1333. [DOI] [PubMed] [Google Scholar]
- 55.Merad M, Collin M, Bromberg J. Dendritic cell homeostasis and trafficking in transplantation. Trends Immunol. 2007;28:353–9. doi: 10.1016/j.it.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Gilliet M, Liu YJ. Generation of human CD8 T regulatory cells by CD40 ligand-activated plasmacytoid dendritic cells. J Exp Med. 2002;195:695–704. doi: 10.1084/jem.20011603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Menges M, Rossner S, Voigtlander C, Schindler H, Kukutsch NA, Bogdan C, et al. Repetitive injections of dendritic cells matured with tumor necrosis factor alpha induce antigen-specific protection of mice from autoimmunity. J Exp Med. 2002;195:15–21. doi: 10.1084/jem.20011341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verginis P, Li HS, Carayanniotis G. Tolerogenic semimature dendritic cells suppress experimental autoimmune thyroiditis by activation of thyroglobulin-specific CD4+ CD25+ T cells. J Immunol. 2005;174:7433–9. doi: 10.4049/jimmunol.174.11.7433. [DOI] [PubMed] [Google Scholar]
- 59.Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, et al. Dendritic cell-expanded, islet-specific CD4+ CD25+ CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J Exp Med. 2007;204:191–201. doi: 10.1084/jem.20061631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moser M. Dendritic cells in immunity and tolerance-do they display opposite functions? Immunity. 2003;19:5–8. doi: 10.1016/s1074-7613(03)00182-1. [DOI] [PubMed] [Google Scholar]
- 61.Battaglia M, Gianfrani C, Gregori S, Roncarolo MG. IL-10-producing T regulatory type 1 cells and oral tolerance. Ann NY Acad Sci. 2004;1029:142–53. doi: 10.1196/annals.1309.031. [DOI] [PubMed] [Google Scholar]
- 62.McGuirk P, McCann C, Mills KH. Pathogen-specific T regulatory 1 cells induced in the respiratory tract by a bacterial molecule that stimulates interleukin 10 production by dendritic cells: a novel strategy for evasion of protective T helper type 1 responses by Bordetella pertussis. J Exp Med. 2002;195:221–31. doi: 10.1084/jem.20011288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manavalan JS, Rossi PC, Vlad G, Piazza F, Yarilina A, Cortesini R, et al. High expression of ILT-3 and ILT-4 is a general feature of tolerogenic dendritic cells. Transpl Immunol. 2003;11:245–58. doi: 10.1016/S0966-3274(03)00058-3. [DOI] [PubMed] [Google Scholar]
- 64.Suciu-Foca N, Manavalan JS, Scotto L, Kim-Schulze S, Galluzzo S, Naiyer AJ, et al. Molecular characterization of allospecific T suppressor and tolerogenic dendritic cells: review. Int Immunopharmacol. 2005;5:7–11. doi: 10.1016/j.intimp.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 65.Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med. 2004;199:1467–77. doi: 10.1084/jem.20040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–64. [PubMed] [Google Scholar]
- 67.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–7. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 68.Fujita S, Seino K, Sato K, Sato Y, Eizumi K, Yamashita N, et al. Regulatory dendritic cells act as regulators of acute lethal systemic inflammatory response. Blood. 2006;107:3656–64. doi: 10.1182/blood-2005-10-4190. [DOI] [PubMed] [Google Scholar]
- 69.Sato K, Yamashita N, Yamashita N, Baba M, Matsuyama T. Regulatory dendritic cells protect mice from murine acute graft-versus-host disease and leukemia relapse. Immunity. 2003;18:367–79. doi: 10.1016/s1074-7613(03)00055-4. [DOI] [PubMed] [Google Scholar]
- 70.Bartz H, Türkel O, Hoffjan S, Rothoeft T, Gonschorek A, Schauer U. Respiratory syncytial virus decreases the capacity of myeloid dendritic cells to induce interferon-gamma in näive T cells. Immunology. 2003;109:49–57. doi: 10.1046/j.1365-2567.2003.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–35. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 72.Smith DE, Renshaw BR, Ketchem RR, Kubin M, Garka KE, Sims JE. Four new members expand the interleukin-1 superfamily. J Biol Chem. 2000;275:1169–75. doi: 10.1074/jbc.275.2.1169. [DOI] [PubMed] [Google Scholar]
- 73.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 74.Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87:2095–147. [PubMed] [Google Scholar]
- 75.Dinarello CA. Blocking IL-1 in systemic inflammation. J Exp Med. 2005;201:1355–9. doi: 10.1084/jem.20050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Steinman RM. Cytokines amplify the function of accessory cells. Immunol Lett. 1988;17:197–202. doi: 10.1016/0165-2478(88)90028-4. [DOI] [PubMed] [Google Scholar]
- 77.Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–86. doi: 10.1084/jem.20050473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9:R28. doi: 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–26. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 80.Ting JP, Kastner DL, Hoffman HM. CATERPILLERs, pyrin and hereditary immunological disorders. Nat Rev Immunol. 2006;6:183–95. doi: 10.1038/nri1788. [DOI] [PubMed] [Google Scholar]
- 81.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–54. [PubMed] [Google Scholar]
- 82.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 83.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 85.Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 86.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 87.Kudo O, Sabokbar A, Pocock A, Itonaga I, Fujikawa Y, Athanasou NA. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone. 2003;32:1–7. doi: 10.1016/s8756-3282(02)00915-8. [DOI] [PubMed] [Google Scholar]
- 88.Keul R, Heinrich PC, Muller-newen G, Muller K, Woo P. A possible role for soluble IL-6 receptor in the pathogenesis of systemic onset juvenile chronic arthritis. Cytokine. 1998;10:729–34. doi: 10.1006/cyto.1997.0343. [DOI] [PubMed] [Google Scholar]
- 89.Woo P, Wilkinson N, Prieur AM, Southwood T, Leone V, Livermore P, et al. Open label phase II trial of single, ascending doses of MRA in Caucasian children with severe systemic juvenile idiopathic arthritis: proof of principle of the efficacy of IL-6 receptor blockade in this type of arthritis and demonstration of prolonged clinical improvement. Arthritis Res Ther. 2005;7:R1281–8. doi: 10.1186/ar1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maini RN, Taylor PC, Szechinski J, Pavelka K, Broll J, Balint G, et al. Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate. Arthritis Rheum. 2006;54:2817–29. doi: 10.1002/art.22033. [DOI] [PubMed] [Google Scholar]
- 91.Hsieh C-S, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphys KM. Development of Th1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–9. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 92.Manetti R, Parronchi P, Giudizi MG, Piccinni M-P, Maggi E, Trinchieri G, et al. Natural killer cell stimulatory factor [interleukin 12 [IL-12]] induces T helper type 1 [Th1]-specific immune responses and inhibits the development of IL-4-producing Th cells. J Exp Med. 1993;177:1199–204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 94.Uhlig HH, McKenzie BS, Hue S, Thompson C, Joyce-Shaikh B, Stepankova R, et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity. 2006;25:309–18. doi: 10.1016/j.immuni.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 95.Kastelein RA, Hunter CA, Cua DJ. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu Rev Immunol. 2007;25:221–42. doi: 10.1146/annurev.immunol.22.012703.104758. [DOI] [PubMed] [Google Scholar]
- 96.Trembleau S, Penna G, Bosi E, Mortara A, Gately MK, Adorini L. Interleukin 12 administration induces T helper type 1 cells and accelerates autoimmune diabetes in NOD mice. J Exp Med. 1995;181:817–21. doi: 10.1084/jem.181.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Segal BM, Shevach EM. IL-12 unmasks latent autoimmune disease in resistant mice. J Exp Med. 1996;184:771–5. doi: 10.1084/jem.184.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, et al. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. 2004;351:2069–79. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 99.Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL-12B and leads to the identification of IL-23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273–90. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–3. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Smits HH, van Beelen AJ, Hessle C, Westland R, de Jong E, Soeteman E, et al. Commensal Gram-negative bacteria prime human dendritic cells for enhanced IL-23 and IL-27 expression and enhanced Th1 development. Eur J Immunol. 2004;34:1371–80. doi: 10.1002/eji.200324815. [DOI] [PubMed] [Google Scholar]
- 102.Reiner SL. Development in motion: helper T cells at work. Cell. 2007;129:33–6. doi: 10.1016/j.cell.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 103.Kikly K, Liu L, Na S, Sedgwick JD. The IL-23/Th(17) axis: therapeutic targets for autoimmune inflammation. Curr Opin Immunol. 2006;18:670–5. doi: 10.1016/j.coi.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 104.Vaknin-Dembinsky A, Balashov K, Weiner HL. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol. 2006;176:7768–74. doi: 10.4049/jimmunol.176.12.7768. [DOI] [PubMed] [Google Scholar]
- 105.Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125–30. doi: 10.1084/jem.20030451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schmidt C, Giese T, Ludwig B, Mueller-Molaian I, Marth T, Zeuzem S, et al. Expression of interleukin-12-related cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19 and interleukin-27p28 in Crohn's disease but not in ulcerative colitis. Inflamm Bowel Dis. 2005;11:16–23. doi: 10.1097/00054725-200501000-00003. [DOI] [PubMed] [Google Scholar]
- 107.Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, et al. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med. 2007;356:580–92. doi: 10.1056/NEJMoa062382. [DOI] [PubMed] [Google Scholar]
- 108.Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–8. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 109.Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, et al. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–4. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 110.Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, et al. Human interleukin-17: a T cell-derived pro-inflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999;42:963–70. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 111.Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–36. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 112.Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–45. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 113.Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–96. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- 114.Victor FC, Gottlieb AB, Menter A. Changing paradigms in dermatology: tumor necrosis factor alpha (TNF-alpha) blockade in psoriasis and psoriatic arthritis. Clin Dermatol. 2003;21:392–7. doi: 10.1016/j.clindermatol.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 115.Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNF-α therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-β. J Exp Med. 2007;204:33–9. doi: 10.1084/jem.20061531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Palucka AK, Blanck JP, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFN-α in autoimmune diseases. Proc Natl Acad Sci USA. 2005;102:3372–7. doi: 10.1073/pnas.0408506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301:5–8. doi: 10.1056/NEJM197907053010102. [DOI] [PubMed] [Google Scholar]
- 118.Ronnblom LE, Alm GV, Oberg KE. Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Ann Int Med. 1991;115:178–83. doi: 10.7326/0003-4819-115-3-178. [DOI] [PubMed] [Google Scholar]
- 119.Bennet L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, et al. Interferon and granulopoieis signatures in Systemic Lupus Erythematosus blood. J Exp Med. 2003;197:711–23. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003;100:2610–5. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Crow MK, Wohlgemuth J. Microarray analysis of gene expression in lupus. Arthritis Res Ther. 2003;5:279–87. doi: 10.1186/ar1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–3. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 123.Blanco P, Pitard V, Viallard JF, Taupin JL, Pellegrin JL, Moreau JF. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52:201–11. doi: 10.1002/art.20745. [DOI] [PubMed] [Google Scholar]
- 124.Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664–78. doi: 10.1002/ana.20464. [DOI] [PubMed] [Google Scholar]
- 125.Tezak Z, Hoffman EP, Lutz JL, Fedczyna TO, Stephan D, Bremer EG, et al. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J Immunol. 2002;168:4154–63. doi: 10.4049/jimmunol.168.8.4154. [DOI] [PubMed] [Google Scholar]
- 126.Bave U, Nordmark G, Lovgren T, Ronnelid J, Cajander S, Eloranta ML, et al. Activation of the type I interferon system in primary Sjogren's syndrome: a possible etiopathogenic mechanism. Arthritis Rheum. 2005;52:1185–95. doi: 10.1002/art.20998. [DOI] [PubMed] [Google Scholar]
- 127.Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren's syndrome. Proc Natl Acad Sci USA. 2006;103:2770–5. doi: 10.1073/pnas.0510837103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-α production. J Exp Med. 2005;202:135–43. doi: 10.1084/jem.20050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Looney RJ, Anolik J, Sanz I. New therapies for systemic lupus erythematosus: cellular targets. Rheum Dis Clin North Am. 2006;32:201–15. doi: 10.1016/j.rdc.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 130.Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, et al. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol. 2006;176:2074–8. doi: 10.4049/jimmunol.176.4.2074. [DOI] [PubMed] [Google Scholar]
- 131.Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–9. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–34. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 133.Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005;115:407–17. doi: 10.1172/JCI23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bave U, Magnusson M, Eloranta ML, Perers A, Alm GV, Ronnblom L. FcγRIIa is expressed on natural IFN-α-producing cells (plasmacytoid dendritic cells) and is required for the IFN-α production induced by apoptotic cells combined with lupus IgG. J Immunol. 2003;171:3296–302. doi: 10.4049/jimmunol.171.6.3296. [DOI] [PubMed] [Google Scholar]
- 135.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 136.Colonna M. Toll-like receptors and IFN-alpha: partners in autoimmunity. J Clin Invest. 2006;116:2319–22. doi: 10.1172/JCI29879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Martin DA, Elkon KB. Autoantibodies make a U-turn: the toll hypothesis for autoantibody specificity. J Exp Med. 2005;202:1465–9. doi: 10.1084/jem.20052228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Deane JA, Bolland S. Nucleic acid-sensing TLRs as modifiers of autoimmunity. J Immunol. 2006;177:6573–8. doi: 10.4049/jimmunol.177.10.6573. [DOI] [PubMed] [Google Scholar]
- 139.Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, et al. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves toll-like receptors 7 and 8. J Exp Med. 2005;202:1575–85. doi: 10.1084/jem.20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lang KS, Recher M, Junt T, Navarini AA, Harris NL, Freigang S, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11:138–45. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 141.Bave U, Alm GV, Ronnblom L. The combination of apoptotic U937 cells and lupus IgG is a potent IFN-alpha inducer. J Immunol. 2000;165:3519–26. doi: 10.4049/jimmunol.165.6.3519. [DOI] [PubMed] [Google Scholar]
- 142.Bave U, Magnusson M, Eloranta ML, Perers A, Alm GV, Ronnblom L. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J Immunol. 2003;171:3296–302. doi: 10.4049/jimmunol.171.6.3296. [DOI] [PubMed] [Google Scholar]
- 143.Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–9. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–28. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 145.Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 2006;54:1906–16. doi: 10.1002/art.21890. [DOI] [PubMed] [Google Scholar]
- 146.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–7. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 147.Wu X, Peng SL. Toll-like receptor 9 signalling protects against murine lupus. Arthritis Rheum. 2006;54:336–42. doi: 10.1002/art.21553. [DOI] [PubMed] [Google Scholar]
- 148.Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–7. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–72. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 150.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–96. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 151.Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1 beta inhibition. N Engl J Med. 2006;355:581–92. doi: 10.1056/NEJMoa055137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364:1779–85. doi: 10.1016/S0140-6736(04)17401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004;50:607–12. doi: 10.1002/art.20033. [DOI] [PubMed] [Google Scholar]
- 154.Kasper LH, Everitt D, Leist TP, Ryan KA, Mascelli MA, Johnson K, et al. A phase I trial of an interleukin-12/23 monoclonal antibody in relapsing multiple sclerosis. Curr Med Res Opin. 2006;22:1671–8. doi: 10.1185/030079906X120931. [DOI] [PubMed] [Google Scholar]
- 155.Gottlieb AB, Cooper KD, McCormick TS, Toichi E, Everitt DE, Frederick B, et al. A phase 1, double-blind, placebo-controlled study evaluating single subcutaneous administrations of a human interleukin-12/23 monoclonal antibody in subjects with plaque psoriasis. Curr Med Res Opin. 2007;23:1081–92. doi: 10.1185/030079907x182112. [DOI] [PubMed] [Google Scholar]
- 156.Lovell DJ, Reiff A, Jones OY, Schneider R, Nocton J, Stein LD, et al. Long-term safety and efficacy of etanercept in children with polyarticular-course juvenile rheumatoid arthritis. Arthritis Rheum. 2006;54:1987–94. doi: 10.1002/art.21885. [DOI] [PubMed] [Google Scholar]
- 157.Osterman MT, Lichtenstein GR. Current and Future Anti-TNF Therapy for Inflammatory Bowel Disease. Curr Treat Options Gastroenterol. 2007;10:195–207. doi: 10.1007/s11938-007-0013-3. [DOI] [PubMed] [Google Scholar]