

Abstract
Increasing evidence indicates that malignant stem cells are important for the pathogenesis of acute myelogenous leukemia (AML) and represent a reservoir of cells that drive the development of AML and relapse. Therefore, new treatment regimens are necessary to prevent relapse and improve therapeutic outcomes. Previous studies have shown that the sesquiterpene lactone, parthenolide (PTL), ablates bulk, progenitor, and stem AML cells while causing no appreciable toxicity to normal hematopoietic cells. Thus, PTL must evoke cellular responses capable of mediating AML selective cell death. Given recent advances in chemical genomics such as gene expression-based high-throughput screening (GE-HTS) and the Connectivity Map, we hypothesized that the gene expression signature resulting from treatment of primary AML with PTL could be used to search for similar signatures in publicly available gene expression profiles deposited into the Gene Expression Omnibus (GEO). We therefore devised a broad in silico screen of the GEO database using the PTL gene expression signature as a template and discovered 2 new agents, celastrol and 4-hydroxy-2-nonenal, that effectively eradicate AML at the bulk, progenitor, and stem cell level. These findings suggest the use of multicenter collections of high-throughput data to facilitate discovery of leukemia drugs and drug targets.
Introduction
Cancer stem cells (CSCs) have recently emerged as a potentially important consideration for studies of basic tumor biology and the development of improved therapies. Like normal stem cells, CSCs are thought to reside at the apex of a developmental hierarchy and are responsible for the continued growth and expansion of bulk tumor populations.1 Consequently, the biological activity of CSCs may contribute to initiation, maintenance, and relapse of at least some forms of cancer. To date, CSCs are best characterized for the blood cancer, acute myelogenous leukemia (AML), where numerous studies have documented the phenotype, cell cycle status, and growth characteristics of malignant stem and progenitor cell types.2–4 Notably, several studies have shown that AML stem cells (AML-SCs) are refractory to commonly used clinical agents such as cytarabine and anthracyclines,5–8 thereby further supporting the hypothesis that malignant stem cells represent a probable reservoir from which disease relapse may occur. Given the central role of AML-SCs in leukemic disease, it is therefore important to identify therapeutic regimens that are capable of eradicating this subpopulation of malignant cells.
To date, preclinical studies have demonstrated that selective ablation of AML-SCs using small molecule-based strategies is possible. For example, in vitro studies demonstrated that the combination of idarubicin and MG-132 (IDR/MG) can effectively eradicate leukemia stem cells via a mechanism involving concomitant inhibition of nuclear factor–κB (NF-κB)–mediated survival signals and induction of oxidative stress.9 Subsequently, it was shown that parthenolide (PTL),7 the bioactive chemical component of the medicinal plant feverfew, could also ablate AML-SCs as a single agent. Again, the mechanism of cell death functioned through combined inhibition of NF-κB and induction of oxidative stress, thus indicating that common biological principles underlie the anti–AML-SC effects of these agents despite their chemical diversity. Based on these findings, one approach to drug discovery might be to perform high-throughput screening of chemical libraries to identify agents that both inhibit NF-κB and induce oxidative stress. However, such approaches are cumbersome and require a large initial investment of time and effort with no guarantee of success. Consequently, exploring alternative means to identify agents with specific biological properties is of considerable interest. Moreover, given the chemical diversity of successful regimens such as IDR/MG and parthenolide, it appears desirable that drug discovery efforts not be limited to specific chemical structures.
As a means to improve discovery of targeted therapies for AML-SCs, in the present study we turned to the use of gene expression signatures to identify agents of interest. To model this strategy, we exploited the natural antileukemia characteristics of PTL, which has been shown to induce very potent and specific effects, mediating rapid death of AML-SCs, but not normal hematopoietic stem and progenitor cells. The global transcriptional response to PTL was determined by microarray analysis and used to find other compounds that produce a similar transcriptional response. Importantly, this approach can be employed without necessarily understanding or characterizing the complex interplay of pathways involved in the cellular response to PTL. Compounds that produce transcriptional responses similar to PTL can simply be tested empirically for anti–AML-SC activity. Notably, previous expression-based strategies have been successfully used to identify differentiation therapies10 and functional similarity between seemingly diverse chemical perturbations,11 a strategy epitomized by the Connectivity Map (CMap).12 The latter studies not only proved the feasibility of gene expression–based drug discovery, but also demonstrated that predictions regarding the functions of compounds can be made across different cell types using gene expression microarrays. This important feature of the approach permits gene expression queries to span a broad range of tissues and not be limited to comparing signatures within a specific cell type.
An important advance of this work is the application of strategies that permit discovery of similar gene expression profiles across any publicly available database and the subsequent discovery of novel anti–AML-SC agents. We model this process using data obtained at multiple centers and deposited in the Gene Expression Omnibus (GEO),13 which contains entries from thousands of experiments in multiple cell types. Surprisingly, using several similarity measures, we discovered a recurring and chemically diverse group of compounds that mimic the PTL gene expression pattern and, like PTL, are capable of ablating AML cells at the bulk, progenitor, and stem-cell level. As with PTL,7 the mechanism of action for these new compounds involves concomitant inhibition of the NF-κB survival signal and induction of oxidative stress, suggesting their general importance in targeting AML stem cells. Moreover, for the first time, we demonstrate the use of geographically dispersed gene expression microarray data sets across multiple tissue types to identify new compounds that are able to target AML stem cells.
Methods
Compounds, primary tissues, and cell culture
Celastrol and hemin were obtained from Sigma-Aldrich (St Louis, MO). PTL was obtained from BioMol (Plymouth Meeting, PA). 4-hydroxy-nonenal (HNE) was obtained from Calbiochem (San Diego, CA). Gedunin was obtained from Gaia Chemical (Gaylordsville, CT). Human primary AML was obtained with informed consent. Cord blood and bone marrow cells were obtained either through consenting donors or from the National Disease Research Interchange (NDRI). Cells were grown at 37°C in serum-free medium as described.7 Studies were performed in accordance with University of Rochester Institutional Review Board, guidelines of the National Institutes of Health, and principles of the Declaration of Helsinki.
Isolation of CD34+ AML total RNA
CD34+ AML was isolated using antibody-coupled magnetic bead separation as previously described.7 Cells were cultured for 1 hour and subsequently treated with 7.5 μM PTL. Total RNA was harvested at 6 hours after treatment using the RNeasy protocol (Qiagen, Valencia, CA) according to the manufacturer's instructions. The final preparation, in RNase-free water, was stored at −80°C. RNA concentration and quality was determined by UV absorbance (Nanodrop, Thermo-Fisher Scientific, Wilmington, DE) and ribosomal RNA integrity (Agilent 2100 Bioanalyzer, Agilent Technologies, Santa Clara, CA). CD34+ AML cells were processed in pairs such that each PTL treatment-control pair (one pair per patient) was treated with the same PTL aliquot after 1 hour in culture. Each patient sample was further processed in the same RNA isolation, labeling, and hybridization batch to minimize nonbiological variability within pairs.
Microarray hybridization and analysis
RNA from 12 patient samples of CD34+ AML treated with PTL or left untreated was amplified and labeled with biotin using the Ovation Biotin RNA Amplification and Labeling System (NuGEN, San Carlos, CA) according to the manufacturer's procedure. The fragmented, biotinylated cDNA was used for hybridization at 45°C for 17 hours with Affymetrix HG-U133 Plus 2.0 GeneChip microarrays (Affymetrix, Santa Clara, CA) per manufacturer's recommendations by the University of Rochester Functional Genomics Core Facility. Differential expression analysis was performed using R (http://www.r-project.org/) and BioConductor (http://www.bioconductor.org/). Raw expression data for differential expression analysis was normalized using Robust Multi-array Averaging (RMA) with quantile normalization.14 Differentially expressed probesets were identified using paired t tests, and P values were adjusted for multiple testing using the Benjamini-Hochberg procedure.15 An adjusted P value less than .01 was considered significant. Categorical analysis of the statistically significant differentially expressed genes was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID).16,17
Acquisition and processing of public microarray data
The public microarray data used in this study was obtained from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) website (http://www.ncbi.nlm.nih.gov/geo). Simple Omminus Format in Text (SOFT) files corresponding to the complete contents of GEO platforms GPL96 (HG-U133A), GPL570 (HG-U133+2.0), GPL571 (HG-U133Av2), and GPL3921 (HT-HG-U133A) were downloaded via file transfer protocol (FTP). A parser extracted the expression data for 16 566 complete GEO experiments from these files, which were imported as matrices for analysis in R. The probesets were filtered according to 22 215 common probesets across these platforms, also excluding all quality control (AFFX) probesets. A list of these probesets is available in the data supplement (available on the Blood website: see the Supplemental Materials link at the top of the online article). This filtered data were rank transformed on a scale from 1 to 22 215.
Query of the GEO data
Feature selection for querying GEO data were performed using a paired t test for the PTL versus control comparison across 12 patients. A total of 150 probesets were selected according to the highest 75 and lowest 75 t statistics, after filtering for the 22 215 common probesets across the 16 566 GEO experiments. MAS 5.0-normalized PTL and untreated expression data were then rank transformed. The mean rank for each probeset was taken separately for the PTL group and the untreated group. The query signatures consisted of 150 probesets and their original ranks in each group (PTL or untreated) on a 1 to 22 215 scale. The rank-transformed GEO data were filtered on these same 150 probesets, maintaining the original rank on the 1 to 22 215 scale. Next, the rank correlation was computed for both PTL-treated and untreated CD34+ AML against each GEO entry by pairwise Pearson correlation of the original ranks. The V-score represents partial correlation coefficients18 given by:
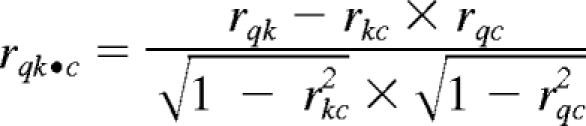 |
where rqk•c (V-score) denotes the partial correlation between the query data, q, and k-th GEO entry, k, given control data, c. When testing the k-th GEO entry for similarity to PTL treatment, q = PTL-treated CD34+ AML and c = untreated CD34+ AML. Conversely, when testing the k-th GEO entry for similarity to CD34+ AML, q = CD34+ AML and c = PTL-treated CD34+ AML.
Classification of GEO data
The R implementation of Random Forest in the randomForest package was trained on rank-transformed global gene expression data for CD34+ AML treated with PTL or left untreated, normalized according to the MAS 5.0 procedure. Both the training data and testing data (GEO) were subset on the same 22 215 probesets as above, and all of these probesets were considered for prediction. The default parameters were used where the number of probesets considered at each split (mtry) was 125 and 500 trees were grown. The optimal mtry parameter was determined using the tuneRF function of the randomForest R library. Test data consisted of 16 566 GEO experiments processed as above. The PTL treatment class was partitioned into type 1 and type 2 responses to account for patient-to-patient variation in PTL responses, reflecting whether marked inhibition of interferon-induced genes (type 2) occurred or not (type 1), which improved the ability to specifically identify known experimentally determined anti–AML-SC agents such as MG-132 and prostaglandin J2 in independent data found in GEO. This determination was made according to a parametric analysis of geneset enrichment-based19 approach, where the z-test was substituted with a t test. The absence of interferon-controlled genesets among the top 10 most significant genesets indicated type 1 class. PTL-like gene expression patterns were discovered in the type 1 PTL class.
Flow cytometric assays
To determine viability, cells were stained using annexin V-FITC (BD Biosciences, San Jose, CA) and 7-aminoactinomycin (7-AAD, Probes-Invitrogen, Carlsbad, CA) to detect phosphatidylserine exposition and cell permeability, respectively, as described.7 Cells were additionally stained with antibodies against phenotypic markers CD34 and CD38 (BD Biosciences) to assess viability in phenotypically defined subpopulations. At least 50 000 events were recorded per condition on an LSR II flow cytometer (BD Biosciences). Data analysis was conducted using FlowJo 8.2 software for Mac OS X (TreeStar, Ashland, OR). Cells that are negative for annexin V and 7-AAD are scored as viable. A gating scheme for ascertaining viability is provided in supplemental data. To determine free thiol content, cells were stained with monobromobimane (mBBr) and DAPI (Probes-Invitrogen).
Fluorescence-activated cell sorting
Cells were stained using CD34-APC– and CD38-PE–conjugated antibodies (BD Biosciences) with DAPI and then sorted under sterile conditions for CD34+ CD38− populations using a FACSAria cell sorter (BD Biosystems). The purity of each sort was greater than 96%. Cells were then placed in serum-free medium at 1 million cells/mL to be later treated with the indicated compounds for 6 hours.
Colony forming unit and NOD/SCID xenotransplant assays
Methylcellulose culture assays were used to assess the colony forming ability of both AML and normal hematopoietic progenitors. Growth conditions and scoring were performed as described7 in the presence or absence of drugs. NOD/SCID xenotransplant assays were performed as described.7
Confocal microscopy
Cells were fixed in methanol at −20°C. The cells were permeabilized with blocking buffer (10% fetal bovine serum and 0.1% Tween 20 in 1× phosphate-buffered saline [pH 7.4]) as described.20 Cells were stained using either rabbit polyclonal anti-p65 (C-20), anti-Nrf2 (C-20) (Santa Cruz Biotechnologies, Santa Cruz, CA), anti-HO1 (GeneTex, San Antonio, TX) in blocking buffer for 2 hours at room temperature. Cells were washed and stained with goat-anti–rabbit Alexa488 secondary antibodies and ToPro3 for nuclear stain (Invitrogen, Carlsbad, CA). Slides were mounted using Fluoromount-G 234 (no. 63 722; Southern Biotech, Birmingham, AL). Slides were left to dry overnight. Fluorescence was observed using a 100× objective (1.4 numeric aperture), on a Leica SP1 inverted scanning confocal microscope (Heidelberg, Germany). Image acquisition was performed using Leica confocal software v.2.6.1 Build 1537.
Electrophoretic mobility shift assays and immunoblots
The electrophoretic mobility shift assay (EMSA) was performed as described6 using nuclear extracts prepared from cells either untreated or treated for 6 hours with 2 μM celastrol, 20 μM gedunin, 30 μM HNE, or 50 μM hemin. For immunoblots, cells were prepared and lysates were submitted to denaturing electrophoresis as described21 and probed with anti–phospho-p65(Ser-536) (Cell Signaling, Beverly, MA) or anti–HO-1 (GeneTex).
Statistical analysis
Statistical significance for cell viability and colony forming unit assays was performed using one-way analysis of variance followed by Tukey multiple comparison test to determine the significance of particular comparisons, except where otherwise indicated. Two-way analysis of variance was used to determine significance in the NOD/SCID xenotransplant assays followed by Bonferroni post-tests. Analyses and graphs where performed using GraphPad Prism software (GraphPad Software, San Diego, CA).
Chemical structures
Chemical structures shown in this report were derived through ChemBank (http://chembank.broad.harvard.edu/).
Publicly deposited data
The microarrays used to generate the PTL gene expression signature are available at GEO (http://ncbi.nlm.nih.gov/geo/) accession GSE7538.
Additional experimental details
Additional experimental details are located in supplemental data.
Results
Generation of parthenolide gene expression signature
To initiate these studies, we developed a gene expression signature capturing PTL-mediated cell death in AML for use as a template for detecting PTL-like biological states. We obtained gene expression profiles resulting from PTL treatment of CD34+ AML cells from 12 patients for 6 hours alongside patient-matched untreated controls on Affymetrix HG-U133+2.0 gene expression microarrays. Six hours of treatment is sufficient to irreversibly commit AML-SCs to die but is a timepoint at which no overt cell death is yet apparent, and thus reflects the transcriptional status of cells programmed to die as a result of PTL exposure.7 A total of 702 probesets, termed genes hereafter, were differentially expressed using a P < .01 statistical cutoff following multiple test correction (Table S1). Categorical analysis of these differentially expressed genes revealed the induction of transcriptional responses to oxidative stress, unfolded proteins, as well as inhibition of NF-κB (Table S2).
Using the expression data derived above, we next initiated a search for other biological states that mimic PTL bioactivity using an extremely comprehensive gene expression based in silico screen. We reasoned that matching gene expression patterns based on all genes in the microarray would result in matches based primarily on cell type and other non–PTL-specific factors. Thus, we reduced our search so as to focus on PTL discriminatory genes defined as the genes with the most extreme positive and negative t statistics in the PTL versus control comparison; a 150-gene signature reflecting the most statistically significant up- and down-regulated genes discriminating the PTL and untreated groups (Figure S3). This signature is indicated in Table S3, details of which are provided in “Methods.” Given the enormity of GEO and its lack of readily identifiable treatment-control pairs as found in the CMap, a correlation-based measure was used. Correlation metrics such as Pearson correlation and partial correlation have well-established use in various microarray analysis applications such as hierarchical clustering and construction of genetic networks,22,23 elucidating sample-wise and gene-wise relationships in microarray data. Moreover, correlation was reported to perform similarly to the gene set enrichment analysis (GSEA)–based metrics of the CMap.12,24 We thus focused our comparison between microarrays on the 150-gene signature and measured the rank correlation of each GEO entry to both PTL-treated and untreated CD34+ AML. Because correlations can still arise from nontreatment-related factors such as cell type, GEO entries highly correlated to both treated and untreated AML are likely less relevant and were penalized to receive lower scores using partial correlation so as to reduce this effect, details of which are in “Methods.” We termed this metric the “V-score.”
Query of public data in GEO identifies candidate agents
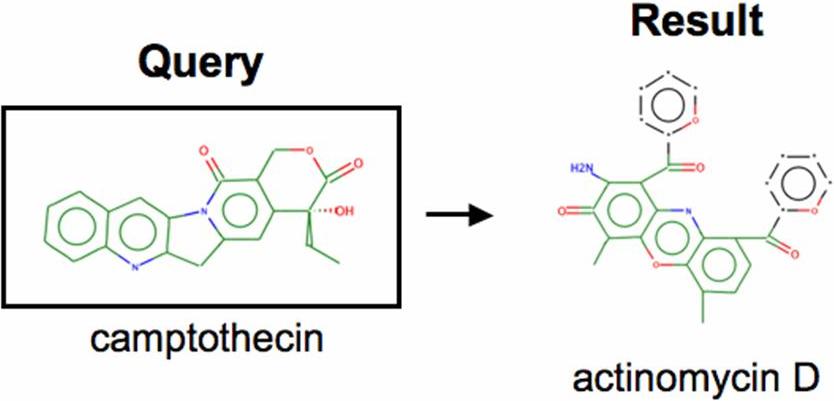
Expression data against which to compare the PTL signature was extracted for a total of 16 566 complete GEO entries performed on the Affymetrix U133-based platforms. We computed V-scores to identify the most PTL-like expression patterns among this GEO data. A number of testable compounds were discovered among the top hits of this approach (Table 1), some of which we have previously shown to be effective anti–AML-SC agents, such as MG-132 and prostaglandin J2 (PGJ2),6,7 providing an immediate validation of the current approach. Importantly, when search parameters were reversed to instead search for GEO expression profiles resembling untreated CD34+ AML, the V-score methodology successfully identified GEO expression profiles derived from primitive CD34+ and CD133+ myeloid leukemia cells (Table S4). As an independent test of the V-score approach, we also performed queries using a camptothecin-based signature and found that the methodology was useful in identifying functionally related compounds (topoisomerase I inhibitors), the details of which are included in supplemental data (Tables S5,S6). Finally, we observed that the several of the compounds identified in Table 1 were also found in the CMap database (celastrol, MG-132, and prostaglandin J2). Upon querying the CMap using its GSEA-based metrics, these compounds were found to be top matches with the PTL signature, thereby further validating the findings of Table 1 (Table S7).
Table 1.
Results of query of GEO data: top V-score search results
| Rank | GEO accession no. | Description | V-score |
|---|---|---|---|
| 1 | GSM127022 | LNCaP cells 20 μM gedunin for 6 hours | 0.62249 |
| 2 | GSM127020 | LNCaP cells 20 μM gedunin for 6 hours | 0.62235 |
| 3 | GSM119204 | MCF7 cells 10 μM 15-delta prostaglandin J2 for 6 hours | 0.61694 |
| 4 | GSM45111 | RKO cells 60 μM 4-hydroxy-2-nonenal (HNE) for 6 hours | 0.60400 |
| 5 | GSM127021 | LNCaP cells 20 μM gedunin for 6 hours | 0.60027 |
| 6 | GSM45099 | RKO cells 60 μM 4-hydroxy-2-nonenal (HNE) for 6 hours | 0.59773 |
| 7 | GSM16536 | K562 cells 50 μM hemin for 72 hours | 0.59411 |
| 8 | GSM16532 | K562 cells 50 μM hemin for 48 hours | 0.59399 |
| 12 | GSM119109 | MCF7 cells 2.5 μM celastrol for 6 hours | 0.58509 |
| 13 | GSM43958 | RKO cells 60 μM 4-hydroxy-2-nonenal (HNE) for 6 hours | 0.58496 |
| 14 | GSM119282 | MCF7 cells 21 μM MG-132 for 6 hours | 0.58341 |
To further corroborate our findings with PTL, we next examined whether similar results as with the V-score in Table 1 were possible using Random Forest class prediction,25 a method independent of the V-score and GSEA that does not require preselection of genes.26 Class predictors have been proposed fairly extensively for the classification of hematologic malignancies into diseases and subtypes (eg, AML vs ALL) based on gene expression patterns captured on microarrays.27,28 The deployment of a multitude of classifiers across all public data for systematic drug discovery is thus an attractive concept, given their ability to operate in the absence of identifiable untreated controls. We generated a Random Forest classifier based on PTL treatment of AML as a template. The resulting classifier identified 19 GEO entries, which comprised 10 independent compounds (Table 2). Several of the compounds identified by the Random Forest classifier overlapped with top hits from the V-score approach: HNE, celastrol, MG-132, and PGJ2. Thus, the same candidate agents were identified using 2 completely separate computational approaches to query the nearly 17 000 GEO entries. Importantly, the compounds demonstrate significant chemical diversity (Figure 1), indicating they are not simply structural analogs.
Table 2.
All expression profiles in GEO classified as “PTL-like” by Random Forest
| GEO Accession # | Description |
|---|---|
| GSM119109 | MCF7 cells celastrol (2.5 μM) for 6 hours |
| GSM119282 | MCF7 cells MG-132 (21 μM) for 6 hours |
| GSM45115 | RKO cells 60 μM 4-hydroxynonenal for 24 hours |
| GSM119247 | MCF7 cells 15-delta prostaglandin J2 (10 μM) for 6 hours |
| GSM119204 | MCF7 cells 15-delta prostaglandin J2 (10 μM) for 6 hours |
| GSM119083 | MCF7 cells 5224221 (12 μM) for 6 hours |
| GSM119153 | MCF7 cells 5224221 (12 μM) for 6 hours |
| GSM119164 | MCF7 cells 5182598 (25 μM) for 6 hours |
| GSM119232 | MCF7 cells 17-dimethylamino-geldanamycin (0.1 μM) for 6 hours |
| GSM119145 | MCF7 cells 17-allylamino-geldanamycin (1 μM) for 6 hours |
| GSM119236 | MCF7 cells 17-allylamino-geldanamycin (1 μM) for 6 hours |
| GSM119242 | MCF7 cells 17-allylamino-geldanamycin (1 μM) for 6 hours |
| GSM119199 | MCF7 cells 17-allylamino-geldanamycin (1 μM) for 6 hours |
| GSM119179 | MCF7 cells 17-allylamino-geldanamycin (1 μM) for 6 hours |
| GSM119186 | MCF7 cells 17-dimethylamino-geldanamycin (0.1 μM) for 6 hours |
| GSM119191 | MCF7 cells 17-allylamino-geldanamycin (1 μM) for 6 hours |
| GSM119201 | MCF7 cells geldanamycin (1 μM) for 6 hours |
| GSM119150 | MCF7 cells monorden (0.1 μM) for 6 hours |
| GSM119192 | MCF7 cells monorden (0.1 μM) for 6 hours |
Figure 1.

Chemical structures of parthenolide and compounds found through a gene expression based search of GEO. A query of GEO was performed using the gene expression signature of cells treated with parthenolide (left panel). The 4 anti–AML-SC compounds identified by the search were celastrol, 4-hydroxynonenal, 15Δ-prostaglandin J2, and MG-132 (right panel).
Agents discovered by in silico screening have properties in common with parthenolide
For further characterization, we selected the 2 agents identified by both computational methods (celastrol and HNE), as well as 2 candidates detected only using the V-score approach (gedunin and hemin). Though anticancer activity has been described for many of these compounds,11,29–33 none of these agents are previously known to demonstrate the ability to specifically target leukemic stem cells. The known biological properties of these compounds are shown in Table 3. Interestingly, the 4 molecules share certain properties. For example, like PTL, celastrol and gedunin are terpenoids with reported NF-κB–inhibiting capabilities34–36 and for which a biological similarity has been established using chemical genomics.11 HNE is a nonterpenoid lipid peroxidation product that also inhibits NF-κB and possesses other activities associated with previously reported anti–AML-SC drugs37,38: proteasome inhibition39 and stress induction with Nrf2 activation.40 Hemin has been shown in various cell systems to produce oxidative injury,41,42 Nrf2 and HO-1 induction,43 and attenuation of NF-κB.44,45 Given this common biology, we next determined whether these compounds were capable of targeting AML bulk, progenitor, and stem cells at concentrations that are not lethal to normal hematopoietic cells.
Table 3.
Features of compounds analyzed in this study
| PTL | Celastrol | Gedunin | HNE | Hemin | |
|---|---|---|---|---|---|
| Terpenoid | + | + | + | − | − |
| NF-κB inhibitor | + | + | + | + | + |
| Proteasome inhibitor | nd | + | nd | + | nd |
| Oxidative stress | + | * | nd | + | + |
nd indicates not determined.
Indicates literature reports antioxidant activity,56 Nrf2 activation in this study suggests induction of oxidative stress for AML.
Celastrol ablates AML at the bulk, progenitor, and stem cell level
Since celastrol and gedunin share structure and NF-κB–inhibiting activity with PTL, we began by investigating the ability of these compounds to ablate AML-SCs. Intriguingly, biological comparison of the 2 compounds demonstrated a striking contrast. Celastrol effectively mimicked PTL-like specific toxicity against primitive AML cells as determined by flow cytometry (Figure 2A). Exposure of CD34+/CD38− AML cells to 2 μM celastrol for 24 hours resulted in only 13% mean survival as determined by FACS analysis of annexin V/7AAD stained cells, well below the 82% mean survival observed for normal controls (P < .01). Celastrol also exhibited toxicity to total and CD34+ AML populations (Figure S1). This selectivity was also observed in AML progenitor cells in colony forming unit (CFU) assays, where mean AML CFU was reduced to 10% with 2 μM celastrol treatment relative to untreated AML CFU. In contrast, reduction of mean erythroid and myeloid colonies from treatment of normal cells with 2 μM celastrol was relatively modest at 75% and 69%, respectively (Figure 2B). We further measured the toxicity against AML-SCs in vivo using the NOD/SCID xenotransplant assay46 in which celastrol significantly (P = .0003) impaired engraftment of AML-SCs into sublethally irradiated mice (Figure 2C). In contrast, gedunin was unable to eradicate total AML, CD34+ AML, or CD34+/CD38− AML cells at concentrations up to 20 μM (Figure 2D). At 6 hours after treatment, both celastrol and gedunin effectively inhibit NF-κB, as indicated by the diminished nuclear localization of NF-κB/p65 determined by confocal immunofluorescence microscopy (Figure 2E top panel). Notably, celastrol, but not gedunin, could activate Nrf2 as determined by its nuclear localization (Figure 2E bottom panel). The ability of both celastrol and gedunin to inhibit NF-κB is further indicated by loss of NF-κB binding in gel shift assays (Figure 2F top panel) and by diminished p65(Ser-536) phosphorylation with both compounds at 6 hours, though gedunin was less effective at altering this phosphorylation than celastrol. Moreover, robust induction of HO-1, an Nrf2-induced cellular stress response protein,47 was only apparent with celastrol treatment but not with gedunin treatment in immunoblots (Figure 2F bottom panel). Thus, in primary AML, gedunin could not effectively induce Nrf2-mediated stress responses and was less effective at NF-κB inhibition compared with the efficacious anti–AML-SC compound celastrol (as evidenced by stronger nuclear localization of NF-κB p65 in confocal microscopy and greater p65 phosphorylation in immunoblots with gedunin treatment, Figure 2D,E). The ability of celastrol to exert NF-κB inhibition and stress induction was then examined in FACS purified CD34+CD38− primary AML cells. Immunoblots of these purified cells revealed that loss of phospho-p65 and induction of HO-1 seen in CD34+ AML cells extends to phenotypically defined CD34+ CD38− AML stem cells (Figure S4). These findings further underscore the importance of simultaneous stress induction and NF-κB inhibition as features of efficacious anti–AML-SC agents, and these features are consistently present in different AML subpopulations.7,9
Figure 2.

Biological characterization of terpenoids identified by gene expression analyses. (A) Bar chart of FACS analysis indicating significant impairment of viability (single asterisk, P < .01; N = 3 patient samples) of primary CD34+ CD38− AML cells versus normal human CD34+ CD38− bone marrow treated with 2 μM celastrol. Alongside, a representative FACS analysis of viability is shown. The lower left quadrant (annexin V negative/7-AAD negative) represents viable cells, whereas events in the lower right and upper right panels represent dying and dead cells, respectively. Viability is represented as a percentage of untreated controls. (B) Methylcellulose colony assays of primary human cells treated with 2 μM celastrol. Normal erythroid, myeloid, and AML colony forming units are shown relative to untreated controls. Error bars represent the standard error of the mean. Black bars are normal cells. White bars are AML. Significant selectivity (asterisk) is indicated by either the AML versus myeloid or AML versus erythroid comparisons with celastrol treatment (single asterisk, P < .01; N = 3 patient samples). Viability is represented as a percentage of untreated controls. (C) Bar charts indicate the percent engraftment of AML cells in NOD/SCID mice after 18 hours in culture with 2 μM celastrol (Cel) versus untreated control (Unt). White bars represent the cohort of animals injected with untreated cells; black bars represent 2 μM celastrol treatment. Significant loss of engraftment (P < .01, single asterisk; N = 3 patient samples, 3-4 mice/sample) in NOD/SCID mice is observed with celastrol treatment. No asterisk, P = .11 (4 mice). Overall P value for the treatment is P = .0003. (D) Bar chart indicating viability of phenotypically defined AML cells 24 hours after treatment with 20 μM gedunin (N = 3 patient samples). Viability is represented as a percentage of untreated controls. (E) Representative confocal microscopy images of primary CD34+ AML cells treated with 2 μM celastrol, 20 μM gedunin, or left untreated. Top panels show overlays labeling for NF-κB p65 (green) and nucleus (red). Bottom panels show overlays for Nrf2 (yellow) and nucleus (blue). (F) Top panel: EMSAs indicating NF-κB binding relative to untreated (UNT) cells with either 2 μM celastrol (CEL) or 20 μM gedunin (GED) 6 hours after treatment of CD34+ AML cells. Bottom panel: immunoblots indicating phospho-p65, HO-1, and β-actin levels at 6 hours in primary CD34+ AML treated with 2 μM celastrol (CEL) treatment, 20 μM gedunin (GED), or left untreated (UNT).
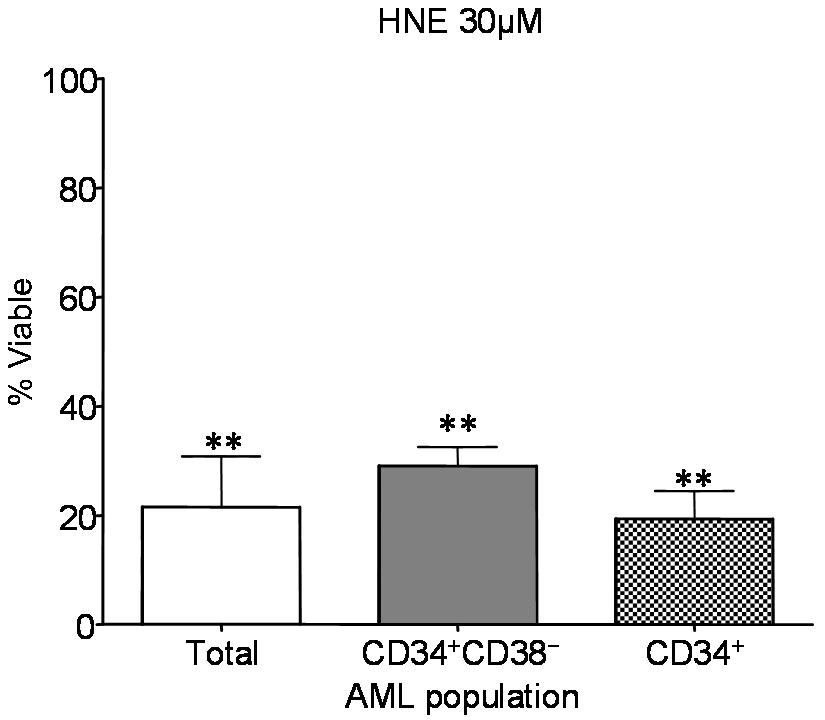
HNE ablates AML at the bulk, progenitor, and stem-cell level
We then proceeded to examine the 2 nonterpenoids discovered by our in silico screen: HNE and hemin. Treatment with 30 μM HNE was able to effect PTL-like selective cell death (Figure 3A). At 24 hours after treatment, CD34+/CD38− AML cells treated with 30 μM HNE were 29.8% viable compared with a mean viability in normal cells of 93.6% (P < .01). HNE also exhibited toxicity to total and CD34+ AML populations (Figure S2). As with celastrol, HNE demonstrated dramatic impairment of AML progenitors in CFU assays such that no colonies whatsoever formed, while not overtly affecting the CFU capacity of normal hematopoietic progenitors (Figure 3B). In addition, we evaluated AML-SC function after exposure to HNE using the NOD/SCID xenotransplant assay46 in which HNE significantly (P < .0001) impaired engraftment of AML-SCs into sublethally irradiated mice (Figure 3C). In contrast, 50 μM hemin did not cause cell death in any AML subpopulation, despite its PTL-like capability as an Nrf2-triggering oxidant (Figure 3D). Consistent with our hypotheses48,49 regarding the characteristics of efficacious agents, hemin demonstrated only modest inhibition of NF-κB and very modest Nrf2-activation in primary AML (Figure 3E,F). The ability of HNE to produce NF-κB inhibition and stress induction was examined in FACS-purified CD34+ CD38− primary AML cells. Immunoblots of these purified cells revealed that loss of phospho-p65 and induction of HO-1 seen in CD34+ AML cells extends to phenotypically defined CD34+ CD38− AML stem cell (Figure S4). Again, simultaneous NF-κB inhibition and stress induction were evident for agents capable of eradicating primitive AML cells.
Figure 3.

Biological characterization of nonterpenoids identified by gene expression analyses. (A) Bar chart of FACS analysis indicating significant impairment of viability (single asterisk, P < .01; N = 3 patient samples) of primary human CD34+ CD38− AML cells versus normal CD34+ CD38− marrow cells treated with 30 μM HNE. Alongside, a representative FACS analysis of viability is shown. The lower left quandrant (annexin V negative/7-AAD negative) represents viable cells, whereas events in the bottom right and top right panels represent dying and dead cells, respectively. Viability is represented as a percentage of untreated controls. (B) Methylcellulose colony assays of primary human cells treated with 30 μM HNE. Normal erythroid, myeloid, and AML colony forming units are shown relative to untreated controls. Error bars represent the standard error of the mean. Black bars are normal cells. White bars are AML. Significant selectivity (double asterisk) is indicated by either the AML versus myeloid or AML versus erythroid comparisons with HNE treatment (P < .001; N = 3 patient samples). Viability is represented as a percentage of untreated controls. (C) Bar charts indicate the percent engraftment of AML cells in NOD/SCID mice after 18 hours in culture with 30 μM HNE versus untreated control (Unt). White bars represent the cohort of animals injected with untreated cells; black bars represent 30 μM HNE treatment. Significant loss of engraftment (P < .001, double asterisk; P < .0001, triple asterisk; N = 3 patient samples, 4 mice/sample) in NOD/SCID mice is observed with HNE treatment. Overall P value for the treatment is P < .0001. (D) 50 μM hemin lacks toxicity to total (white bar), CD34+/CD38− (gray bar), and CD34+ (shaded bar) primary human AML populations. Viability is represented as a percentage of untreated controls. (E) Representative confocal microscopy images of primary CD34+ AML left untreated or treated with 30 μM HNE or 50 μM hemin. Top panels show overlays labeling for NF-κB p65 (green) and nucleus (red). Bottom panels show overlays for Nrf2 (yellow) and nucleus (blue). (F) Top panel: EMSAs of NF-κB binding relative to untreated (UT) cells with either 30 μM HNE or 50 μM hemin at 6 hours after treatment in CD34+ AML. Bottom panel: immunoblots of phospho-p65, HO-1, and β-actin levels at 6 hours in primary CD34+ AML treated with 30 μM HNE treatment, 50 μM hemin, or left untreated (UT).
Celastrol and HNE demonstrate molecular properties that are comparable to parthenolide
Next, we compared celastrol and HNE alongside PTL. It is established that PTL eradicates the majority of AML cells within 24 hours at the bulk, progenitor, and stem cell level.7 Again, previous studies in other cell systems have established that PTL toxicity is mediated through inhibition of NF-κB and oxidative stress via depletion of intracellular thiol content.49 To examine the involvement of these established mechanisms in the cell death observed as a consequence of exposure to celastrol and HNE, primary CD34+ AML cells were treated with either celastrol or HNE at multiple doses. For all agents compared, inhibition of NF-κB (loss of phospho-p65) and depletion of intracellular thiols (mBBr) were evident and demonstrated dose dependency. Furthermore, maximal toxicity was achieved at concentrations where NF-κB inhibition was apparent alongside thiol depletion (Figure S5), further establishing the importance of these biological features across multiple anti-AML stem cell agents.
Discussion
The results of parthenolide-based queries of GEO data identified 2 previously unknown compounds, HNE and celastrol, as anti–AML-SC agents. Intriguingly, nothing previously known about these agents would likely have predicted their use for this purpose. The only commonly known characteristic of either compound that is strongly associated with AML-SC drugs is NF-κB inhibition. However, as previously demonstrated by molecular genetic and chemical analyses, inhibition of NF-κB alone is not sufficient to eradicate AML-SCs. This point is further supported by the fact that gene expression–based queries in the present study did not identify many known NF-κB inhibitors. Indeed, as proposed in earlier studies, it appears that while inhibition of NF-κB is a consistent feature of compounds that modulate AML-SCs, additional properties such as stress induction are also essential.
With respect to the methodology employed in this study, several aspects of the empirical findings are noteworthy. First, as described in the CMap studies, it is quite feasible to obtain gene expression signatures from a specific cell type and use that data to query distinctly different cell types. The CMap database employs breast, leukemia, and prostate cancer cell lines to generate gene expression profiles and was successfully used for discovery of drugs across multiple tissue types. Our study extends the principle across all human cell types present in GEO and successfully found similarities from AML cells in lines derived from breast (MCF7) and colorectal (RKO) cancer cells (Tables 1,2). While this cross-tissue analysis will certainly miss some cell-type specific properties, it is nonetheless a powerful approach and forms a central tenet of gene expression–based drug discovery. Second, for our objective of finding agents relevant to AML-SCs, it was not necessary to use purified populations of AML-SCs. Again, using CD34+ AML blasts was sufficient for the purpose of identifying agents of interest in a chemical genomic screen, as clearly demonstrated through the biological validation studies in Figures 2 and 3. As noted for the principle of cross-tissue comparison mentioned above, it is possible that some properties of AML-SCs were not captured in the analysis and that use of AML-SCs may have yielded improved results. However, given the logistical difficulty in obtaining sufficient numbers of AML-SCs from primary patient specimens for microarray-based studies, we suggest that the success using AML blasts is a very promising finding. However, the general use of using CD34+ AML blasts as a surrogate for CD34+ CD38− AML stem cells in chemical genomic studies will require additional studies. Though the effects of PTL and the new PTL-like drugs were conserved in all AML compartments tested, it is not clear whether this level of conservation is generalizable to all anti–AML-SC agents. PTL and the PTL-like agents discovered here perturb stress and survival signals that are present in all subpopulations, thereby mediating cell death in the entire AML population. Furthermore, the non–PTL-like novel agent, TDZD-8, also perturbs conserved features of AML subpopulations and thereby effects cross-compartmental cell death.50 Despite these observations, newly discovered agents must still be functionally tested for the ability to target enriched AML-SCs. Finally, no attempt was made to determine a “cutoff” value for the V-score approach, for instance, a numerical value for the metric above which the results are more valid. Rather, the top hits were simply validated empirically. While it is possible that a specific V-score may provide a useful cutoff value, it is much more likely that such a value will be highly context dependent and vary as a function of the specific type of query. In the future, with much additional use of the present methodology and other methodologies that prove useful for querying GEO and other public databases, it should be possible to better clarify this issue. While the repositories and methodologies employed here are potentially useful for addressing a range of biological questions, their ultimate utility for drug discovery will be proven by further empirical testing as well as by further depositions of pharmacogenomic and molecular genetic data in GEO.
Although we have focused on identifying compounds that eradicate AML-SCs, the approaches described herein represent a potential strategy and proof-of-principle by which therapies and therapeutic targets may be discovered using publicly deposited data as it is deposited into the public domain in “real time.” The present effort underscores the benefits of open access to data and the annotation standards advocated by the Microarray Gene Expression Data (MGED) Society and others.51–53 We expect that additional classifiers and/or similarity metrics will also prove to be of use for querying public data; a subject of ongoing investigation. Given the current paucity of established anti–AML-SC agents, our ability to enhance the discovery of anti–AML-SC conditions in public data are expected to improve as additional anti–AML-SC drugs are discovered and biologically validated.
A potentially confounding issue going forward is defining the lingua franca for gene expression data. Here, we made use of the most widely available human gene expression platform for our analyses: the Affymetrix U133 family of microarrays. However, in addition to usual random variation, experimental inconsistencies and normalization differences between GEO expression profiles likely affected our ability to detect certain GEO entries, considering the reported ability for normalization methods to alter the within-array correlation structure of genes.54 Detection of truly “like” perturbations is also likely to be affected by experiment-specific parameters including time point, medium, and cell type. Therefore, it is probable that certain perturbations were missed as a result of these experimental factors. Finally, well-intended recent efforts to improve data quality such as the remapping of Affymetrix probes to new probesets55 can fragment the public data by altering probe names and the corresponding transcripts to which they hybridize. Thus, implementation of consistent microarray normalization and annotation conventions will greatly enhance the future use of the collective knowledge contained in repositories like GEO.
In summary, these studies demonstrate that all public gene expression data are potentially useful for drug discovery and can be accessed by any investigator with the appropriate computational tools. As public resources expand, we expect the present approach to find additional compounds or genetic perturbations of interest that may help us identify efficacious compounds or better drug targets to improve the management of leukemia.
Supplementary Material
Acknowledgments
We gratefully acknowledge Dr Jackie Howitt, Caroline Burtner CNM, and Joan Brenner CNM at Lake Affect Ob/Gyn and Midwifery Care for obtaining human cord blood specimens and Dr Stephen Welle and Michelle Zanche of the University of Rochester Functional Genomics Core Facility.
We also acknowledge support from the Douglas Kroll research foundation, the Leukemia and Lymphoma Society (6099-06), the National Cancer Institute (R01CA90446), and the Samuel Waxman Cancer Research Foundation. C.T.J. is a Scholar of the Leukemia and Lymphoma Society. D.C.H. was supported in part by NIH 5T32-CA09363 and a grant from the James P. Wilmot Cancer Center.
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.C.H. designed research, performed experiments, developed methods, wrote software, analyzed data, and wrote the paper. M.L.G. designed research, performed experiments, analyzed data, and wrote the paper. C.C. and X.L. performed experiments. R.A. wrote software. F.Y., J.L.L., and M.C. provided vital reagents. C.T.J. designed research, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Craig T. Jordan, 601 Elmwood Ave, Box 703, University of Rochester School of Medicine and Dentistry, Rochester, NY; e-mail: craig_jordan@urmc.rochester.edu.
References
- 1.Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 3.Jordan CT, Guzman ML. Mechanisms controlling pathogenesis and survival of leukemic stem cells. Oncogene. 2004;23:7178–7187. doi: 10.1038/sj.onc.1207935. [DOI] [PubMed] [Google Scholar]
- 4.Krause DS, Van Etten RA. Right on target: eradicating leukemic stem cells. Trends Mol Med. 2007;13:470–481. doi: 10.1016/j.molmed.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 6.Guzman ML, Neering SJ, Upchurch D, et al. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–2307. doi: 10.1182/blood.v98.8.2301. [DOI] [PubMed] [Google Scholar]
- 7.Guzman ML, Rossi RM, Karnischky L, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163–4169. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costello RT, Mallet F, Gaugler B, et al. Human acute myeloid leukemia CD34+/CD38− progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000;60:4403–4411. [PubMed] [Google Scholar]
- 9.Guzman ML, Swiderski CF, Howard DS, et al. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci U S A. 2002;99:16220–16225. doi: 10.1073/pnas.252462599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stegmaier K, Ross KN, Colavito SA, O'Malley S, Stockwell BR, Golub TR. Gene expression-based high-throughput screening(GE-HTS) and application to leukemia differentiation. Nat Genet. 2004;36:257–263. doi: 10.1038/ng1305. [DOI] [PubMed] [Google Scholar]
- 11.Hieronymus H, Lamb J, Ross KN, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell. 2006;10:321–330. doi: 10.1016/j.ccr.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 12.Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 13.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 15.Benjamini Y, H Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 16.Dennis G, Jr, Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 17.Hosack DA, Dennis G, Jr., Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4:R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cramer H. Mathematical Methods of Statistics. Princeton, NJ: Princeton University Press; 1946. [Google Scholar]
- 19.Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Topisirovic I, Guzman ML, McConnell MJ, et al. Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol Cell Biol. 2003;23:8992–9002. doi: 10.1128/MCB.23.24.8992-9002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14:1777–1784. doi: 10.1038/sj.leu.2401903. [DOI] [PubMed] [Google Scholar]
- 22.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toh H, Horimoto K. Inference of a genetic network by a combined approach of cluster analysis and graphical Gaussian modeling. Bioinformatics. 2002;18:287–297. doi: 10.1093/bioinformatics/18.2.287. [DOI] [PubMed] [Google Scholar]
- 24.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Breiman L. Random forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- 26.Diaz-Uriarte R, Alvarez de Andres S. Gene selection and classification of microarray data using random forest. BMC Bioinformatics. 2006;7:3. doi: 10.1186/1471-2105-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 28.Yeoh EJ, Ross ME, Shurtleff SA, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–143. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 29.Cerbone A, Toaldo C, Laurora S, et al. 4-Hydroxynonenal and PPARgamma ligands affect proliferation, differentiation, and apoptosis in colon cancer cells. Free Radic Biol Med. 2007;42:1661–1670. doi: 10.1016/j.freeradbiomed.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 30.Borovic S, Cipak A, Meinitzer A, et al. Differential sensitivity to 4-hydroxynonenal for normal and malignant mesenchymal cells. Redox Rep. 2007;12:50–54. doi: 10.1179/135100007X162194. [DOI] [PubMed] [Google Scholar]
- 31.Vizio B, Poli G, Chiarpotto E, Biasi F. 4-hydroxynonenal and TGF-beta1 concur in inducing antiproliferative effects on the CaCo-2 human colon adenocarcinoma cell line. Biofactors. 2005;24:237–246. doi: 10.1002/biof.5520240128. [DOI] [PubMed] [Google Scholar]
- 32.Nagase M, Oto J, Sugiyama S, Yube K, Takaishi Y, Sakato N. Apoptosis induction in HL-60 cells and inhibition of topoisomerase II by triterpene celastrol. Biosci Biotechnol Biochem. 2003;67:1883–1887. doi: 10.1271/bbb.67.1883. [DOI] [PubMed] [Google Scholar]
- 33.Sethi G, Ahn KS, Pandey MK, Aggarwal BB. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood. 2007;109:2727–2735. doi: 10.1182/blood-2006-10-050807. [DOI] [PubMed] [Google Scholar]
- 34.Crombie L. The cathedulin alkaloids. Bull Narc. 1980;32:37–50. [PubMed] [Google Scholar]
- 35.Lee JH, Koo TH, Yoon H, et al. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol. 2006;72:1311–1321. doi: 10.1016/j.bcp.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 36.Penido C, Costa KA, Costa MF, Pereira Jde F, Siani AC, Henriques MG. Inhibition of allergen-induced eosinophil recruitment by natural tetranortriterpenoids is mediated by the suppression of IL-5, CCL11/eotaxin and NFkappaB activation. Int Immunopharmacol. 2006;6:109–121. doi: 10.1016/j.intimp.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 37.Camandola S, Poli G, Mattson MP. The lipid peroxidation product 4-hydroxy-2,3-nonenal inhibits constitutive and inducible activity of nuclear factor kappa B in neurons. Brain Res Mol Brain Res. 2000;85:53–60. doi: 10.1016/s0169-328x(00)00234-5. [DOI] [PubMed] [Google Scholar]
- 38.Page S, Fischer C, Baumgartner B, et al. 4-Hydroxynonenal prevents NF-kappaB activation and tumor necrosis factor expression by inhibiting IkappaB phosphorylation and subsequent proteolysis. J Biol Chem. 1999;274:11611–11618. doi: 10.1074/jbc.274.17.11611. [DOI] [PubMed] [Google Scholar]
- 39.Conconi M, Friguet B. Proteasome inactivation upon aging and on oxidation-effect of HSP 90. Mol Biol Rep. 1997;24:45–50. doi: 10.1023/a:1006852506884. [DOI] [PubMed] [Google Scholar]
- 40.Siow RC, Ishii T, Mann GE. Modulation of antioxidant gene expression by 4-hydroxynonenal: atheroprotective role of the Nrf2/ARE transcription pathway. Redox Rep. 2007;12:11–15. doi: 10.1179/135100007X162167. [DOI] [PubMed] [Google Scholar]
- 41.Goldstein L, Teng ZP, Zeserson E, Patel M, Regan RF. Hemin induces an iron-dependent, oxidative injury to human neuron-like cells. J Neurosci Res. 2003;73:113–121. doi: 10.1002/jnr.10633. [DOI] [PubMed] [Google Scholar]
- 42.Gonzales S, Erario MA, Tomaro ML. Heme oxygenase-1 induction and dependent increase in ferritin: a protective antioxidant stratagem in hemin-treated rat brain. Dev Neurosci. 2002;24:161–168. doi: 10.1159/000065686. [DOI] [PubMed] [Google Scholar]
- 43.Attuwaybi BO, Kozar RA, Moore-Olufemi SD, et al. Heme oxygenase-1 induction by hemin protects against gut ischemia/reperfusion injury. J Surg Res. 2004;118:53–57. doi: 10.1016/j.jss.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 44.Belcher JD, Mahaseth H, Welch TE, Otterbein LE, Hebbel RP, Vercellotti GM. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Invest. 2006;116:808–816. doi: 10.1172/JCI26857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasaki T, Takahashi T, Maeshima K, et al. Heme arginate pretreatment attenuates pulmonary NF-kappaB and AP-1 activation induced by hemorrhagic shock via heme oxygenase-1 induction. Med Chem. 2006;2:271–274. doi: 10.2174/157340606776930781. [DOI] [PubMed] [Google Scholar]
- 46.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 47.Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- 48.Guzman ML, Jordan CT. Considerations for targeting malignant stem cells in leukemia. Cancer Control. 2004;11:97–104. doi: 10.1177/107327480401100216. [DOI] [PubMed] [Google Scholar]
- 49.Guzman ML, Jordan CT. Feverfew: weeding out the root of leukaemia. Expert Opin Biol Ther. 2005;5:1147–1152. doi: 10.1517/14712598.5.9.1147. [DOI] [PubMed] [Google Scholar]
- 50.Guzman ML, Li X, Corbett CA, et al. Rapid and selective death of leukemia stem and progenitor cells induced by the compound 4-benzyl, 2-methyl, 1,2,4-thiadiazolidine, 3,5 dione (TDZD-8). Blood. 2007;110:4436–4444. doi: 10.1182/blood-2007-05-088815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ball CA, Brazma A, Causton H, et al. Submission of microarray data to public repositories. PLoS Biol. 2004;2:E317. doi: 10.1371/journal.pbio.0020317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stoeckert CJ, Jr., Causton HC. Ball CA Microarray databases: standards and ontologies. Nat Genet. 2002;32(suppl):469–473. doi: 10.1038/ng1028. [DOI] [PubMed] [Google Scholar]
- 53.Fellenberg K, Hauser NC, Brors B, Hoheisel JD, Vingron M. Microarray data warehouse allowing for inclusion of experiment annotations in statistical analysis. Bioinformatics. 2002;18:423–433. doi: 10.1093/bioinformatics/18.3.423. [DOI] [PubMed] [Google Scholar]
- 54.Qiu X, Brooks AI, Klebanov L, Yakovlev N. The effects of normalization on the correlation structure of microarray data. BMC Bioinformatics. 2005;6:120. doi: 10.1186/1471-2105-6-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai M, Wang P, Boyd AD, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:1341–1357. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}