

Abstract
Transgenic mice that overexpress mutant human amyloid precursor protein (APP) exhibit one hallmark of Alzheimer’s disease pathology, namely the extracellular deposition of amyloid plaques. Here, we describe significant deposition of amyloid β (Aβ) in the cerebral vasculature [cerebral amyloid angiopathy (CAA)] in aging APP23 mice that had striking similarities to that observed in human aging and Alzheimer’s disease. Amyloid deposition occurred preferentially in arterioles and capillaries and within individual vessels showed a wide heterogeneity (ranging from a thin ring of amyloid in the vessel wall to large plaque-like extrusions into the neuropil). CAA was associated with local neuron loss, synaptic abnormalities, microglial activation, and microhemorrhage. Although several factors may contribute to CAA in humans, the neuronal origin of transgenic APP, high levels of Aβ in cerebrospinal fluid, and regional localization of CAA in APP23 mice suggest transport and drainage pathways rather than local production or blood uptake of Aβ as a primary mechanism underlying cerebrovascular amyloid formation. APP23 mice on an App-null background developed a similar degree of both plaques and CAA, providing further evidence that a neuronal source of APP/Aβ is sufficient to induce cerebrovascular amyloid and associated neurodegeneration.
The pathological hallmarks of Alzheimer’s disease (AD) are the deposition of amyloid β (Aβ) in the neural parenchyma (plaques) and the formation of neuronal tangles. Although not a criteria for diagnosis of AD, the deposition of Aβ in the cerebral vasculature can be detected in 90% of AD patients (1). However, cerebral amyloid angiopathy (CAA) can occur in the absence of AD pathology and vice versa. For example, patients with hereditary cerebral hemorrhage with amyloidosis-Dutch type, an autosomal-dominant severe form of CAA caused by a point mutation in amyloid precursor protein (APP), do not seem to develop significantly more amyloid plaques or neurofibrillary tangles than the normal elderly (2). CAA also occurs as a sporadic disorder, evident in ≈30% of people over 60 years of age and in 50% at age 90 (1). The deposition of Aβ in cerebral vessels leads to severe vascular pathology and is a significant risk factor for cerebral hemorrhage (1, 3). Vascular amyloid deposition can lead to degeneration of vascular smooth muscle cells and disruption of the blood–brain barrier (1, 4).
Progress in CAA research has been slow because of the paucity of animal models. Past studies have largely been based on models of naturally occurring CAA in some aged nonhuman primate species and dogs (5). The discovery of mutations in APP that cause AD has led to the development of transgenic mouse models that, as they age, develop amyloid plaques similar to those found in AD brain (6–8). We have recently shown that, in one such mouse model, the APP23 mouse, the formation of amyloid plaques is related to neuron loss, synaptic bouton loss, alterations in the cholinergic system, and a marked glial reaction (refs. 9 and 10; M.E.C., A.L.P., P. H. Kelly, K.-H.W., D.A., C.S.-P., A. Enz, B.S., M.S., and M.J., unpublished work). These mouse models offer a unique opportunity to study mechanisms of both amyloid formation and related neurodegeneration.
Here, we report that, in addition to amyloid plaques, significant deposition of cerebrovascular amyloid occurs in APP23 mice as they age, and we demonstrate that neurodegeneration and microhemorrhage are associated with CAA. The sole neuronal source of APP in this model is sufficient to lead to vascular amyloid and high cerebrospinal fluid (CSF) Aβ levels. These results suggest transport and drainage pathways rather than local production or blood uptake of Aβ as a primary mechanism underlying cerebrovascular amyloid formation in these mice.
Materials and Methods
Animals.
Generation of the B6,D2-TgN(Thy1-APPSwe) transgenic mouse line (APP23) is described elsewhere (6). The founder mice were back-crossed (n > 4) with C57BL/6 (B6) mice. A total of 32 (15 hemi- and 7 homozygous transgenic; 10 littermate controls) adult male mice 14–21 months of age were used for histological and quantitative analysis, and 2 additional aged hemizygous mice were used for electron microscopy. Aβ concentration in CSF and blood was measured in 8- and 24-month-old hemizygous mice. APP23 mice were bred with App-null mice (C.S.-P. and B.S., unpublished work) producing mice hemizygous for human APP and lacking endogenous mouse APP [C57BL/6-App-TgN(Thy1-APPSwe)]. Twelve- and eighteen-month-old animals from this breeding were used.
Tissue Processing.
Mice were anesthetized and perfused with 4% paraformaldehyde in PBS. The left hemisphere was processed for paraffin embedding (11), and the right hemisphere was postfixed, cryoprotected, and frozen (12). Serial coronal sections were cut through both hemispheres.
Cresyl-violet, Congo red, Thioflavin-S, Berlin Blue for iron (13), and nuclear-red staining were performed according to standard protocols. Immunohistochemistry was done according to previously published protocols (11, 12) by using the ABC-method with diaminobenzidine and/or Vector SG as chromogens (Vector Laboratories). The following antibodies were used: NT-11 to Aβ (6), AS42/14, which recognizes amino acid 42 of Aβ (6), A4CT to the C-terminal 100 amino acid of APP (ref. 14; courtesy of K. Beyreuther, Center for Molecular Biology, Heidelberg), β-dystroglycan (ref. 15; Novocastra, Newcastle upon Tyne, U.K.), synaptophysin (ref. 11; Dako), and CD11b (MAC-1) (ref. 10; Serotec). In situ hybridization and electron microscopy were done as described (10, 16).
Quantification of Vascular Amyloid.
CAA rating, mean diameter of affected vessels, and percent of vessel surface area covered by congophilic amyloid was assessed as detailed in the supplemental material on the PNAS web site, www.pnas.org.
CSF and Blood Collection for Biochemical Analyses.
A retro-orbital blood sample was collected in anesthetized animals by using heparin-coated capillary tubes and was immediately frozen. The cisternae magna was then surgically exposed and cleaned of blood, and a custom-made, calibrated glass pipette was inserted through the covering membranes into the cisterna magna. A slight suction was applied, yielding a CSF sample of 3–8 μl that was immediately frozen on dry ice. Any CSF samples contaminated with the slightest trace of blood were discarded. Human CSF samples were taken by lumbar puncture (courtesy of C. Hock, Univ. of Basel) (17).
SDS/PAGE and Western Blot Analysis.
Protein electrophoresis was performed with 0.75-mm bicine gels (18). Amounts corresponding to 1 or 2 μl of pure CSF were loaded, electrophoresed, and transferred to an immobilon-P membrane (Millipore), which was then boiled in PBS. Mouse monoclonal antibody 6E10 specific for human Aβ (ref. 19; courtesy of K. Kim and H. Wisniewski, New York State Institute for Basic Research in Developmental Disability, New York) was followed by peroxidase and chemiluminescence. Synthetic Aβ1–40 and Aβ1–42 peptides were obtained from Bachem. Cortex samples were from a homogenate of dissected neocortex, and 1 or 2 μl were loaded at a dilution of 1:44 (1 mg in 44 μl buffer). Some blots were stripped and reincubated with a polyclonal antibody (C8) against the 20 C-terminal amino acids of APP.
Results
Vascular Amyloid in APP23 Mice Exhibits Characteristics Similar to Human CAA.
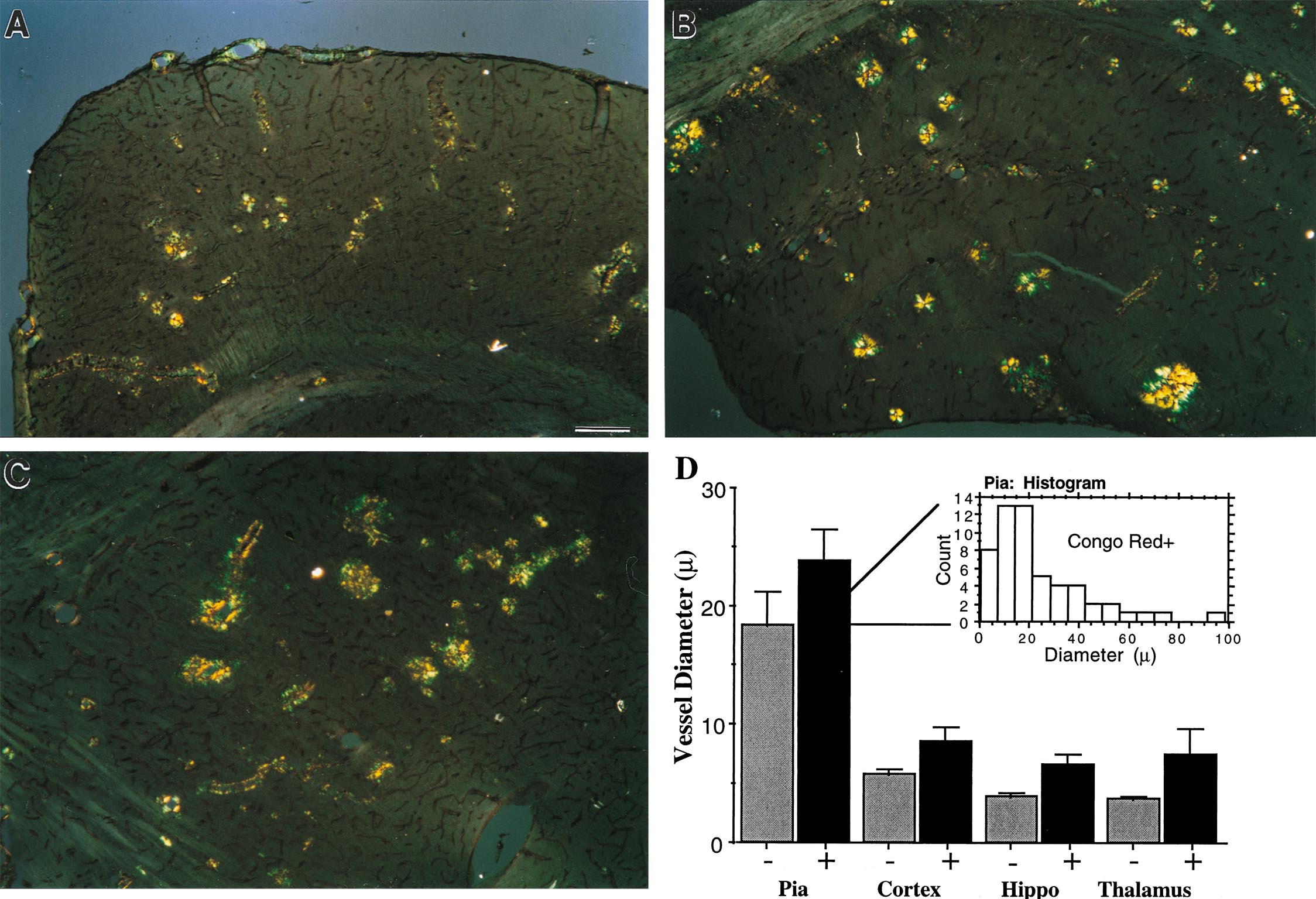
APP23 mice develop significant vascular amyloid deposits primarily in pial, thalamic, cortical, and hippocampal vessels as they age. In a subset of cortical (Fig. 1B), thalamic (Fig. 1A), and hippocampal vessels, Aβ immunostaining revealed penetration of the amyloid into the neuropil. Such dyshoric amyloid, which occurred most often in capillaries, increased proportionally to the number of vessels affected with amyloid. Vascular deposits could also be stained with antibodies specific for Aβ1–42, although this staining was less prominent (not shown). As in humans, individual vessels had a varying extent of amyloid deposition, and, within the same region, even vessels of the same subtype exhibited a wide heterogeneity of severity. Vessels stained with Thioflavin-S exhibited a pattern of fluorescence in concentric rings, and Congo red staining of affected vessels revealed typical yellow–green birefringence, both similar to humans and indicative of the presence of amyloid fibrils (Fig. 1 C and D). Inspection of Aβ staining on semi-thin sections revealed initial Aβ localization within the outer vessel wall (Fig. 1E). Based on anatomical inspection, arteries appeared to be more frequently affected than veins (Fig. 1F).
Figure 1.

CAA in APP23 mouse brain. (A and B) CAA is predominantly found in pial, cortical, hippocampal, and thalamic vessels. Shown is Aβ-immunostaining in the thalamus (A) and cortex (B) of a 16-month-old homozygous APP transgenic mouse. Vascular amyloid often infiltrates the nearby neuropil (arrowheads). (C) Thioflavin-S staining verified that the amyloid was fibrillary and the ring-like staining was similar to that seen in human CAA. (D) Congo red birefringence was also apparent in this section, which was double-stained for β-dystroglycan (brown) to visualize all vessels. Pathology ranged from amyloid confined to the vessel wall (arrows) to vessels with amyloid fibrils extending into the neuropil (arrowhead). (E) Semithin section of a vessel with Aβ-immunoreactivity confined to the outer vessel wall. (F) Side-by-side view of a Congo red-positive artery (arrowhead) and a Congo red-negative vein (arrow) at the pial surface. [Bars = 70 μm (A), 30 μm (B), 20 μm (C), 50 μm (D), 10 μm (E), and 25 μm (F).]
To verify the fibrillar nature of the amyloid and further characterize its localization, affected vessels were examined by electron microscopy (Fig. 2). Consistent with light microscopic observations and ultrastructural observations in AD (20), amyloid fibrils were most often found in association with the abluminal portion of the vessel wall. In more affected vessels, amyloid often formed a continuous ring within the vessel wall (Fig. 2A). High-magnification micrographs revealed amyloid fibrils similar in localization and appearance to those reported in human CAA (21). In less affected vessels, the basement membrane appeared to be intact, but, as pathology progressed, amyloid fibrils could be observed continuously from the endothelial cells into the neuropil, where the fibrils appeared to be surrounded by cytoplasm with a dense granular appearance similar to that of microglia or pericytes (Fig. 2C). In some cases, the normally tight endothelial cell junctions seemed to be disrupted.
Figure 2.

Ultrastructure of vascular amyloid in APP23 mice. (A) Cortical vessel of a 20-month-old hemizygous APP23 mouse that is surrounded by amyloid (arrowheads). Boxed area is shown in B. (B) High power view of amyloid fibrils (AF) between the endothelial cells (E) and the neuropil. (C) Fine radiating amyloid fibrils often infiltrate the nearby neuropil (arrowheads). Such infiltrating fiber-bundles are surrounded by a dense granular cytoplasm. [Bars = 5 μm (A) and 1 μm (B and C).]
CAA Increases with Age and Depends on APP Expression Level.
We have previously shown that the extent of amyloid plaque deposition in APP23 mice depends on the level of transgenic APP expression and increases with age (M.E.C., A.L.P., P. H. Kelly, K.-H.W., D.A., C.S.-P., A. Enz, B.S., M.S., and M.J., unpublished work). Here we have found that homozygous mice have significantly more vascular amyloid than hemizygous mice of the same age (Fig. 3A). With aging, a pronounced increase in vascular amyloid was noted (Fig. 3B), with detectable amyloid accumulation in vessels occurring slightly later than the first plaques. These observations suggest a common onset and progression of these two amyloid pathologies. However, after controlling for age, no significant relationship was observed between CAA grade and plaque load, as shown in Fig. 3C. Thus, although age and APP expression level are clear risk factors for amyloid deposition in both the parenchyma and the cerebral vasculature, our data point to the importance of other independent factors or stochastic events.
Figure 3.

Effects of APP expression, age, and plaque load on CAA. (A) Quantification of CAA grade indicates that homozygous mice (+/+) have significantly more vascular amyloid than hemizygous mice (+/−) within the same age-range. No vascular amyloid is observed in wild-type (wt) mice. Error bars indicate SEM. (B) CAA development is age-dependent, as shown for the hemizygous mice with ages ranging from 14–21 months. (C) Because the development of parenchymal amyloid (plaques) is also age-dependent, we used partial correlation to examine the relationship between amyloid plaque load and CAA grade with age held constant. Individual CAA grade and plaque load values were adjusted by a linear formula inverse to their respective age regression lines (age held constant at the mean of 18.3 months). The resulting graph (C) and partial correlation statistics (r = 0.21) failed to indicate a significant relationship between plaques and CAA.
Vascular Amyloid Occurred in a Wide Range of Vessels and Covers a Significant Portion of the Vessel Surface.
As summarized in Table 1 and detailed in the supplemental material, stereological quantitation of the percent vessel surface covered by amyloid indicated that a majority of pial vessels were affected, and a significant percentage of vessel surface area in thalamus, cortex, and hippocampus also contained fibrillary amyloid. The size distribution of affected vessels revealed that larger vessels were more likely to contain fibrillary amyloid, although vessels of all sizes, from capillaries to large arteries, were affected to some degree (see supplemental material).
Table 1.
Quantification of the percent of total vessel surface area that was Congo red-positive in various regions of three mice with the highest CAA grade
| Vascular amyloid | Pia | Cortex | Hippocampus | Thalamus |
|---|---|---|---|---|
| Percent of Congo red-positive surface area | 61 ± 1.3% | 8 ± 1.8% | 8 ± 2.6% | 13 ± 2.2% |
The mean plaque load of these three mice was 20%. Values are shown ± SEM.
Neurodegeneration Is Associated with Amyloid-Bearing Vessels.
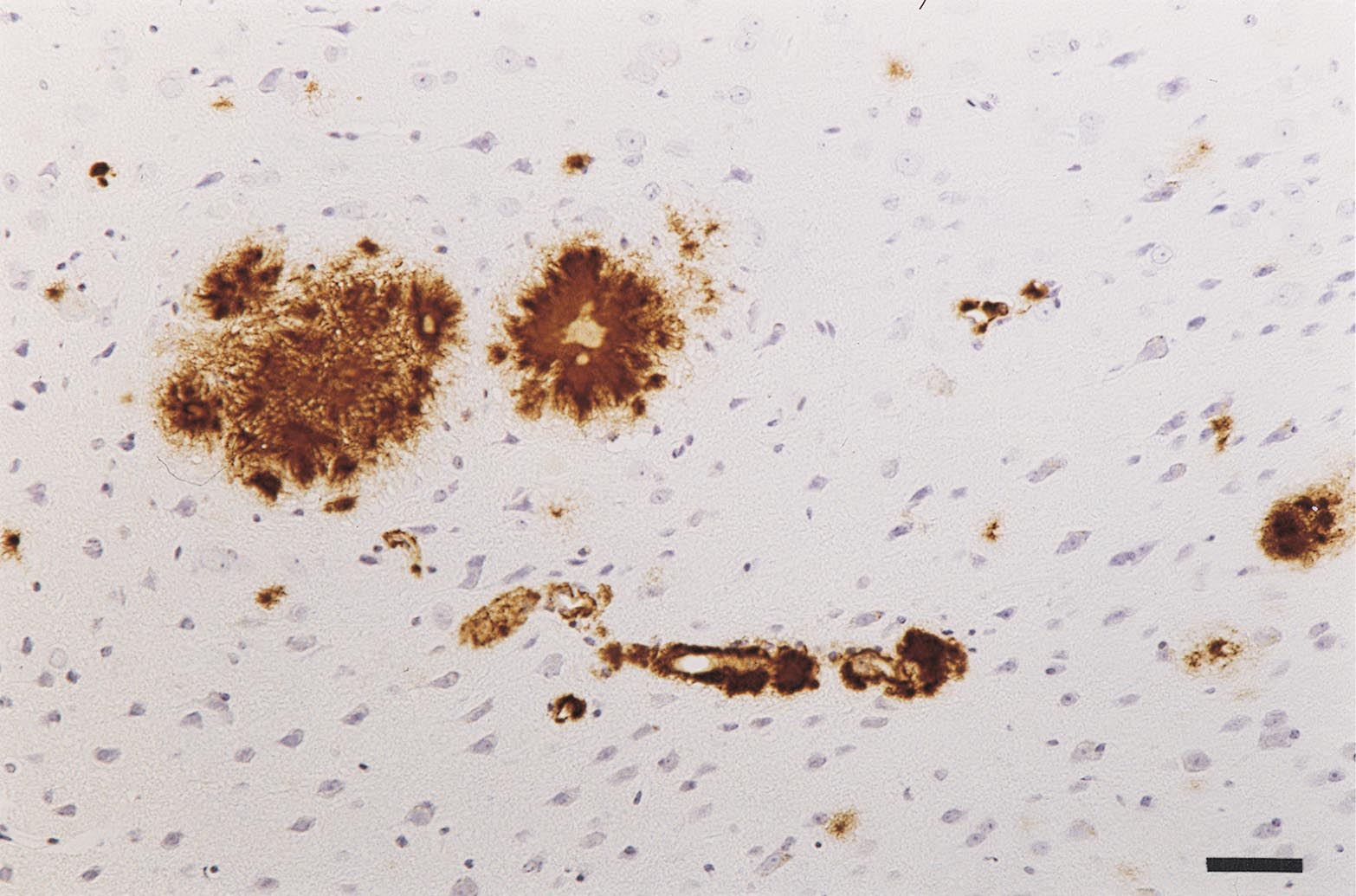
Qualitative evidence for neuron loss was frequently observed when amyloid fibrils extended from the vessels into the neuropil (Fig. 4A), suggesting either a toxic effect of Aβ or that nutritive substances can no longer be supplied from such vessels (local ischemia). In every example in which amyloid was present in the neuropil (plaques included), there was also evidence of activated microglia (Fig. 4B) and large, dystrophic synaptic boutons stained for synaptophysin (Fig. 4C).
Figure 4.

Neurodegeneration associated with vascular amyloid in APP23 mice. (A) Cresyl violet staining suggests neuron loss surrounding vessels with extensive vascular amyloid. (B) Activated microglia were observed when amyloid was present in the neuropil either in the form of plaques (asterisk) or dyshoric amyloid-containing vessels (arrowheads). (C) Synaptophysin-labeling reveals dystrophic boutons (arrowheads) around vascular amyloid infiltrating the neuropil. (D) Staining for iron (blue) shows microglia that have incorporated products from the blood—an indication of microhemorrhage. [Bars = 40 μm (A), 60 μm (B), 13 μm (C), and 10 μm (D).]
It is still unclear what factors lead to blood–brain barrier breakdown or cerebral hemorrhage in patients with CAA. When microhemorrhages occur, microglial cells in the vicinity are activated and internalize the various substances that are not normally found in the neuropil. To detect occurrence of microhemorrhages, we performed routine Berlin Blue staining for iron. In all of the animals that had significant vascular amyloid (CAA grade >30; n = 5), several examples of vessels surrounded by iron-positive microglia were apparent (Fig. 4D). The frequency of this observation was, however, much lower than the frequency of amyloid-bearing vessels, suggesting that microhemorrhages are a rare event and that other cofactors must be present for hemorrhages to occur (3). No evidence of iron-positive microglia were found in either control mice or mice with relatively little vascular amyloid (CAA grade <10). The presence of neuron loss, dystrophic synaptic boutons, activated microglia, and microhemorrhages suggest that CAA can lead to significant neurodegeneration.
Transport or Circulation of APP/Aβ Play a Role in Cerebral Amyloidosis.
In brain and other tissues, APP is normally ubiquitously expressed, including expression by all cell types within the vasculature (22, 23). In APP23 mice, the use of a murine Thy-1 promoter element limited transgenic APP expression to brain and, more specifically, to neurons (Fig. 5). Furthermore, within the brain, neuronal expression was limited to specific subregions (particularly the cortex, hippocampus, and amygdala). Substantial development of amyloid plaques and CAA occurred in subregions with prominent APP expression and also in regions without detectable expression levels of the transgene, most notably the thalamus (Fig. 5 and Table 1). The development of amyloid fibrils is concentration-dependent in vitro (24), and, for plaques and CAA to form in regions with low levels of expression, APP or Aβ must either be transported to that location (25) or must circulate through another mechanism: for instance, CSF (17), brain interstitial fluid (ISF) (26), or blood (27).
Figure 5.

Regional and neuron-specific expression of human APP in APP23 mice. (A) In situ hybridization for human APP reveals labeling in neocortex, hippocampus, and amygdala. Other regions, such as the thalamus, had no detectable APP expression. (B) Similarly, immunohistochemistry with an antibody specific to human APP reveals neuron labeling in the same regions. However, labeling of dystrophic boutons in the thalamus is also apparent (arrowheads). (C) Immunohistochemistry with an antibody to Aβ reveals vascular amyloid (arrowheads) and amyloid plaques in neocortex, hippocampus, amygdala, and also thalamus. (D) A high magnification view of a thalamic vessel stained for human APP indicates no expression of the transgene within the vasculature. (E and F) Amyloid-laden vessels are shown at high magnification for comparison of human APP expression (brown) and Aβ deposition (blue–gray) between thalamus (E) and neocortex (F). In the cortex, clear localization of APP is apparent within neurons and in dystrophic neurites around amyloid. In the thalamus, in contrast, no neuronal labeling is apparent, although APP is also localized within dystrophic neurites (arrowhead). [Bars = 500 μm (A–C), 10 μm (D), and 30 μm (E and F).]
In APP23 mice, where the thalamus contains significant pathology without significant transgene expression, a synaptic source of APP/Aβ must be considered due to prominent corticothalamic projections. However, amyloid fibrils are first apparent within the outer vessel wall, an area lacking significant synaptic contacts or human APP expression (Fig. 5D). In addition, the presence and early deposition of vascular amyloid within the pia, hippocampal fissure, and white-matter tracks argue against synaptic deposits as the primary mechanism by which amyloid is accumulated within the vessel wall, although transport from vasomotor nuclei cannot be excluded as a synaptic source of Aβ. An intriguing observation is prominent immunostaining for human APP in dystrophic boutons around perivascular amyloid, even in the thalamus and white-matter, which lack APP expression (Fig. 5 E and F). Thus, the most likely explanation is that human APP/Aβ is axonally transported and secreted and then diffuses or is further transported into the vessel proper, where fibrils start to form. As pathology progresses and amyloid fibrils extend into the neuropil, dystrophic boutons form, increasing the release of APP/Aβ and thus accelerating amyloid deposition in a feed-forward manner.
The Presence of High Levels of Aβ in the CSF Indicate that the Flow of Aβ from Neurons to CSF Facilitates Amyloid Deposition.
In humans, the presence of Aβ circulating in blood and CSF has been consistently described (17, 28, 29). Furthermore, Aβ injected into brain and/or CSF is cleared into blood (30), and Aβ injected into blood is sequestered by microvessels and is present in the CSF and brain parenchyma, where it can bind to existing amyloid deposits (27, 31). To further examine mechanisms of vascular amyloid formation, we investigated the presence of human Aβ in CSF and blood of APP23 mice. Results indicate that high levels of the neuronally expressed human Aβ are detectable in CSF by Western blotting (≈40 pg/μl; Fig. 6A). Levels of Aβ1–40 were higher than Aβ1–42, similar to that in brain parenchyma. CSF Aβ levels were ≈10× higher in APP23 mice than in humans (Fig. 6 B and C). Using the same techniques, no detectable Aβ was present in blood of APP23 mice (Fig. 6A), although trace amounts of Aβ were apparent using immunoprecipitation (data not shown). Thus, the flow of Aβ from neurons to CSF must be considered as a factor in the formation of Aβ deposits in the vasculature.
Figure 6.

High levels of human Aβ in CSF of APP23 mice. (A) Western blot for human Aβ in CSF (1 μl) from a nontransgenic control [wild-type (Wt)], APP23, and APP23 × App-null mouse, with cortex from an APP23 mouse and synthetic Aβ shown for comparison. The wild-type mice showed no reactivity to human Aβ. In contrast, APP23 and APP23 × App-null showed CSF levels of Aβ1–40 of ≈40 pg/μl. Aβ1–42 was also detectable; however, levels were much lower than Aβ1–40. APP (secreted and full length) was also detectable, and, in CSF, the ratio of Aβ to APP was much higher than in cortex, indicating the presence of cellular APP forms in cortex. When the blot was incubated with a C-terminal antibody (C8; not shown), the APP and Aβ bands from the CSF samples were no longer present, indicating that the APP band in CSF represents secreted APP. A nonspecific band (*) was observed in all CSF samples, which showed no relationship to either APP or Aβ levels and was highly variable from experiment to experiment. A 2-μl plasma sample from an APP23 mouse is shown with no detectable levels of Aβ indicated. (B) Comparison of Aβ and APP levels between mouse and human CSF. Again, high levels of Aβ were present in CSF (2 μl) from the two additional APP23 mice shown, which was not seen in the WT mouse. CSF samples from two AD patients and two aged human controls had much lower Aβ levels than the transgenic mice. Interestingly, the human CSF had a higher ratio of APP to Aβ, perhaps because of the Swedish mutation used in APP23 mice. (C) To better measure Aβ levels in human CSF, five times the volume was loaded (10 μl) and indicated values <5 pg/μl.
Amyloid Deposition and High CSF Aβ Levels Are also Present in APP23 Mice on an App-Null Background.
The endogenous mouse Aβ is produced by multiple cell types, and the relative contribution of the transgenic versus endogenous peptides is difficult to determine. Although no amyloid deposition is observed in nontransgenic mice, it is possible that human Aβ acts as a seed on which mouse Aβ is progressively deposited (24) and/or that human Aβ stimulates endogenous Aβ production in cells of the vessel wall that in turn may be locally deposited. Thus, we performed breeding between APP23 mice and App-null mice to produce mice expressing human APP on a null APP background, thereby eliminating endogenous mouse APP/Aβ from the system. In these mice, prominent vascular amyloid deposition was also observed, and observations at both the light and ultrastructural level were indistinguishable from those for the APP23 mice alone (see supplemental material). In addition, Aβ was present in CSF of these mice at similar levels (Fig. 6A), confirming that neuronally produced APP is further processed into Aβ and subsequently drains or is transported into CSF. Thus, the endogenous mouse Aβ does not seem to be necessary for formation of vascular amyloid deposits and further demonstrates that a neuronal source of APP is sufficient for induction of amyloid deposition, even in regions quite distant from the source of APP production.
Discussion
We have described a mouse model that, in addition to amyloid plaques, develops cerebrovascular accumulation of Aβ. The CAA and associated pathologies in this mouse exhibited a striking similarity to that observed in aged individuals and AD patients. CAA in APP23 mice led to focal neuron loss, dystrophic synaptic boutons, and activation of microglia, providing evidence that CAA leads to neurodegeneration. The presence of microhemorrhages in APP23 mice provides further evidence that CAA is a significant risk factor for these pathologies.
Mechanisms and Pathways of Vascular Aβ Deposition.
As has been described in naturally occurring CAA (1, 5), vascular amyloid deposition in APP23 mice is most frequently found in arteries/arterioles and occurs most often in vessels outside the brain parenchyma proper (e.g., pia, fissures). The arterial predilection suggests that an anatomical difference between arteries and veins (e.g., presence of significant amounts of smooth muscle) could contribute to the development of CAA. Indeed, it has been hypothesized that Aβ is deposited by vascular smooth muscle cells and/or perivascular microglia, and APP expression and Aβ production by vascular cells have been well documented (22, 23). These reports and the observation that vessels apart from the neuropil are more often affected strongly implicate local production or circulating Aβ as an important source, although these hypotheses fail to explain the exclusive localization of CAA to cerebral vessels. In the present study, through examination of APP transgenic mice on an App-null background, we have shown that a neuronal source of APP is sufficient to induce cerebrovascular amyloid, demonstrating that production of APP within the vasculature is not a necessary event for the formation of CAA.
The blood hypothesis for the mechanism of CAA suggests Aβ uptake from the blood and blood–brain barrier transport (27, 31). The lack of significant levels of Aβ in the blood of APP23 mice argues against this mechanism in our mice. Furthermore, other reports have demonstrated that transgenic mice that have high blood levels of Aβ and develop peripheral amyloid pathology do not develop cerebral amyloidosis (32, 33). Infusion studies have also suggested that, under physiological conditions, minimal amounts of Aβ are transported across the blood–brain barrier (34).
Elimination of vascular production and blood circulation as potential sources of Aβ in our mouse model led us to examine the pathway whereby neuronally produced APP travels to CSF from the brain parenchyma. Weller and colleagues (26, 35) have described a brain ISF drainage pathway along parenchymal vessels, arteries supplying the brain, olfactory epithelium, and, finally, the cervical lymph nodes. Furthermore, it has been suggested that significant amounts of Aβ drain along this pathway in humans (26). In APP23 mice, the preferential localization of CAA matched vessels implicated in ISF drainage. Arterioles supplying the regions with the highest levels of Aβ (cortex and adjacent regions) were most likely to be affected. Although the blood supply to the thalamus is not derived from regions of high APP expression, corticothalamic axonal transport and release of Aβ is a possible mechanism by which Aβ reaches the thalamus before draining along the vessels.
The unique neuronal source of APP in this model illustrates that Aβ circulates from the brain parenchyma into CSF and that this pathway may facilitate the formation of amyloid deposits within the vasculature. The ISF drainage pathway in the perivascular space is in close proximity to smooth muscle cells, which may facilitate amyloid deposition (36, 37). In addition, substances thought to bind amyloid and increase fibrilization such as heparan sulfate proteoglycans are also localized in the basement membrane (38).
APP23 mice express APP and Aβ at significantly higher levels than humans. Mechanisms of cerebrovascular amyloid deposition in these mice may therefore be different from mechanisms in humans, although our morphological observations suggest a similar pathogenesis. There may be other contributing factors in humans that are independent of Aβ level. For instance, age-related pathological changes, such as thickening of the vascular basement membrane, atherosclerosis, and hypertension could comprise the normal flow of ISF and accelerate Aβ accumulation.
Murine Models of CAA.
A mouse model of CAA offers distinct advantages over the existing models of naturally occurring CAA in squirrel monkeys and dogs (5). In this report, we have described a rodent model exhibiting striking similarities to human CAA and have made progress toward understanding significance and mechanisms of CAA. Another mouse model has been reported that exhibits vascular pathology, including a thickening of the basement membrane through overexpression of transforming growth factor β1 (39). Interestingly, when this mouse was crossed with an APP transgenic mouse, accumulation of Aβ was also observed in cerebral vessels (39), further indicating that breakdown of vascular processes can lead to Aβ accumulation, although the presence of neurodegeneration and other similarities to human CAA have not been reported in this model.
Although our results support the involvement of APP/Aβ transport and ISF drainage pathways in the pathogenesis of CAA, additional studies will be necessary to further isolate underlying factors and mechanisms responsible for CAA and related neurodegeneration. In addition, the expression of hereditary cerebral hemorrhage with amyloidosis-Dutch type mutant APP in transgenic mice may lead preferentially to CAA, allowing differentiation of the factors responsible for Aβ deposition in plaques versus vessels. Therapeutic interventions aimed at changing the level, drainage, or transport of Aβ may also lead to a better understanding of cerebrovascular amyloidosis. These new avenues of research are in the position to address several long-standing questions in the pathogenesis of AD and CAA.
Supplementary Material
Acknowledgments
The authors thank C. Hock for human CSF samples; K. Beyreuther, H. Wisniewski, and K. Kim for antibodies; T. Hartmann for experimental help; L. Walker for comments to the manuscript; D. Ingram for statistical advice; C. Bauer, H. Zysset, and S. Ipsen for photography and histology assistance; and T. O’Reilly and M. Krebs for advice regarding mouse CSF samples. This work was supported by grants from the Swiss National Science Foundation, the Roche Research Foundation, and the AETAS Foundation (Geneva).
Abbreviations
- AD
Alzheimer’s disease
- Aβ
amyloid β
- APP
amyloid precursor protein
- CAA
cerebral amyloid angiopathy
- CSF
cerebrospinal fluid
- ISF
interstitial fluid
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Vinters H V. Stroke. 1987;18:311–324. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- 2.Levy E, Carman M D, Fernandez-Madrid I J, Power M D, Lieberburg I, van Duinen S G, Bots G T, Luyendijk W, Frangione B. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 3.Itoh Y, Yamada M, Hayakawa M, Otomo E, Miyatake T. J Neurol Sci. 1993;116:135–141. doi: 10.1016/0022-510x(93)90317-r. [DOI] [PubMed] [Google Scholar]
- 4.Kawai M, Kalaria R N, Cras P, Siedlak S L, Velasco M E, Shelton E R, Chan H W, Greenberg B D, Perry G. Brain Res. 1993;623:142–146. doi: 10.1016/0006-8993(93)90021-e. [DOI] [PubMed] [Google Scholar]
- 5.Walker L C. Brain Res Brain Res Rev. 1997;25:70–84. doi: 10.1016/s0165-0173(97)00017-9. [DOI] [PubMed] [Google Scholar]
- 6.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold K H, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti P A, et al. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 8.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Nature (London) 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 9.Calhoun M E, Wiederhold K H, Abramowski D, Phinney A L, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M. Nature (London) 1998;395:755–756. doi: 10.1038/27351. [DOI] [PubMed] [Google Scholar]
- 10.Stalder M, Phinney A L, Probst A, Sommer B, Staufenbiel M, Jucker M. Am J Pathol. 1999;154:1673–1684. doi: 10.1016/S0002-9440(10)65423-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calhoun M E, Kurth D, Phinney A L, Long J M, Hengemihle J, Mouton P R, Ingram D K, Jucker M. Neurobiol Aging. 1998;19:599–606. doi: 10.1016/s0197-4580(98)00098-0. [DOI] [PubMed] [Google Scholar]
- 12.Jucker M, Walker L C, Schwarb P, Hengemihle J, Kuo H, Snow A D, Bamert F, Ingram D K. Neuroscience. 1994;60:875–889. doi: 10.1016/0306-4522(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 13.Gomori Am J Pathol. 1936;12:655–663. [PMC free article] [PubMed] [Google Scholar]
- 14.Dyrks T, Weidemann A, Multhaup G, Salbaum J M, Lemaire H G, Kang J, Muller-Hill B, Masters C L, Beyreuther K. EMBO J. 1988;7:949–957. doi: 10.1002/j.1460-2075.1988.tb02900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tian M, Jacobson C, Gee S H, Campbell K P, Carbonetto S, Jucker M. Eur J Neurosci. 1996;8:2739–2747. doi: 10.1111/j.1460-9568.1996.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 16.Sola C, Mengod G, Probst A, Palacios J M. Neuroscience. 1993;53:267–295. doi: 10.1016/0306-4522(93)90304-x. [DOI] [PubMed] [Google Scholar]
- 17.Hock C, Golombowski S, Muller-Spahn F, Naser W, Beyreuther K, Monning U, Schenk D, Vigo-Pelfrey C, Bush A M, Moir R, et al. Eur Neurol. 1998;39:111–118. doi: 10.1159/000007917. [DOI] [PubMed] [Google Scholar]
- 18.Klafki H W, Wiltfang J, Staufenbiel M. Anal Biochem. 1996;237:24–29. doi: 10.1006/abio.1996.0195. [DOI] [PubMed] [Google Scholar]
- 19.Kim K S, Miller D L, Sapienza V J, Chen C M J, Bai C, Grundke-Iqbal I, Currie J R, Wisniewski H M. Neurosci Res Commun. 1988;2:121–130. [Google Scholar]
- 20.Yamaguchi H, Yamazaki T, Lemere C A, Frosch M P, Selkoe D J. Am J Pathol. 1992;141:249–259. [PMC free article] [PubMed] [Google Scholar]
- 21.Wisniewski H M, Wegiel J, Wang K C, Lach B. Acta Neuropathol. 1992;84:117–127. doi: 10.1007/BF00311383. [DOI] [PubMed] [Google Scholar]
- 22.Natte R, de Boer W I, Maat-Schieman M L, Baelde H J, Vinters H V, Roos R A, van Duinen S G. Brain Res. 1999;828:179–183. doi: 10.1016/s0006-8993(99)01361-x. [DOI] [PubMed] [Google Scholar]
- 23.Kalaria R N, Premkumar D R, Pax A B, Cohen D L, Lieberburg I. Brain Res Mol Brain Res. 1996;35:58–68. doi: 10.1016/0169-328x(95)00180-z. [DOI] [PubMed] [Google Scholar]
- 24.Lansbury P T., Jr Neuron. 1997;19:1151–1154. doi: 10.1016/s0896-6273(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 25.Buxbaum J D, Thinakaran G, Koliatsos V, O’Callahan J, Slunt H H, Price D L, Sisodia S S. J Neurosci. 1998;18:9629–9637. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weller R O, Massey A, Newman T A, Hutchings M, Kuo Y M, Roher A E. Am J Pathol. 1998;153:725–733. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackic J B, Weiss M H, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic B V. J Neurochem. 1998;70:210–215. doi: 10.1046/j.1471-4159.1998.70010210.x. [DOI] [PubMed] [Google Scholar]
- 28.Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, Sandbrink R, Masters C L, Beyreuther K. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- 29.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, et al. Nature (London) 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 30.Ghersi-Egea J F, Gorevic P D, Ghiso J, Frangione B, Patlak C S, Fenstermacher J D. J Neurochem. 1996;67:880–883. doi: 10.1046/j.1471-4159.1996.67020880.x. [DOI] [PubMed] [Google Scholar]
- 31.Poduslo J F, Curran G L, Haggard J J, Biere A L, Selkoe D J. Neurobiol Dis. 1997;4:27–34. doi: 10.1006/nbdi.1997.0132. [DOI] [PubMed] [Google Scholar]
- 32.Kawarabayashi T, Shoji M, Sato M, Sasaki A, Ho L, Eckman C B, Prada C M, Younkin S G, Kobayashi T, Tada N, et al. Neurobiol Aging. 1996;17:215–222. doi: 10.1016/0197-4580(95)02061-6. [DOI] [PubMed] [Google Scholar]
- 33.Fukuchi K, Ho L, Younkin S G, Kunkel D D, Ogburn C E, LeBoeuf R C, Furlong C E, Deeb S S, Nochlin D, Wegiel J, et al. Am J Pathol. 1996;149:219–227. [PMC free article] [PubMed] [Google Scholar]
- 34.Shayo M, McLay R N, Kastin A J, Banks W A. Life Sci. 1997;60:PL115–PL118. doi: 10.1016/s0024-3205(96)00685-6. [DOI] [PubMed] [Google Scholar]
- 35.Weller R O. J Neuropathol Exp Neurol. 1998;57:885–894. doi: 10.1097/00005072-199810000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Van Nostrand W E, Melchor J P, Ruffini L. J Neurochem. 1998;70:216–223. doi: 10.1046/j.1471-4159.1998.70010216.x. [DOI] [PubMed] [Google Scholar]
- 37.Urmoneit B, Prikulis I, Wihl G, D’Urso D, Frank R, Heeren J, Beisiegel U, Prior R. Lab Invest. 1997;77:157–166. [PubMed] [Google Scholar]
- 38.Snow A D, Mar H, Nochlin D, Kimata K, Kato M, Suzuki S, Hassell J, Wight T N. Am J Pathol. 1988;133:456–463. [PMC free article] [PubMed] [Google Scholar]
- 39.Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L. Nature (London) 1997;389:603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}