
Figure. Signaling pathways involved osteoclastogenesis in disease states leading to activation of NFATc1.
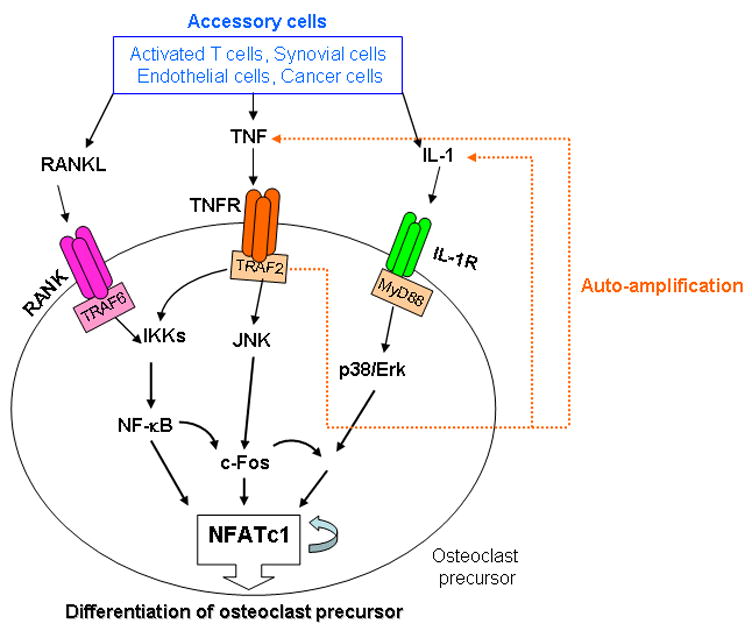
In inflammatory conditions, such as rheumatoid arthritis, the numbers of immune and accessory cells are increased in affected joints. Some of these cells produce RANKL in response to locally elevated levels of pro-inflammatory cytokines and other inflammatory mediators. RANKL binds to RANK on the surface of osteoclast precursors and recruits the adapter protein, TRAP6, leading to activation of NF-κB through phosphorylation and inactivation of inhibitory kappa kinases (IKKs) and NF-κB inhibitory kinase (not shown here). This induces activation of c-Fos. NF-κB and c-Fos interact with the NFATc1 promoter to trigger NFATc1 auto-amplification of NFATc1 and the transcription of genes, which mediate completion of the differentiation process. In addition to RANKL expression, these cells as well as macrophage/monocytes and osteoclasts themselves produce large amounts of TNF. TNF binds to the TNF receptor and this also activates c-Fos through both the NF-κB and JNK pathways in osteoclast precursors. TNF also stimulates its own expression and that of IL-1 by OCPs (orange lines). IL-1 does not activate c-Fos, but in OCPs in which c-Fos has been activated, for example by RANKL or TNF, IL-1 can induce osteoclastogenesis directly. This leads to more osteoclast formation using the same NFATc1 activated mechanism as RANKL. In this model, RANKL/RANK signaling is essential for OCP differentiation under both physiologic (through osteoblastic cells) and pathologic conditions (through accessory cells and OCP themselves), while TNF signaling appears to play a major role in inflammatory bone diseases.