

Abstract
Reactive oxygen intermediates (ROI) play a critical role in the defense of plants against invading pathogens. Produced during the “oxidative burst,” they are thought to activate programmed cell death (PCD) and induce antimicrobial defenses such as pathogenesis-related proteins. It was shown recently that during the interaction of plants with pathogens, the expression of ROI-detoxifying enzymes such as ascorbate peroxidase (APX) and catalase (CAT) is suppressed. It was suggested that this suppression, occurring upon pathogen recognition and coinciding with an enhanced rate of ROI production, plays a key role in elevating cellular ROI levels, thereby potentiating the induction of PCD and other defenses. To examine the relationship between the suppression of antioxidative mechanisms and the induction of PCD and other defenses during pathogen attack, we studied the interaction between transgenic antisense tobacco plants with reduced APX or CAT and a bacterial pathogen that triggers the hypersensitive response. Transgenic plants with reduced capability to detoxify ROI (i.e., antisense APX or CAT) were found to be hyperresponsive to pathogen attack. They activated PCD in response to low amounts of pathogens that did not trigger the activation of PCD in control plants. Our findings support the hypothesis that suppression of ROI-scavenging enzymes during the hypersensitive response plays an important role in enhancing pathogen-induced PCD.
Localized activation of programmed cell death (PCD) in response to microbial attack is thought to act as a defense mechanism that inhibits the growth of pathogens within infected plant tissues (1). By killing cells at and around the site of infection this process generates a physical barrier composed of dead plant cells and limits the availability of nutrients to the pathogen because of rapid dehydration that accompanies tissue death (2, 3). Also termed the hypersensitive response (HR), this cell death response is accompanied by the induction of numerous antimicrobial defenses. Among these are pathogenesis-related (PR) proteins, such as glucanases and chitinases, and phytoalexins (4). It is believed that the coordinated activation of PCD and defense mechanisms at the site of pathogen entry provides the plant with an efficient defense response that prevents pathogen proliferation and its possible consequence: systemic infection (1–6).
PCD that occurs during the HR is accompanied by an increase in the production of reactive oxygen intermediates (ROI) (7–9). Recent studies indicated that ROI in the form of H2O2 and O2−· may be key mediators of PCD during the HR (10–12). ROI also were implicated as signal transduction agents that lead to the induction of other defense mechanisms such as PR proteins (4), salicylic acid (SA), biosynthesis (13), and systemic acquired resistance (SAR; ref. 14). The production of O2−· was shown to activate PCD in an Arabidopsis cell death mutant (10), and H2O2 was shown to induce PCD and defense mechanisms in bean and tobacco (11, 12). Likewise, inhibitors of ROI production, or low oxygen pressure, were found to suppress HR cell death (10–12, 15). It is presumed that the recognition of an invading pathogen results in the activation of a plasma membrane-associated NAD(P)H oxidase and the excess production of O2−· (16). Subsequent spontaneous or superoxide dismutase (SOD)-catalyzed dismutation of O2−· leads to the accumulation of H2O2 that diffuses into cells and causes the activation of defense mechanisms and PCD (11, 12). In accordance with this model, ROI scavenging mechanisms such as ascorbate peroxidase (APX) and catalase (CAT) were shown to be suppressed during the HR, a process that was suggested to potentiate the induction of PCD and other defenses because of the increase in ROI levels that results from the decreased capability of cells to scavenge H2O2 during the HR (17, 18).
Although the relationship between ROI production and HR cell death is well documented, the majority of studies supporting this relationship were of a pharmacological nature. In addition, although it was suggested that the suppression of antioxidative mechanisms enhances pathogen-induced PCD, direct genetic evidence for this model was not presented. To examine the relationship between ROI production during the HR, the suppression of antioxidative mechanisms, and PCD, we examined the activation of PCD and defense mechanisms in transgenic tobacco plants with reduced capability to detoxify ROI. For this purpose we used tobacco plants that express antisense RNA for cytosolic APX (cAPX; ref. 19) or CAT (20–22). We found that such transgenic plants with reduced capability to detoxify ROI are hyperresponsive to pathogen attack. They activated PCD in response to low amounts of pathogens that did not trigger the activation of PCD in control plants. Our findings support the hypothesis that suppression of ROI-scavenging enzymes during the HR plays an important role in enhancing pathogen-derived PCD.
Materials and Methods
Plant Material.
The production of transgenic tobacco plants (Nicotiana tabacum cv. Bel W3) expressing antisense RNA for cAPX (antisense-APX) and their corresponding controls [WT(GUS)] were described previously (19). The production and characterization of transgenic tobacco plants (Nicotiana tabacum cv SR1) that express antisense RNA for CAT1 has been reported (AS1; refs. 20–22). Growth of plants and experiments were conducted under controlled environmental conditions at 22–24°C. Continuous illumination was provided by cool-white fluorescent lamps (150–200 μmol⋅m−2⋅sec−1). To ensure that HR cell death in plants was not affected by different environmental factors that may induce SAR or PR gene expression, thereby influencing the rate or intensity of the HR, the expression of PR-1 in leaves of transgenic and control plants was examined before pathogen infection.
Bacterial Infection.
Fully expanded leaves of 4- to 5-week-old plants were inoculated with Pseudomonas syringae pv. phaseolicola (NPS3121) or P. syringae pv tabaci (ATCC 11528) according to Mittler et al. (17). Mock-infected plants were infiltrated with water. Mock and pathogen-infected plants were kept at 22–24°C under continuous illumination (150–200 μmol⋅m−2⋅sec−1). At different times after infection, leaves were sampled, photographed, and analyzed for PCD induction and expression of the PR protein, PR-1, as described below. Additionally, bacteria were extracted from infected leaves and plated, as described previously (17), to assay for viability and in planta growth. In accordance with previous reports, P. syringae pv. phaseolicola (NPS3121) was unable to grow in planta in tobacco (17).
Paraquat Treatment.
Fully expanded leaves of 4- to 5-week-old control and antisense plants were infiltrated with different concentrations of paraquat (methyl viologen; Sigma) as described by Mittler et al. (23). Plants were incubated for 5 h at 24°C under continuous illumination (200 μmol⋅m−2⋅sec−1), sampled, and assayed for cell death as described below.
Measurement of Ion Leakage from Leaf Discs.
Cell death was assayed by measuring ion leakage from leaf discs (17, 23–25). For each measurement, five leaf discs (9-mm diameter) were floated abaxial side up on 5 ml of distilled water for 3 h at room temperature. After incubation, the conductivity of the bathing solution was measured with a Consort conductivity meter (model K511; Belgium), referred to as value A. The leaf discs then were returned to the bathing solution, introduced into sealed tubes, and incubated with the bathing solution at 95°C for 25 min. After cooling to room temperature the conductivity of the bathing solution again was measured; this is referred to as value B. For each measurement ion leakage was expressed as percent leakage, i.e., (value A/value B) × 100.
Field-Inversion Gel Electrophoresis (FIGE).
Fragmentation of nuclear DNA, a hallmark of apoptotic cell death, was detected via FIGE as described by Mittler et al. (25). FIGE was performed with a Hoefer Switchback Pulse Controller by using 1% agarose gels and 0.5× TBE buffer. Electrophoretic separation was performed at 6.9 V/cm by using a reverse mode with a forward/reverse ratio of 3.0:1 and a multiple run mode: RUN1, 10-min run-in followed by 8 h at a pulse time of 1–20 sec; RUN2, 8 h at a pulse time of 0.8–1.5 sec. After electrophoretic separation, the gels were stained with ethidium bromide and photographed. A λ DNA ladder (Promega) was used to determine the size of DNA fragments. After staining, DNA was transferred from gels to a nylon membrane and hybridized with a mixture of probes corresponding to different tobacco nuclear genes as described by Mittler et al. (25).
Isolation of Protein and Protein Blot Analysis
Immunodetection of cAPX (26), PR-1, rbcL, and HSP70 was performed by protein blot analysis of total leaf protein with a chemiluminescence detection system (SuperSignal; Pierce).
RNA Isolation and Analysis.
Total RNA was isolated as described previously (17) and subjected to RNA gel blot analysis. RNA gel blots were hybridized first with probes for PR-1 or cAPX and then with a probe for 18S rRNA to ensure equal loading of RNA. RNA gel blot hybridization and membrane washing were performed as described by Mittler et al. (17).
Treatment of Tobacco Leaves with High Oxygen Pressure.
Untreated, pathogen-, or mock-infected leaves were enclosed in gas-tight, temperature-controlled (22°C), transparent chambers (total volume, 1 liter), as described by Shulaev et al. (27); the rest of the plant remained exposed to ambient air. The atmosphere within the chambers was replaced by flushing them with a steady flow (100 ml/min) of ambient air or pure oxygen (high oxygen pressure).
Results
Characterization of Antisense-APX Plants.
The construction of cAPX antisense-expressing plants was described previously (19). As shown in Fig. 1A, F1 progenies of transgenic plants with reduced levels of cAPX mRNA contained no detectable levels of cAPX protein. As described previously, these plants showed no apparent phenotype when grown under controlled environmental conditions. During growth under controlled conditions the activities of other ROI-scavenging enzymes such as catalase, SOD, or glutathione reductase, as well as the level of ascorbic acid, were not significantly higher in these plants compared with control plants (data not shown). Although they lacked a major H2O2-scavenging enzyme and, therefore, may accumulate H2O2, PR-1 expression was not induced in untreated antisense-APX plants (Fig. 1A). This finding suggested that SAR was not activated in these plants and that SA levels were not elevated during normal growth under controlled conditions. As shown in Fig. 1B, antisense cAPX plants were more sensitive to treatment with paraquat (methyl viologen), a herbicide that elevates the cellular production of ROI. This result provided further support for the hyperresponsiveness of antisense-APX plants to treatments associated with enhanced levels of ROI (19). Normal growth under controlled conditions, hyperresponsiveness to ROI production, and the lack of an SAR marker (PR-1) during normal growth made these plants an ideal subject for the study of ROI involvement in the process of PCD during the HR.
Figure 1.

Analysis of transgenic tobacco plants with reduced cAPX protein. (A) A protein blot of transgenic and control plants showing that the level of cAPX protein is reduced dramatically in transgenic plants that express antisense RNA for cAPX compared with control plants. The level of PR-1 is shown not to be induced in untreated transgenic or control plants. As a positive control we used application of SA (1 mM; ref. 23). This treatment induced the expression of PR-1 in control plants (SA). The level of HSP70 and RbcL was used to demonstrate equal loading of protein. Analysis was performed on two individual transgenic plants and two individual control plants. (B) Hyperresponsiveness of transgenic plants with reduced levels of cAPX to oxidative stress induced by paraquat. The graph shows enhanced cell death in antisense-APX plants exposed to different concentrations of paraquat compared with control plants.
Response of Antisense-APX Plants to Pathogen Attack.
It was reported previously that cAPX expression is posttranscriptionally suppressed during the HR of tobacco plants infected with viral or bacterial pathogens (17). To examine the contribution of APX suppression to the induction of PCD during pathogen infection, we compared the bacteria-induced HR of antisense-APX plants with that of control plants.
Because HR cell death is thought to play an active role in inhibiting pathogen growth, the extent of plant cell death induced by a specific pathogen may be affected by the rate of pathogen growth. To compare the extent of pathogen-induced cell death between antisense-APX and control plants, in a manner that is independent of the rate of pathogen growth, we used a bacterial strain that induces the HR, but is unable to grow in planta in tobacco (P. syringae pv. phaseolicola NPS3121; ref. 15). By infiltrating leaves of control and transgenic plants with the same amount of bacteria we could compare the extent of cell death between the different plants. As shown in Fig. 2, compared with control plants, the extent of bacteria-induced cell death was higher in transgenic tobacco plants with reduced cAPX protein. This difference was especially notable at low titers of bacteria (i.e., a bacterial titer of OD600 = 0.1), suggesting that the low level of ROI produced at these titers was sufficient to trigger the HR in antisense plants. In contrast, control plants, capable of more efficient ROI removal, did not activate the HR when infiltrated with the same low titer of bacteria. As a control for PCD induced by bacteria, leaves of control and antisense plants were infiltrated with a Hrp− derivative of NPS3121 (NPS4000). These bacteria did not induce a HR in control or transgenic plants (data not shown).
Figure 2.

Enhanced HR cell death in transgenic tobacco plants expressing antisense RNA for cAPX. (A) Photograph showing that infiltration of antisense-APX leaves (Right) with a low titer of bacteria (OD600 = 0.1) induces HR cell death. In contrast, infiltration of control leaves (Left) with the same titer failed to induce PCD. Infiltration of leaves with a high titer of bacteria (OD600 = 1) induced cell death in both plants. Mock infiltration (i.e., OD600 = 0) was performed with water. (B) A graph showing enhanced HR cell death in antisense-APX plants infiltrated with different concentrations of the HR-inducing bacteria, P. syringae pv. phaseolicola (NPS3121), compared with control plants infiltrated with the same amount of bacteria. An OD600 of 1.0 is the equivalent of 500,000 colony-forming unit (cfu)/ml.
As shown in Fig. 3A, induction of cell death in transgenic plants with reduced levels of cAPX occurred at about 12 h postinfection (using a bacterial titer of OD600 = 0.1). Under these conditions very little cell death was observed in control plants. To examine the effect that the increase in ROI levels, and the enhanced cell death, had on the activation of defense gene expression, we measured the increase in PR-1 steady-state mRNA level in transgenic and control plants during bacteria-induced HR (Fig. 3A). As shown in Fig. 3B, PR-1 mRNA accumulated in transgenic as well as control plants in a similar manner in response to infection with bacteria (OD600 = 0.1). Thus, although cell death was not enhanced in control plants (Fig. 3A), PR-1 gene expression was activated. This finding suggested that HR cell death can be uncoupled from defense gene activation, at least under the experimental conditions that we used.
Figure 3.
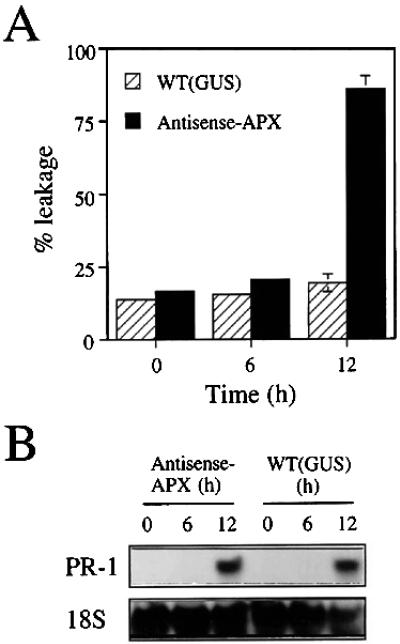
Time course of cell death and PR-1 induction during bacteria-triggered HR. (A) Induction of HR cell death in transgenic antisense-APX plants compared with control plants. Bacteria (OD600 = 0.1) was infiltrated into leaves of control and antisense plants. Leaves were sampled at different times and analyzed for the induction of HR cell death. (B) Induction of PR-1 mRNA in transgenic and control plants infected with bacteria. Leaf samples were obtained as described in A and analyzed for the level of PR-1 mRNA.
Inhibition of Pathogen-Induced Cell Death in Antisense-APX Plants.
To confirm that the enhanced HR cell death observed in transgenic cAPX antisense plants resulted from their inability to scavenge the increased levels of ROI produced during the HR, we tested whether antioxidants or inhibitors of ROI production would suppress the enhanced cell death observed in these plants in response to infection with low titers of bacteria (shown in Figs. 2 and 3A). As shown in Fig. 4A, infiltration of leaves with bacteria and a mixture of antioxidants [20 mM reduced ascorbic acid and 10 mM reduced glutathione or 1,000 units/ml CAT] suppressed bacteria-induced cell death. Extraction of bacteria from inoculated leaves, followed by a viability assay, confirmed that the inhibition of PCD did not result from an adverse effect of the antioxidants on bacteria viability (data not shown). As shown in Fig. 4B, a similar observation was made when bacteria were infiltrated into leaves with 10 μM of diphenyleneiodonium (DPI), an inhibitor of the plasma membrane-associated NAD(P)H oxidase complex that is thought to generate O2−· during bacteria-induced HR (10, 11).
Figure 4.

Antioxidants and an inhibitor of ROI generation during the HR suppress bacteria-induced cell death in transgenic plants with reduced cAPX protein. (A) Inhibition of HR cell death by a mixture of antioxidants (ASC+GSH; 20 mM reduced ASC and 10 mM reduced GSH) or by CAT (1,000 units/ml). CAT or antioxidants were infiltrated into leaves of control and transgenic plants together with bacteria (OD600 = 0.1). Twelve hours after inoculation, leaves were sampled and analyzed for cell death. (B) Inhibition of HR cell death by an inhibitor of the NAD(P)H oxidase complex (DPI; 10 μM). DPI was infiltrated into leaves of control and transgenic plants together with bacteria (OD600 = 0.1). Twelve hours after inoculation leaves were sampled and analyzed for cell death. (C) Inhibition of bacteria-induced nuclear DNA fragmentation in transgenic plants with reduced cAPX protein by DPI. Total DNA was extracted from duplicate leaf samples that were obtained as described in B. DNA fragmentation was assayed by FIGE.
The appearance of bacteria-induced HR, i.e., lesions, that developed during infiltration of antisense cAPX leaves with a low titer of bacteria was similar to the appearance of lesions that developed on control plants at high titers of bacteria (Fig. 2A). It was reported previously that bacteria-induced PCD in plants is accompanied by the fragmentation of nuclear DNA to large (50-kb) fragments (25), a process that is one of the hallmarks of apoptosis (28). To examine whether cell death that occurred in cAPX antisense plants infected with low titers of bacteria was accompanied by a similar process and to test whether this process depends on ROI production, we extracted DNA from control and treated plants and subjected this DNA to analysis via FIGE. As shown in Fig. 4C, cell death that occurred in antisense plants was accompanied by fragmentation of nuclear DNA, indicating that this process of cell death is similar to PCD that occurred during the HR of control plants infected with a high titer of bacteria (OD600 = 1.0; see also Fig. 5C). Moreover, cell death (Fig. 4B) as well as fragmentation of nuclear DNA (Fig. 4C) were inhibited by DPI (10 μM), an inhibitor of ROI production during the HR (10–12).
Figure 5.

Enhanced pathogen-induced PCD in transgenic tobacco plants with reduced catalase (AS1). (A) A protein blot showing that PR-1 protein does not accumulate in AS1 plants growing under controlled conditions. PR-1 expression is shown to be induced after infection of plants with bacteria (P. syringae pv. phaseolicola, NPS3121; OD600 = 0.1). (B) A graph showing enhanced HR cell death in AS1 plants infiltrated with different concentrations of bacteria compared with wild-type plants infiltrated with the same amount of bacteria. (C) Cell death induced in AS1 plants by a low titer of bacteria is accompanied by fragmentation of nuclear DNA. Wild-type and AS1 plants were infiltrated with a high (OD600 = 1) or low (OD600 = 0.1) titer of bacteria. At 18 h postinfection total DNA was extracted from duplicate leaf samples and DNA fragmentation was assayed by FIGE. (D) Rapid development of necrotic lesions in AS1 plants in response to infiltration with the bacterial pathogen P. syringae pv. tabaci. Wild-type and AS1 plants were infiltrated with water (Mock) or P. syringae pv. tabaci (OD600 = 1) and photographed 18 h after inoculation. An OD600 of 1.0 is the equivalent of 500,000 cfu/ml.
Response of Transgenic Tobacco Plants with Reduced CAT to Pathogen Attack.
CAT is a key H2O2-scavenging enzyme in plants (21). It was reported previously that CAT is suppressed during the interaction of plants with invading pathogens. Two different modes of CAT inhibition were suggested to occur during the HR: inhibition of CAT activity by SA (29) and suppression of CAT expression at the level of steady-state mRNA level (18). It was suggested that CAT suppression results in the augmentation of pathogen-induced PCD and the activation of defense mechanisms (12, 20, 29, 30). To test this hypothesis we challenged transgenic tobacco plants with reduced CAT1 mRNA and protein (AS1) with a bacterial pathogen that induces the HR (as shown in Figs. 2–4 for antisense-APX plants).
AS1 transgenic tobacco plants have been characterized extensively (20–22). They are hyperresponsive to treatments that result in the excess production of ROI (21). Moreover, the application of some of these treatments to AS1 plants results in the induction of PR proteins such as PR-1 (20, 22), indicating that in these plants enhanced ROI levels may activate PR expression and the SAR-signaling pathway. Before pathogen infection we examined whether AS1 leaves grown under controlled conditions contain detectable levels of PR-1 protein. As shown in Fig. 5A, leaves of AS1 plants grown under controlled conditions did not contain PR-1 protein. Expression of PR-1 was induced in plants by infiltration of leaves with a low titer of bacteria (NPS3121; OD600 = 0.1). These results indicated that at least under the controlled growth conditions that we used, SAR was not induced in transgenic AS1 plants and that these plants could be used for the study of ROI involvement in the induction of PCD during the HR.
To examine the extent of pathogen-induced PCD in AS1 plants we infected control and AS1 plants with different titers of bacteria. As shown in Fig. 5B, AS1 plants were hyperresponsive to the application of bacteria. As with antisense APX-plants (Fig. 2B), the hyperresponsiveness of AS1 plants was especially notable at low titers of bacteria. These findings support the hypothesis that reduced capability of cells to scavenge ROI results in an enhanced pathogen-induced PCD. To confirm that the enhanced cell death observed at a low bacterial titer (OD600 = 0.1) is similar to the PCD response of plants to a high titer (OD600 = 1), we compared the mode of DNA fragmentation between control and AS1 plants infected with low and high titers of bacteria. As shown in Fig. 5C, cell death initiated by infection of AS1 plants with a low titer of bacteria resulted in a similar pattern of DNA fragmentation as cell death induced by infiltration of control or AS1 leaves with a high titer of bacteria. This observation suggested that the cell death observed in AS1 plants at low titers of bacteria resulted from the activation of PCD. PCD induced in AS1 plants infected with a low titer of bacteria was inhibited when bacteria was infiltrated with a mixture of antioxidants or CAT (data not shown), suggesting that ROI play a direct role in the induction of PCD under these conditions.
AS1 plants, grown under controlled conditions, were challenged with the bacterial pathogen P. syringae pv. tabaci. As shown in Fig. 5D, they were more susceptible to attack by this pathogen and developed necrotic lesions at a rate that was faster than wild-type plants. AS1 plants were also hyperresponsive to the fungal elicitor cryptogein (A. C. Allan and R. Fluhr, personal communication).
Effect of Changes in Plant ROI Metabolism on HR Cell Death.
Our findings provide strong support for the involvement of ROI and ROI metabolism in HR cell death. To examine further the role ROI may play in mediating HR cell death we tested whether increasing the production of ROI during infection of plants with bacteria stimulated PCD. As shown in Fig. 6A, infection of plants with bacteria under conditions that enhanced the formation of ROI (i.e., incubation of infected leaves with an atmosphere of pure oxygen; ref. 15) resulted in an enhanced activation of PCD. Because treatment at high oxygen pressure enhances the production of ROI [indicated by the induction of mRNAs encoding SOD and cAPX in mock-inoculated leaves incubated at high oxygen pressure (15); data not shown, see also Fig. 6C], the enhanced PCD that accompanies this treatment may mimic the enhanced cell death observed in CAT or APX antisense plants infected with a low titer of bacteria.
Figure 6.

Enhancement or suppression of HR cell death by alterations in ROI metabolism. (A) Enhanced HR cell death in tobacco plants infected with bacteria at high oxygen pressure. Leaves of wild-type Bel-W3 plants were infiltrated with bacteria and treated with high oxygen pressure. Twelve hours after inoculation, leaves were sampled and analyzed for cell death. (B) Suppression of pathogen-induced PCD by pretreatment of leaves with high oxygen pressure. Leaves of wild-type Bel-W3 plants were treated with high oxygen pressure for 5 h. After treatment, leaves were taken out of the chambers, incubated for 30 min at ambient conditions, and challenged with different concentrations of bacteria. Twelve hours after inoculation, leaves were sampled and analyzed for cell death. (C) RNA gel blot showing that treatment of leaves with high oxygen pressure for 5 h results in the induction of cAPX mRNA. An OD600 of 1.0 is the equivalent of 500,000 cfu/ml.
Because suppressing the ROI-scavenging capability of plants via transgene technology resulted in an enhanced HR cell death, we examined whether inducing the ROI-scavenging mechanisms of the plant before pathogen infection will result in the suppression of PCD. As shown in Fig. 6B, pretreatment (5 h) of plants with high oxygen pressure resulted in the suppression of HR cell death in response to subsequent infection with bacteria. As shown in Fig. 6C, pretreatment with oxygen for 5 h resulted in the induction of anti-ROI defense mechanisms such as APX. The finding that induction of ROI-scavenging mechanisms before pathogen challenge results in the suppressing of HR cell death suggests that these mechanisms may interfere with the PCD response of plants, unless properly suppressed. Thus, the suppression of ROI-scavenging systems during the HR may be a critical step in mounting an adequate defense response.
Discussion
ROI appear to play a crucial role in plants. They are involved in a diverse array of biological processes such as response to environmental stress, injury, or pathogen infection (i.e., the HR). Interestingly, ROI may play different or even opposing roles during different processes. During the HR they are actively produced to trigger different defenses such as PCD. On the other hand, their production during environmental stress is thought to be a byproduct of stress metabolism and a danger to the well being of the cell. The steady-state level of ROI within cells is determined by an interplay between the activity of ROI-generating mechanisms such as the NAD(P)H oxidase complex or different electron transfer mechanisms and the activity of ROI-detoxifying enzymes such as SOD, APX, or CAT. During environmental stresses when the production of ROI poses a danger to cells, ROI-scavenging mechanisms are induced to remove ROI and protect cells. However, during the HR when ROI production is enhanced and ROI are required to trigger the HR, the expression of ROI-scavenging mechanisms is thought to be suppressed to amplify the accumulation of ROI. It was shown previously that suppression of ROI-scavenging mechanisms such as cAPX or CAT results in greater sensitivity of plants to different environmental stresses (19, 21). These findings provided direct evidence for the importance of ROI removal and ROI-removal mechanisms to the protection of plants during stress. Here we present evidence that the suppression of ROI-scavenging mechanisms such as cAPX or CAT during the HR plays an important role in the activation of the HR. Thus, a reduction in the ROI-scavenging capability of cells enhances the cell death response that is induced upon pathogen attack.
Jabs et al. (10) presented key evidence for the involvement of an O2−·-generating system in the propagating cell death response of lsd1, an Arabidopsis cell death mutant that is deficient in the negative control of HR cell death (31). Their work provided strong genetic evidence for the previously established link between ROI such as O2−· or its dismutated product, H2O2, and pathogen-induced PCD (7–9). The biological phenomenon of pathogen-induced HR cell death that is mediated by ROI led to the prediction that for PCD to occur in an efficient manner, host mechanisms for ROI removal such as APX and CAT should be suppressed. It was found that during pathogen-induced HR the expression of CAT mRNA was down-regulated (18). In addition, the translation of mRNA encoding cAPX was found to be inhibited during the HR (17). The potential of SA that is produced during the HR to inactivate CAT and APX activities also was suggested to contribute to the enhancement of HR cell death (12, 29, 32). Here we report that transgene suppression of antiperoxidative enzymes such as cAPX or CAT results in an enhanced PCD in response to pathogen attack. Our findings provide direct evidence for the involvement of ROI in mediating pathogen-induced PCD and support the hypothesis that suppression of anti-ROI mechanisms during the HR contributes to the enhancement of ROI accumulation and the subsequent induction of pathogen-induced PCD.
In support of our findings that transgenic plants unable to properly remove ROI during the HR are hyperresponsive to pathogen attack, we found that infection of plants under conditions that potentiate the production of ROI (i.e., high oxygen pressure) resulted in the enhancement of PCD. Thus, increasing the production of ROI during the HR resulted in an enhanced rate of PCD. Enhanced production of ROI in transgenic plants that express a mutant calmodulin (VU-3) also was reported recently to result in an enhanced HR cell death in response to infection with a bacterial pathogen (33). In a previous study we found that tobacco mosaic virus (TMV)-induced HR was not affected by a treatment with high oxygen pressure (15). This finding may suggest that, in tobacco, the mechanism of ROI production during bacteria-induced HR is different from that activated during TMV-induced HR. In support of this possibility, DPI that was found to inhibit bacteria-induced HR in tobacco (Fig. 4) did not inhibit TMV-induced HR in tobacco (23). It is possible that in tobacco different mechanisms for ROI production are activated by different pathogens as suggested by Allan and Fluhr (34).
In contrast to the enhancement of the cell death response that occurred when plants were infected and immediately subjected to high oxygen pressure, pretreatment with high oxygen pressure for 5 h before pathogen infection resulted in the suppression of PCD. Pretreatment with high oxygen pressure resulted in the induction of ROI-scavenging mechanisms. These may scavenge the ROI produced during the initial phases of the HR and thereby prevent or suppress the initiation of the HR cascade. However, in this particular experimental design we cannot rule out the possibility that pretreatment with high oxygen pressure resulted in the induction of SAR or a related mechanism that enhanced resistance to the pathogen and thereby avoided the HR. Although it did not result in the induction of PR-1 (data not shown), it is possible that high oxygen treatment still induced other defense mechanisms. An alternative might have been using transgenic plants with elevated levels of ROI-scavenging mechanisms. However, in contrast to the induction of ROI-scavenging mechanisms by high oxygen pressure, which is likely to include multiple ROI-scavenging systems, overexpression of a particular ROI-scavenging enzyme may not provide the plant with an efficient ROI-removing capability. Indeed, there was no statistically significant difference between bacteria-induced cell death in wild-type plants and transgenic tobacco plants that overexpress cAPX or SOD (S. Ajit and B. Zilinskas, personal communication).
Our findings with transgenic plants that express antisense RNA for cAPX or CAT may have biotechnological implications. Antisense transgenes may be used to enhance the response of plants to pathogens and to facilitate the rate of pathogen-induced PCD in infected plant tissue. Such a control over PCD activation or propagation may be especially critical for plant–pathogen interactions in which PCD is activated upon pathogen challenge but at a rate that is not rapid enough to block pathogen proliferation (1). The expression of the antisense transgenes can be controlled via an inducible promoter such as a pathogen-responsive promoter (35). This promoter will be activated upon pathogen infection and cause a reduction in the ROI-scavenging capability of infected cells. As described above, this reduction will result in an enhanced PCD and the inhibition of pathogen spread. Because other organisms, including mammals, use ROI as a trigger for the activation of PCD (16), this strategy may also be used to fight infection or disease in other organisms (using antisense constructs that will target the key ROI-detoxifying enzymes of the particular organism).
In our study we used transgenic plants with reduced cAPX or CAT. However, during the HR of a wild-type plant infected with an avirulent pathogen, the expression and activity of both enzymes are suppressed. Because we find that the suppression of only one enzyme is sufficient to facilitate PCD, it is likely that during the HR in plants the suppression of both will have a dramatic effect on the ROI-removing capability of cells. As demonstrated in the current report, this suppression plays a key role in the development of an efficient defense response in plants by enhancing pathogen-induced PCD. ROI were shown recently to act as systemic signals for the induction of SAR and antioxidative mechanisms (36, 37). It would be interesting to examine whether suppression of ROI-scavenging mechanisms also is involved in the transmission of these signals.
Acknowledgments
We thank Drs. Robert Fluhr, Daniel Klessig, Eric Lam, Peter Lindgren, Rachel Nechushtai, and Barbara A. Zilinskas for gifts of plasmids, antibodies, and bacterial strains. We also thank Dr. Leonora Reinhold for critical comments. We gratefully acknowledge Drs. Barbara A. Zilinskas, Robert Fluhr, Seena Ajit, and Andrew C. Allan for sharing unpublished results. This work was supported by funding provided by The Yigal Alon Fellowship, The Hebrew University Intramural Research Fund Basic Project Awards, and The Natural Sciences and Engineering Research Council of Canada.
Abbreviations
- APX
ascorbate peroxidase
- cAPX
cytosolic APX
- CAT
catalase
- HR
hypersensitive response
- PCD
programmed cell death
- PR
pathogen related
- ROI
reactive oxygen intermediates
- SA
salicylic acid
- SAR
systemic acquired resistance
- SOD
superoxide dismutase
- FIGE
field-inversion gel electrophoresis
- DPI
diphenyleneiodonium
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Goodman R N, Novacky A J. The Hypersensitive Response Reaction in Plants to Pathogens: A Resistance Phenomenon. St. Paul, MN: Am. Phytopathol. Soc.; 1994. [Google Scholar]
- 2.Dangl J L, Dietrich R A, Richberg M H. Plant Cell. 1996;8:1793–1807. doi: 10.1105/tpc.8.10.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenberg J T. Proc Natl Acad Sci USA. 1996;93:12094–12097. doi: 10.1073/pnas.93.22.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Y, Shah J, Klessig D F. Gen Dev. 1997;11:1621–1639. doi: 10.1101/gad.11.13.1621. [DOI] [PubMed] [Google Scholar]
- 5.Mittler R, Lam E. Trends Microbiol. 1996;4:10–15. doi: 10.1016/0966-842x(96)81499-5. [DOI] [PubMed] [Google Scholar]
- 6.Pennel R I, Lamb C. Plant Cell. 1997;9:1157–1168. doi: 10.1105/tpc.9.7.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mehdy M C. Plant Physiol. 1994;105:467–472. doi: 10.1104/pp.105.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker C J, Orlandi E W. Annu Rev Phytopathol. 1995;33:299–321. doi: 10.1146/annurev.py.33.090195.001503. [DOI] [PubMed] [Google Scholar]
- 9.Hammond-Kosack K E, Jones J D G. Plant Cell. 1996;8:1773–1791. doi: 10.1105/tpc.8.10.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jabs T, Dietrich R A, Dangl J. Science. 1996;273:1853–1856. doi: 10.1126/science.273.5283.1853. [DOI] [PubMed] [Google Scholar]
- 11.Levine A, Tenhaken R, Dixon R A, Lamb C. Cell. 1994;79:583–593. doi: 10.1016/0092-8674(94)90544-4. [DOI] [PubMed] [Google Scholar]
- 12.Draper J. Trends Plant Sci. 1997;2:162–165. [Google Scholar]
- 13.Leon J, Lawton M A, Raskin I. Plant Physiol. 1995;108:1673–1678. doi: 10.1104/pp.108.4.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryals J A, Neuenschwander U H, Willits M G, Molina A, Steiner H Y, Hunt M D. Plant Cell. 1996;8:1809–1819. doi: 10.1105/tpc.8.10.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mittler R, Shulaev V, Seskar M, Lam E. Plant Cell. 1996;8:1991–2001. doi: 10.1105/tpc.8.11.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jabs T. Biochem Pharmacol. 1999;57:231–245. doi: 10.1016/s0006-2952(98)00227-5. [DOI] [PubMed] [Google Scholar]
- 17.Mittler R, Feng X, Cohen M. Plant Cell. 1998;10:461–474. doi: 10.1105/tpc.10.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dorey S, Baillieul F, Saindrenan P, Fritig B, Kauffmann S. Mol Plant Microb Inter. 1998;11:1102–1109. [Google Scholar]
- 19.Orvar B L, Ellis B E. Plant J. 1997;11:1297–1305. doi: 10.1046/j.1365-313x.1997.11020203.x. [DOI] [PubMed] [Google Scholar]
- 20.Chamnongpol S, Willekens H, Langebartels C, Van Montagu M, Inze D, Van Camp W. Plant J. 1996;10:491–503. [Google Scholar]
- 21.Willekens H, Chamnongpol S, Davey M, Schraudner M, Langebartels C, Van Montagu M, Inze D, Van Camp W. EMBO J. 1997;16:4806–4816. doi: 10.1093/emboj/16.16.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chamnongpol S, Willekens H, Moeder W, Langebartels C, Sandermann H, Jr, Van Montagu M, Inze D, Van Camp W. Proc Natl Acad Sci USA. 1998;95:5818–5823. doi: 10.1073/pnas.95.10.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mittler R, Shulaev V, Lam E, Cohen M. Plant Mol Biol. 1998;39:1025–1035. doi: 10.1023/a:1006110223774. [DOI] [PubMed] [Google Scholar]
- 24.Greenberg J T, Ausubel F M. Plant J. 1993;4:327–342. doi: 10.1046/j.1365-313x.1993.04020327.x. [DOI] [PubMed] [Google Scholar]
- 25.Mittler R, Simon L, Lam E. J Cell Science. 1997;110:1333–1344. doi: 10.1242/jcs.110.11.1333. [DOI] [PubMed] [Google Scholar]
- 26.Mittler R, Zilinskas B A. Plant Physiol. 1991;97:962–968. doi: 10.1104/pp.97.3.962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shulaev V, Leon J, Raskin I. Plant Cell. 1995;7:1691–1701. doi: 10.1105/tpc.7.10.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peitsch M C, Polzar B, Stephan H, Crompton T, MacDonald H R, Mannherz H G, Tschopp J. EMBO J. 1993;12:371–377. doi: 10.1002/j.1460-2075.1993.tb05666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z, Silva H, Klessig D F. Science. 1993;262:1883–1886. doi: 10.1126/science.8266079. [DOI] [PubMed] [Google Scholar]
- 30.Takahashi H, Chen Z, Du H, Liu Y, Klessig D F. Plant J. 1997;11:993–1005. doi: 10.1046/j.1365-313x.1997.11050993.x. [DOI] [PubMed] [Google Scholar]
- 31.Dietrich R A, Richberg M H, Schmidt R, Dean C, Dangl J L. Cell. 1997;88:685–694. doi: 10.1016/s0092-8674(00)81911-x. [DOI] [PubMed] [Google Scholar]
- 32.Durner J, Klessig D F. Proc Natl Acad Sci USA. 1995;92:11312–11316. doi: 10.1073/pnas.92.24.11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harding S A, Roberts D M. Planta. 1998;206:253–258. [Google Scholar]
- 34.Allan A C, Fluhr R. Plant Cell. 1997;9:1559–1572. doi: 10.1105/tpc.9.9.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strittmatter G, Janssens J, Opsomer C, Botterman J. Bio/Technology. 1995;13:1085–1089. [Google Scholar]
- 36.Alvarez M E, Pennell R I, Meijer P J, Ishikawa A, Dixon R A, Lamb C. Cell. 1998;92:773–784. doi: 10.1016/s0092-8674(00)81405-1. [DOI] [PubMed] [Google Scholar]
- 37.Karpinski S, Reynolds H, Karpinska B, Wingsle G, Creissen G, Mullineaux P. Science. 1999;284:654–657. doi: 10.1126/science.284.5414.654. [DOI] [PubMed] [Google Scholar]