

Abstract
Within hours after the ingestion of a blood meal, the mosquito midgut epithelium synthesizes a chitinous sac, the peritrophic matrix. Plasmodium ookinetes traverse the peritrophic matrix while escaping the mosquito midgut. Chitinases (EC 3.2.1.14) are critical for parasite invasion of the midgut: the presence of the chitinase inhibitor, allosamidin, in an infectious blood meal prevents oocyst development. A chitinase gene, PgCHT1, recently has been identified in the avian malaria parasite P. gallinaceum. We used the sequence of PgCHT1 to identify a P. falciparum chitinase gene, PfCHT1, in the P. falciparum genome database. PfCHT1 differs from PgCHT1 in that the P. falciparum gene lacks proenzyme and chitin-binding domains. PfCHT1 was expressed as an active recombinant enzyme in Escherichia coli. PfCHT1 shares with PgCHT1 a substrate preference unique to Plasmodium chitinases: the enzymes cleave tri- and tetramers of GlcNAc from penta- and hexameric oligomers and are unable to cleave smaller native chitin oligosaccharides. The pH activity profile of PfCHT1 and its IC50 (40 nM) to allosamidin are distinct from endochitinase activities secreted by P. gallinaceum ookinetes. Homology modeling predicts that PgCHT1 has a novel pocket in the catalytic active site that PfCHT1 lacks, which may explain the differential sensitivity of PfCHT1 and PgCHT1 to allosamidin. PfCHT1 may be the ortholog of a second, as yet unidentified, chitinase gene of P. gallinaceum. These results may allow us to develop novel strategies of blocking human malaria transmission based on interfering with P. falciparum chitinase.
As control of Anopheles mosquito populations in malaria-endemic regions becomes less effective, drug-resistant malaria continues to spread throughout the tropical world (1). A malaria vaccine designed to protect human populations at risk for malaria has yet to be developed (2). New strategies to control malaria are needed.
A number of investigators have proposed transmission-blocking vaccines as one component of an overall program of malaria control (3). Such vaccines are designed to induce antibodies in humans that, when ingested by the mosquito along with a Plasmodium-containing blood meal, interfere with the development of the parasite within the mosquito midgut. Animal models of transmission-blocking vaccines, based primarily on two P. falciparum zygote/ookinete surface proteins, Pfs25 and Pfs28, have demonstrated proof of principle (4), but results of human clinical trials have not been reported to date.
A Plasmodium ookinete-secreted enzyme, chitinase, has been demonstrated to be another target of blocking malaria transmission from humans to mosquitoes (5). The potential importance of chitinase in malaria parasite biology was first suggested by a transmission electron micrograph showing the P. gallinaceum ookinete penetrating and appearing to focally degrade the chitinous peritrophic matrix (PM) in the Aedes aegypti midgut (6). The PMs of the Plasmodium vectors Anopheles gambiae (which carries human malaria parasites) and A. aegypti (which carries avian malaria parasites) are composed of chitin, a β-1,4-linked polymer of GlcNAc, with intercalated proteins including trypsins and peritrophins (7–9). P. gallinaceum ookinetes secrete active chitinase (refs. 10 and 11; J.V., J. Valenzuela, L. Aravind, J. Ribeiro, and D. Kaslow, unpublished data). Although chitinases are found throughout the prokaryote and eukaryote kingdoms, where they play a role in cell wall modification and generating sources of nitrogen and carbon (12), the biological function of Plasmodium chitinases must be different, because ookinetes do not contain chitin and there is no evidence that ookinetes use chitin or mono- or oligomers of GlcNAc as a carbon source. Chitinases are critical for allowing the parasite to escape the mosquito midgut, as evidenced by the observation that addition of the chitinase inhibitor allosamidin to a blood meal prevents oocyst development (5). Both P. gallinaceum in A. aegypti and P. falciparum in A. freeborni fail to develop into oocysts in the presence of this inhibitor. P. gallinaceum chitinases recently have been characterized in detail (J.V., J. Valenzuela, L. Aravind, J. Ribeiro, and D. Kaslow, unpublished data), and a P. gallinaceum chitinase gene, PgCHT1, the only apicomplexan chitinase gene identified to date, encodes an active endochitinase (C.S., R. Langer, and J.V., unpublished data).
To date, the presence of a P. falciparum ookinete-secreted chitinase has only been inferred, because it has not been possible to produce sufficient quantities of P. falciparum ookinetes for direct biochemical studies (5). We describe here the identification and characterization of the product of a P. falciparum chitinase gene. Our findings suggest novel approaches toward the goal of blocking malaria transmission.
Materials And Methods
Chemicals and Reagents.
Routine chemicals were from Amresco (Euclid, OH) or Sigma. Molecular biology reagents were from Life Technologies (Gaithersburg, MD). 4-Methylumbelliferone (4MU) substrates were from Sigma. Native chitin oligosaccharides (GlcNAc1–6) were from Calbiochem.
Identification, Cloning, and Sequencing of PfCHT1.
Sense and antisense primers derived from PfCHT1, spanning nucleotides 154–178 and 546–570, respectively, were used to generate a 417-bp digoxigenin (dig)-labeled probe by PCR, which was used to screen a phage library of P. falciparum genomic DNA (Thai isolate K1) (13). The probe contained the most highly conserved regions of the gene, including the substrate-binding and catalytic sites. Blots were developed by using an anti-dig detection system and chemiluminescence (Roche Molecular Biochemical, Indianapolis, IN). A HindIII fragment of DNA prepared from one positive plaque was subcloned into pUC19; this construct was called pUC19–4.2PfCHT1. The DNA sequence of PfCHT1 was established by automated sequencing.
Southern Blotting to Detect PfCHT1.
Southern blots (containing 1 μg of P. falciparum 3D7 strain DNA per lane) were probed as described above by using the same 417-bp fragment. Final wash conditions were 2× SSC/0.5% SDS at 45°C.
Chromosomal Localization of PfCHT1.
PCR, using two independent sets of PfCHT1-specific primers, was performed on individual chromosomes isolated from pulse-field electrophoresis-separated P. falciparum (XP5 clone; a progeny of the HB3 × Dd2 genetic cross) as described (14).
Expression and Preparation of Recombinant P. falciparum Chitinase.
The expression construct was prepared by PCR-amplifying the native coding region of PfCHT1 using pUC19–4.2PfCHT1 as template. The NcoI-containing 5′ primer was GCG CCA TGG GTC ATC GAG CAC GAC CAG GTG AA. The XhoI-containing 3′ primer was CGCG CTC GAG ATG TAA AGA TTC TAC GAA ATA TTC. The construct began immediately after the predicted signal peptide cleavage site (15). PCR products were restriction-digested, ligated into the NcoI and XhoI restriction sites of the pET32b expression vector (Novagen), and transfected into DH10B Escherichia coli cells (Life Technologies). The correct construct was verified by restriction digestion and automated sequencing and used to transform the E. coli strain AD494 (DE3) (Novagen) for expression.
Recombinant bacteria were grown in a fermentation system (BioFlo IV; New Brunswick Scientific), in LB with ampicillin at 100 μg/ml, as follows: (i) growth to OD600 = 0.800 at 37°C; (ii) addition of isopropyl β-d-thiogalactoside to 0.1 mM; and (iii) growth at 18°C for 16 hr. Pellets were treated with 3 ml/g pellet of lysozyme (0.5 mg/ml) in 20 mM Tris, pH 8.0/200 mM NaCl/0.1 mM PMSF and stirred at 22°C for 20 min. Triton X-100 was added to a final concentration of 0.1% (vol/vol), the mixture was incubated for 5 min at 22°C, and 0.1 g/ml DNase I was added with a further 10-min incubation. The cell suspension was centrifuged at 30,000 × g for 1.5 hr, and the supernatant was directly run over a nickel-Sepharose column (AKTA Explorer System; Pharmacia). Recombinant PfCHT1 (rPfCHT1) was cleaved with recombinant enterokinase (Novagen) and amino-terminal sequencing of the PfCHT1 product done in the University of Texas Medical Branch Protein Chemistry Core Facility with an Applied Biosystems 494/HT Procise Sequencing System.
Assessment of Chitinase Activity.
rPfCHT1 activity was assayed in glycol chitin gels by microfluorimetry and by TLC as described previously (C.S., R. Langer, and J.V., unpublished data).
Determination of Substrate Specificity, pH Profile, and Allosamidin Inhibition Curves.
Using a citrate/sodium phosphate buffer system with pH ranging from 4.0 to 7.0 in 0.5 pH unit increments, rPfCHT1 was incubated with native chitin oligosaccharides (GlcNAc1–6) or 4MU-labeled GlcNAc1–4 substrates and analyzed by TLC or microfluorimetry as described (C.S., R. Langer, and J.V., unpublished data). Microfluorimetry results are reported as initial rates of substrate hydrolysis in relative fluorescence units.
Detection of PfCHT1 Transcripts in Mosquito Midgut Stages.
P. falciparum sexual stages were produced by feeding infectious gametocytes (3D7 clone) to Anopheles freeborni (16). Mosquito midguts containing ookinete stages were collected 24 hr postfeed. Total RNA was extracted from a pool of 70 midguts by using Trizol (Life Technologies). After treatment with DNase I, first-strand cDNA was generated by using a mixture of oligo(dT) and random hexamers. PCR amplification was performed in the presence or absence of reverse transcriptase (Superscript Preamplification System; Life Technologies) to control for genomic DNA contamination. Amplification of PfCHT1 was performed by using the primers 5′ ATT ATG CTT TTA TCT CTT GGA GG and 5′ AGT CTT TAC AAA ATC ACC AAT GG. As a control, a fragment of the P. falciparum gene, Pfs28, expressed by retort and ookinete stages in the mosquito midgut (17), was amplified by using the primers 5′ CAT AAC GTT GAA TAA GGC TCG GG and 5′CTA TAT GAT GTA TCA GCC TGG TCC.
Molecular Modeling of PfCHT1.
Homology models were constructed by using a previously reported sequence alignment (J.V., J. Valenzuela, L. Aravind, J. Ribeiro, and D. Kaslow, unpublished data) and energy was minimized by using sybyl (Tripos Associates, St. Louis). The atomic coordinates of hevamine complexed with allosamidin (Protein Data Bank Code 1LLO) and Serratia marscences chitinase Chi A (Protein Data Bank Code 1CTN) were used as templates.
Results
Characterization of the Full-Length P. falciparum Chitinase Gene, PfCHT1.
A partial sequence of PfCHT1 was identified initially in the P. falciparum genome project (J.V., J. Valenzuela, L. Aravind, J. Ribeiro, and D. Kaslow, unpublished data). A 4.2-kb fragment containing the full-length PfCHT1 gene was obtained from screening a phage library of P. falciparum genomic DNA. PfCHT1 has a predicted single-exon ORF of 1,134 bp, with a 71.2% A/T content. Sequence translation predicts a protein of 378 aa and an expected molecular mass of 42,792 Da. The 15 bp 5′ to the predicted translational initiation site are (−15) AATAAATATATAAAC (−1), consistent with sequences reported to flank the translational start sites of yeast, P. falciparum, and other protozoa (18–20). A secretory signal peptide sequence of 28 aa is predicted to be present at the amino terminus (Fig. 1) (15), which further supports the assignment of the translational initiation codon. In the 400 bp of 90% AT-rich DNA 5′ to the predicted start codon, no intron splice sites, exons, or start methionines are identifiable but stop codons are present in all three reading frames. PfCHT1 contains substrate binding and catalytic sites typical of family 18 chitinases (J.V., J. Valenzuela, L. Aravind, J. Ribeiro, and D. Kaslow, unpublished data). Comparison of PfCHT1 with PgCHT1 (Fig. 1) suggests that PfCHT1 lacks a chitin-binding domain. An encoded chitin-binding domain is not detectable within 1,500 bp of sequence downstream of the predicted stop codon within the 4.2-kb genomic clone that contains PfCHT1. In contrast to PgCHT1, PfCHT1 does not contain an amino acid sequence consistent with a proenzyme domain or sequences homologous to the NT1 or NT2 loops found in PgCHT1 (Fig. 1).
Figure 1.

Comparison of amino acid sequences of PfCHT1 and PgCHT1. The predicted signal peptides are underlined. Putative proenzyme, catalytic, and chitin-binding domains are indicated; substrate-binding and catalytic active sites are overlined. NT1 and NT2 delineate the two secreted forms of PgCHT1.
Hybridization of a Southern blot with the same 417-bp digoxigenin-labeled fragment used to screen the genomic phage library demonstrated a single band on genomic DNA digested by five separate restriction enzymes (Fig. 2a). This is consistent with a single- or low-copy-number gene. Genomic DNA restricted by BglII yielded one predominant band of ≈8.2 kb and two smaller, less prominent, bands of ≈6–6.5 kb of uncertain significance. PfCHT1 has been localized to chromosome 12 (Fig. 2b).
Figure 2.

Restriction mapping, chromosomal localization, and transcriptional activity of the PfCHT1 gene. (a) Southern blot of P. falciparum strain 3D7. Restriction enzymes used to digest the DNA are shown across the top; molecular sizes are indicated in bp at left. (b) Chromosomal localization of PfCHT1 by PCR on pulse-field gel-electrophoresis-separated P. falciparum chromosomal DNA. Gene-specific primers demonstrate that PfCHT1 is located on chromosome 12. Std, 500-bp DNA ladder, shown at left. Chromosome numbers are at the top. Dd2, genomic DNA template from P. falciparum strain Dd2 used as a positive control. (c) Reverse transcription–PCR (RT-PCR) analysis to determine the presence of PfCHT1 message in total RNA extracted from P. falciparum-infected A. freeborni midguts. The same preparation of RNA was used for RT-PCR of both PfCHT1 and Pfs28. RT-PCR of Pfs28 mRNA, which encodes an P. falciparum zygote/ookinete surface protein, was included as a positive control. The 100-bp ladder is indicated; +RT and −RT, with and without reverse transcriptase.
To date, it has not been possible to obtain sufficient quantities of in vitro transformed P. falciparum ookinetes to study chitinase proteins directly or to detect PfCHT1 messenger RNA by Northern analysis. To determine whether stages of P. falciparum developing within the mosquito midgut express PfCHT1, total RNA was prepared and analyzed for the presence of PfCHT1 transcript by reverse transcription–PCR (Fig. 2c). At 24 hr postblood meal, a PfCHT1 message was detectable in P. falciparum-infected mosquito midguts. Sequencing of the amplicons showed that they were identical to PfCHT1 (data not shown). A PfCHT1 message was not detected in midguts taken from mosquitoes 24 hr after a noninfectious blood meal (data not shown). These findings demonstrate that P. falciparum ookinetes within the mosquito midgut transcribe PfCHT1.
We have examined PfCHT1 transcription in cDNA libraries of other P. falciparum stages. PfCHT1 and the zygote/ookinete marker Pfs25 were both detected in gametocyte cDNA as well as in two asexual blood-stage cDNA libraries known to contain gametocyte transcripts (data not shown). These results with Pfs25 are consistent with previous observations, which also noted that Pfs25 protein expression was delayed until after exflagellation and fertilization in the mosquito midgut (21). Zygotes of P. gallinaceum also contain PgCHT1 mRNA but undetectable PgCHT1 protein or chitinase enzymatic activity (C.S., R. Langer, and J.V., unpublished data). No PfCHT1 mRNA was detected in a sporozoite cDNA library (22).
Expression of Enzymatically Active PfCHT1.
rPfCHT1 was expressed as a thioredoxin (trx) fusion protein in the expression plasmid pET32b by using as host cells the E. coli mutant nonreducing strain AD494, which allows for intracytoplasmic formation of disulfide bonds. The trx-rPfCHT1 fusion protein was constructed with a hexahistidine (His6) tag at both amino and carboxyl termini. The expressed chitinase began at the amino acid immediately after the predicted signal peptide cleavage site and included the remainder of the ORF. Chitinase activity was readily found in crude, soluble extracts of induced recombinant bacteria, as detected by hydrolysis of 4MU-chitotrioside (data not shown). When the same construct was expressed in E. coli BL21 cells, chitinase activity was not detected, despite a comparable total quantity of recombinant protein produced (data not shown). Chitinase activity, by the measures described here, was not detectable in E. coli strains AD494 or BL21 transformed with the pET32b vector with no insert. Cell lysates from a 16-liter fermentation of rPfCHT1 were chromatographed with an imidazole step gradient on a nickel-Sepharose column (Fig. 3a), yielding a trx-rPfCHT1 fusion protein of >95% purity as determined by Coomassie blue staining (Fig. 3b). Western immunoblot and amino-terminal sequencing confirmed the identity of the recombinant protein (data not shown). The amino-terminal His6 tag was found to be responsible for binding to nickel Sepharose; the carboxyl terminal His6 sequence was found not to bind to nickel Sepharose (data not shown). Therefore, any chitinase activity eluting from the nickel Sepharose column at 250 mM imidazole must have an intact amino-terminal His6 tag, indicating that no unexpected proteolytic degradation of trx-rPfCHT1 had occurred to yield a shorter-length, enzymatically active rPfCHT1. The fused trx sequence was released by treatment with enterokinase, which yielded rPfCHT1 with a molecular mass of 39 kDa (Fig. 3c). The final yield was ≈7 mg of enterokinase-cleaved rPfCHT1 from a 16L fermentation run. This 39-kDa protein had robust chitinase activity as determined by hydrolysis of 4MU-GlcNAc3. Amino-terminal sequencing of this 39-kDa band gave the amino acid sequence ARPGE, the amino terminus of rPfCHT1 as designed in the expression construct. The trx-rPfCHT1 fusion protein and its enterokinase-cleaved product rPfCHT1 had similar enzymatic activity (data not shown). These experimental results, in conjunction with the comparison of the primary structures of PfCHT1 and PgCHT1 (Fig. 1), strongly suggest that PfCHT1 is not produced as a zymogen. This finding contrasts with previous suggestions that P. gallinaceum ookinete-secreted chitinase activity is synthesized as a zymogen that is activated by ookinete protease(s) (5, 23–25).
Figure 3.

Purification of enzymatically active rPfCHT1. (a) A lysate of induced bacteria cell mass was clarified, run over a nickel-Sepharose column, and eluted with a step gradient of imidazole in 1 M NaCl/20 mM Tris, pH 8.0. Fractions were assayed for chitinase activity by microfluorimetry with 4MU-GlcNAc3. (b) Samples were analyzed by SDS/PAGE; the gel was stained with Coomassie blue. (c) Treatment of pooled fractions 21 and 22 with enterokinase (EK) after dialysis of the protein against 20 mM Tris, pH 7.5/50 mM NaCl/2 mM CaCl2.
Substrate Specificity of rPfCHT1.
rPfCHT1 was found to digest polymeric chitin efficiently in a nondenaturing polyacrylamide activity gel into which glycol chitin had been incorporated (C.S., R. Langer, and J.V., unpublished data) (data not shown). TLC was used to further characterize the action of rPfCHT1 on native and 4MU-derivatized chitin oligosaccharide substrates (Fig. 4). rPfCHT1 had no hydrolytic action on GlcNAc2 or GlcNAc3 but some activity on GlcNAc4. rPfCHT1 had markedly more activity against the longer native chitin oligosaccharide substrates, GlcNac5 and GlcNAc6 (Fig. 4a). rPfCHT1 did not cleave 4MU-GlcNAc or 4MU-GlcNAc2 substrates, but cleaved the longer substrates (Fig. 4b). 4MU-GlcNAc3 was hydrolyzed at only one glycosidic linkage, yielding 4-MU and GlcNAc3. Cleavage of 4MU-GlcNAc3 was rapid, about three times faster than from 4MU-GlcNAc4, as measured by microfluorimetry (Fig. 4c). The likely reason for the slower release of 4MU from 4MU-GlcNAc4 is that the enzyme also cleaves two other glycosidic bonds, yielding 4MU-GlcNAc and 4MU-GlcNAc2, which cannot be hydrolyzed (Fig. 4b). These data indicate that rPfCHT1 acts as an endochitinase, similar to P. gallinaceum ookinete-secreted chitinases (C.S., R. Langer, and J.V., unpublished data).
Figure 4.

Analysis of the action of rPfCHT1 on native chitin oligosaccharide substrates and 4MU derivatives of chitin oligosaccharides. (a) TLC analysis of end products produced by rPfCHT1 on native GlcNAc1–6. (b) TLC analysis of end products produced by rPfCHT1 on 4MU-GlcNAc1–4. Oligosaccharide substrate lengths are indicated at the top, and reaction products are shown at right. The origins at which the reaction mixtures were spotted are as indicated. Free 4MU fluoresces 12 times more than 4MU derivatives of GlcNAc oligomers; thus, the weaker fluorescent bands corresponding to the mono-, di-, and tetra-MU cannot be compared stoichiometrically to the free 4MU. The origins at which the reaction mixtures were spotted are indicated. S, 4-MU GlcNAc1–4 standards. (c) Microfluorimetry analysis of initial rates of fluorescence produced by rPfCHT1 on 4MU substrates, as a measure of relative initial reaction rates of chitinase activity.
pH Profile and Allosamidin Sensitivity of rPfCHT1.
pH-dependent activity profiles and allosamidin inhibitory concentration curves for rPfCHT1 were determined by microfluorimetry (Fig. 5). The sensitivity of rPfCHT1 to allosamidin inhibition increases with rising pH; no difference in the IC50 curve is seen above pH 6.0. The IC50 of rPfCHT1 to allosamidin is 40 nM, less than that of PgCHT2 (300 nM); both are distinctly different from the IC50 of rPgCHT1 (12 μM) (C.S., R. Langer, and J.V., unpublished data). The allosamidin concentration (0.1–1.0 mM) sufficient to block oocyst development in vivo (5) far exceeds the IC50 of PfCHT1 (40 nM) for allosamidin in vitro, consistent with the hypothesis that PfCHT1 is involved in allowing the ookinete to penetrate the PM.
Figure 5.

pH activity profile and pH-dependent inhibition of rPfCHT1 by allosamidin. (a) Relative rates of rPfCHT1 activity at different pH levels. Shaded bar indicates that at pH 4.5, initial enzyme activity is linear for 10 min at 37°C, but then slows and is irreversibly gone at 20 min. rPfCHT1 has no chitinase activity at pH 3.5 or 4.0. Data are displayed as the mean of three separate experiments; errors (not shown) are 5–7%. (b) Relative rates of allosamidin inhibition of rPfCHT1 activity at pH 5.0 (■) and pH 6.0 and 7.0 (♦). 4MU-GlcNAc3 was used as substrate for both sets of experiments.
Molecular Modeling of PfCHT1.
PfCHT1 is predicted to have an (αβ)8 triose isomerase barrel structure typical of family 18 chitinases (26). A majority of the active-site residues of PfCHT1 are common to either hevamine or Serratia marscesens chitinase ChiA, for which crystal structures are available (26, 27). The Plasmodium chitinases are unique in that they have a Gly for Phe/Met (hevamine/ChiA, respectively) change at a position (353 for PfCHT1, 405 for PgCHT1) that is highly conserved among other family 18 chitinases. This position is in a critical area at the base of the catalytic site (Fig. 6) and may impart a unique structure. To explore further the potential implication of this position as a site for selective drug targeting, homology models were built for PfCHT1, PgCHT1, and human chitotriosidase (28) (Fig. 6). Although the Gly for Phe/Met change substantially enlarges the base of the catalytic pocket in PgCHT1, a complementary Tyr309 in PfCHT1 on the β-7 strand compensates for the missing volume, resulting in an almost perfect overlap of the catalytic pocket with that of human chitotriosidase. This model suggests that it may be difficult to develop a PfCHT1-selective inhibitor. In contrast, the I361 change in the PgCHT1 β-7 strand does not fully compensate for the Gly for Phe/Met change. The resulting unique pocket distinguishes PgCHT1 from PfCHT1 and may explain the differential sensitivity of PfCHT1 and PgCHT1 to allosamidin. In the model, allosamidin does not appear to contact Gly405 of PgCHT1 but does appear to contact Tyr309 in PfCHT1. Crystal structure determination of PfCHT1 and PgCHT1 will be necessary to confirm these predictions.
Figure 6.
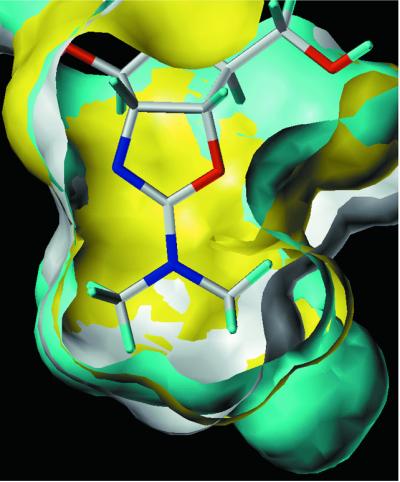
Homology model depicting the overlapping catalytic sites of PfCHT1, PgCHT1, and human chitotriosidase complexed with allosamidin (yellow, PfCHT1; blue, PgCHT1; white, human chitotriosidase). The three active sites are almost perfectly superimposable, with the exception of a novel pocket found in PgCHT1, seen at lower right. The models were built by using the structure of hevamine complexed with allosamidin as a template.
Discussion
Plasmodium ookinetes face two physical barriers as they invade the mosquito midgut: the peritrophic matrix and the epithelium. With the identification of PfCHT1, we can begin to elucidate the precise biochemical mechanisms by which P. falciparum ookinetes penetrate and traverse the PM. We have demonstrated that PfCHT1 encodes an endochitinase, expressed by parasites within the mosquito midgut, with a marked preference for longer chitin oligosaccharide substrates, consistent with the predicted biological function of this enzyme. This substrate specificity is, to date, unique to Plasmodium chitinases (C.S., R. Langer, and J.V., unpublished data). The natural substrate for PfCHT1 has yet to be demonstrated although the enzyme efficiently cleaves polymeric glycol chitin. Also, it remains to be shown formally whether allosamidin inhibition of the Plasmodium chitinase, rather than allosamidin inhibition of the mosquito-expressed midgut chitinase (8), is responsible for preventing ookinetes from escaping the PM. This issue could be addressed with inhibitors that distinguish PfCHT1 or P. gallinaceum chitinases from mosquito midgut chitinases or by the development of a chitinase knockout transgenic mosquito.
Our data provide evidence that Plasmodium ookinetes are likely to secrete products of more than one chitinase gene. The pH profile of rPfCHT1 and its sensitivity to allosamidin closely correspond to a second P. gallinaceum ookinete-secreted chitinase activity that we provisionally have called PgCHT2 (C.S., R. Langer, and J.V., unpublished data). The pH activity profiles of both rPfCHT1 and PgCHT2 are shifted 0.5 pH units toward the acid range compared with both recombinant and native PgCHT1; rPfCHT1 and PgCHT2 also become irreversibly inactivated at 0.5 pH units lower than native and recombinant PgCHT1. rPfCHT1 has a sensitivity to allosamidin much closer to that of PgCHT2 than to PgCHT1 (C.S., R. Langer, and J.V., unpublished data). PfCHT1 has other features in common with PgCHT2: its size (molecular mass of secreted protein of 39 kDa vs. ≈35 kDa for PgCHT2; ookinete-secreted native PgCHT1 has a molecular mass of ≈55 kDa) and the apparent lack of a chitin-binding domain. It appears that PfCHT1 is the ortholog of PgCHT2, and the ortholog of PgCHT1 in P. falciparum has yet to be identified. If Plasmodium ookinetes do indeed secrete products of more than one chitinase gene, different chitinase proteins may work in concert to allow the ookinete to cross the PM. The contribution of PfCHT1 to invasion of the peritrophic matrix currently is being addressed by gene disruption studies.
As a target of blocking malaria parasite transmission to mosquitoes, chitinase differs from previously identified surface antigens of sexual stage parasites in that it has a well characterized biochemical activity at which nonimmunological interventions can be aimed. In addition to the traditional transmission-blocking vaccine approach, computational and structural biology-driven rational drug design to search for chitinase-inhibitory drugs that block transmission may be fruitful. The major challenge will be to develop a nontoxic, inexpensive, highly potent, bioavailable chitinase inhibitor with a long half-life in vivo that can be administered widely to human populations in endemic regions. There are precedents for administering pharmacological compounds to human populations in the water or food supplies: fluoridation of water supplies, iodination of salt, diethylcarbamazine in salt (29), and even quinine in tonic water.
Finally, Plasmodium chitinase could be used as a target in generating transgenic mosquitoes (30–32) that are refractory to ookinete invasion by secreting a chitinase inhibitory peptide into the midgut. Such an approach would be independent of the genetic mapping and sequencing of the A. gambiae genome, currently being pursued, in part, to search for mosquito refractoriness genes (33). In principle, an oligopeptide chitinase inhibitor selective for P. falciparum chitinase could be obtained from screening combinatorial phage display libraries. A synthetic gene encoding such a peptide under the control of a gut-specific, blood meal-inducible promoter such as that of carboxypeptidase (34) or late trypsin (29) could be used, in an appropriate construct, to transform Anopheles mosquitoes to a refractoriness phenotype.
Acknowledgments
We thank Dr. Thomas Templeton for his kind gift of P. falciparum ookinete stage first-strand cDNA, Dr. Xin-zhuan Su for chromosomal localization, and Xiao Lian Liang for fermentation. We thank Drs. David Kaslow, Louis Miller, and Irwin D. Kuntz for ongoing advice and support of this project and Drs. Barbara Doughty, Rebecca Langer, Robert Shope, and Robert Tesh for critically reading the manuscript. C.A.S. was supported by GM-54380 and GM-31318 from the National Institutes of Health. K.A.B. was supported by National Institutes of Health F32 Postdoctoral Training Grant AI-10416. The University of Texas Medical Branch (UTMB) Protein Expression and Purification Laboratory is supported in part by a grant from National Institute on Environmental Health Sciences P30 ES-06676 and the UTMB Sealy Center for Structural Biology. Funding from the Charles E. Culpeper Foundation Scholarships in Medical Science and the UTMB Sealy Center for Structural Biology Collaborative Grants Program (to J.M.V.) is gratefully acknowledged.
Abbreviations
- rPfCHT1
recombinant P. falciparum chitinase
- PM
peritrophic matrix
- 4MU
4-methylumbelliferone
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database [accession no. AF172445 (PfCHT1)].
References
- 1.White N. Br Med Bull. 1998;54:703–715. doi: 10.1093/oxfordjournals.bmb.a011721. [DOI] [PubMed] [Google Scholar]
- 2.Miller L, Hoffman S. Nat Med. 1998;4,Suppl.:520–524. doi: 10.1038/nm0598supp-520. [DOI] [PubMed] [Google Scholar]
- 3.Kaslow D C. Int J Parasitol. 1997;27:183–189. doi: 10.1016/s0020-7519(96)00148-8. [DOI] [PubMed] [Google Scholar]
- 4.Gozar M, Price V, Kaslow D. Infect Immunol. 1998;66:59–64. doi: 10.1128/iai.66.1.59-64.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shahabuddin M, Toyoshima T, Aikawa M, Kaslow D. Proc Natl Acad Sci USA. 1993;90:4266–4270. doi: 10.1073/pnas.90.9.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sieber K, Huber M, Kaslow D, Banks S, Torii M, Aikawa M, Miller L. Exp Parasitol. 1991;72:145–156. doi: 10.1016/0014-4894(91)90132-g. [DOI] [PubMed] [Google Scholar]
- 7.Perrone J, Spielman A. Cell Tissue Res. 1988;252:473–478. doi: 10.1007/BF00214391. [DOI] [PubMed] [Google Scholar]
- 8.Shen Z, Jacobs-Lorena M. J Biol Chem. 1997;272:28895–28900. doi: 10.1074/jbc.272.46.28895. [DOI] [PubMed] [Google Scholar]
- 9.Shen Z, Jacobs-Lorena M. J Biol Chem. 1998;273:17665–17670. doi: 10.1074/jbc.273.28.17665. [DOI] [PubMed] [Google Scholar]
- 10.Huber M, Cabib E, Miller L. Proc Natl Acad Sci USA. 1991;88:2807–2810. doi: 10.1073/pnas.88.7.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vinetz J, Kaslow D. Exp Parasitol. 1998;90:199–202. doi: 10.1006/expr.1998.4322. [DOI] [PubMed] [Google Scholar]
- 12.Flach J, Pilet P-E, Jolles P. Experientia. 1992;48:701–716. doi: 10.1007/BF02124285. [DOI] [PubMed] [Google Scholar]
- 13.Goman M, Langsley G, Hyde J E, Yankovsky N K, Zolg J W, Scaife J G. Mol Biochem Parasitol. 1982;5:391–400. doi: 10.1016/0166-6851(82)90012-3. [DOI] [PubMed] [Google Scholar]
- 14.Su X, Wellems T. Exp Parasitol. 1999;91:367–369. doi: 10.1006/expr.1998.4390. [DOI] [PubMed] [Google Scholar]
- 15.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 16.Templeton T, Keister D, Muratova O, Procter J, Kaslow D. J Exp Med. 1998;187:1599–1609. doi: 10.1084/jem.187.10.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duffy P, Kaslow D. Infect Immunol. 1997;65:1109–1113. doi: 10.1128/iai.65.3.1109-1113.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamauchi K. Nucleic Acids Res. 1991;19:2715–2717. doi: 10.1093/nar/19.10.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saul A, Battistutta D. Mol Biochem Parasitol. 1990;42:55–62. doi: 10.1016/0166-6851(90)90112-y. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton R, Watanabe C, A de Boer H. Nucleic Acids Res. 1987;15:3581–3595. doi: 10.1093/nar/15.8.3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaslow D C, Quakyi I A, Syin C, Raum M G, Keister D B, Coligan J E, McCutchan T F, Miller L H. Nature (London) 1988;333:74–76. doi: 10.1038/333074a0. [DOI] [PubMed] [Google Scholar]
- 22.Fidock D, Nguyen T, Dodemont H, Eling W, James A. Exp Parasitol. 1998;89:125–128. doi: 10.1006/expr.1998.4262. [DOI] [PubMed] [Google Scholar]
- 23.Shahabuddin M, Criscio M, Kaslow D. Exp Parasitol. 1995;80:212–219. doi: 10.1006/expr.1995.1026. [DOI] [PubMed] [Google Scholar]
- 24.Shahabuddin M, Lemos F, Kaslow D, Jacobs-Lorena M. Infect Immunol. 1996;64:739–743. doi: 10.1128/iai.64.3.739-743.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shahabuddin M. Parasitology. 1998;116,Suppl.:S83–S93. doi: 10.1017/s0031182000084973. [DOI] [PubMed] [Google Scholar]
- 26.Terwisscha van Scheltinga A, Hennig M, Dijkstra B. J Mol Biol. 1996;262:243–257. doi: 10.1006/jmbi.1996.0510. [DOI] [PubMed] [Google Scholar]
- 27.Perrakis A, Tews I, Dauter Z, Oppenheim A, Chet I, Wilson K, Vorgias C. Structure. 1994;2:1169–1180. doi: 10.1016/s0969-2126(94)00119-7. [DOI] [PubMed] [Google Scholar]
- 28.Boot R, Renkema G, Strijland A, Zonneveld A, Aerts J. J Biol Chem. 1995;270:26252–26256. doi: 10.1074/jbc.270.44.26252. [DOI] [PubMed] [Google Scholar]
- 29.Gelband H. Am J Trop Med Hyg. 1994;50:655–662. doi: 10.4269/ajtmh.1994.50.655. [DOI] [PubMed] [Google Scholar]
- 30.Besansky N, Collins F. Parasitol Today. 1992;8:186–192. doi: 10.1016/0169-4758(92)90262-z. [DOI] [PubMed] [Google Scholar]
- 31.Coates C, Jasinskiene N, Miyashiro L, James A. Proc Natl Acad Sci USA. 1998;95:3748–3751. doi: 10.1073/pnas.95.7.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jasinskiene N, Coates C, Benedict M, Cornel A, Rafferty C, James A, Collins F. Proc Natl Acad Sci USA. 1998;95:3743–3747. doi: 10.1073/pnas.95.7.3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collins F, Zheng L, Paskewitz S, Kafatos F. Ann Trop Med Parasitol. 1997;91:517–521. doi: 10.1080/00034989760888. [DOI] [PubMed] [Google Scholar]
- 34.Edwards M J, Lemos F J, Donnelly-Doman M, Jacobs-Lorena M. Insect Biochem Mol Biol. 1997;27:1063–1072. doi: 10.1016/s0965-1748(97)00093-3. [DOI] [PubMed] [Google Scholar]