

Abstract
Ferrochelatase catalyzes the terminal step in heme biosynthesis, the insertion of ferrous iron into protoporphyrin to form protoheme IX. The crystal structures of human ferrochelatase both with and without protoporphyrin substrate bound have been determined previously. The substrate-free enzyme has an open active site pocket while in the substrate-bound enzyme the active site pocket is closed around the porphyrin macrocycle and a number of active site residues have reoriented side chains. In order to understand how and why these structural changes occur we have made amino acid substitutions in three residues (H263, H341, and F337) whose side chains occupy different spatial positions in the substrate-free vs. substrate-bound ferrochelatases. The catalytic and structural properties of ferrochelatases containing the amino acid substitutions H263C, H341C or F337A were examined. It was found that in the H263C and H341C variants, but not the F337A variant enzymes, the side chains of N75, M76, R164, H263, F337, H341 and E343 are oriented in a fashion similar to what is found in ferrochelatase with bound porphyrin substrate. However, all of the variant forms possess open active site pockets as is found in the structure of porphyrin-free ferrochelatase. Thus, while the interior walls of the active site pocket are remodeled in these variants, the exterior lips remain unaltered in position. One possible explanation for this collective reorganization of active site side chains is the presence of a hydrogen bond network among H263, H341, and E343. This network is disrupted in the variants by alteration of H263C or H341C. In the substrate-bound enzyme the formation of a hydrogen bond between H263 and a pyrrole nitrogen results in disruption of the network. The possible role of this network in catalysis is discussed.
Keywords: heme synthesis, ferrochelatase, metal chelation, structure-function
Ferrochelatase (protoheme ferrolyase E.C. 4.99.1.1) catalyzes the terminal step in protoheme biosynthesis, the insertion of ferrous iron into protoporphyrin IX (1). This enzyme was the first protein identified and characterized whose catalytic role is the insertion of a metal ion into an organic molecule (2). The enzyme is widely distributed in nature and while it is clearly similar in structure and gross catalytic properties among all sources, there is less than 10% sequence identity between the enzyme in higher eukaryotes and bacteria (3, 4). At the present time there are published crystal structures for three ferrochelatases: one bacterial (5), one yeast (6), and human (7). While these three protein structures are clearly related they differ in that the Bacillus subtilis enzyme is a soluble monomeric protein and the eukaryotic enzymes are both inner mitochondrial membrane-associated homodimers. Animal and a few bacterial ferrochelatases also possess a [2Fe-2S] cluster (4, 8, 9) while the Saccharomyces cerevisiae and B. subtilis ferrochelatases do not.
Properties of the enzyme have been reviewed recently (1). The overall proposed enzymatic reaction involves iron acquisition and desolvation, followed by porphyrin binding, macrocycle distortion prior to removal of two pyrrolic protons, and finally metal chelation. While there is general acceptance that metallation is facilitated by macrocycle distortion, there is controversy about the stereochemistry of metallation, and no clear understanding of the catalytic mechanism. For each of the three ferrochelatases listed above there exist structures with bound putative substrate metals (6, 7, 10). For the bacterial enzyme there is also a structure with N-methylmesoporphyrin, a tight binding, competitive inhibitor of ferrochelatase (11), along with the metal copper (12) and a structure of the enzyme with bound iron (13). No published structures are currently available for the bacterial enzyme with bound protoporphyrin IX or protoheme IX.
Recently the first structure of any ferrochelatase with bound protoporphyrin IX substrate was determined (14). This substrate-bound human ferrochelatase differs substantially from the substrate-free human enzyme in two ways. First, the active site pocket, or “mouth,” is closed around the porphyrin macrocycle leaving only a small open hole over one pyrrole ring. Second, a select set of active site residues possess an altered spatial orientation in comparison to the substrate-free enzyme.
In the current study three active site residues have been altered by site-directed mutagenesis and the structures and kinetic properties of these variant forms of human ferrochelatase have been examined. The data demonstrate that reorientation of active site residues and closing of the active site are separable events. Furthermore, reorientation of active site residues occurs in a concerted manner that appears to involve a complex hydrogen bond network. It is proposed that realignment of this hydrogen bond network is the initial catalytic trigger that is necessary, but not sufficient for macrocycle distortion.
Materials and Methods
Recombinant human ferrochelatase was produced and purified as previously described (15). All studies reported herein employ a R115L variant of human ferrochelatase that has normal activity and has had its crystallographic structure determined previously (7). Production of recombinant human ferrochelatases H263C and H341C variants have been described previously (15). The N75A, M76A and F337A variants were produced and verified using the same techniques as described previously (16).
Enzyme assays were performed using a continuous direct spectroscopic assay (17) at 25°C. Kinetic parameters were determined from data sets of ten assays.
Structure determination
Crystals of the H263C/R115L and H341C/R115L variants were obtained using conditions published for the R115L variant (16) with seeding. Crystals for the F337A variant were grown by the microbatch under oil (70:30 paraffin/silicone oil mixture) method using 2 μL drops containing equal volumes of protein solution (64 mg/ml in 50 mM Tris MOPs, 0.1M KCl, 1% Na-cholate, 250 mM imidazole, pH 8.1) and precipitant solution (0.2 M sodium acetate tri hydrate, 0.1 M sodium cacodylate, pH 6.5 in 30%w/v polyethylene glycol 8000 (Hampton Crystal Screen I-28, Hampton Research). The drops were incubated at 18°C and crystals suitable for mounting were observed after 4 days.
Crystals for the H263C/R115L, H341C/R115L and F337A/R115L variants were harvested and mounted using the loop mounting technique of Teng (18) and flash frozen in liquid nitrogen. No cryoprotection was necessary.
For the H263C/R115L and H341C/R115L variants, data were collected under cryogenic conditions (T = 100K) on an Raxis IV image plate detector using mirror focused (MSC Blue confocal optics) 5kW X-rays. The data were indexed, integrated and scaled using HKL 1.9.1 (19). For the F337A/R115L variant, data were collected under cryogenic conditions (T = 105K) on beamline 22BM (SER-CAT) Advanced Photon Source, Argonne National Laboratory using a MarResearch 225 CCD detector. The resulting data were indexed, integrated, and scaled using the HKL2000 program suite (19). Data collection and processing details are collected in Table 1.
Table 1.
Statistics from the crystallographic analysis
| Crystal | H263C/R115L | H341C/R115L | F337A/R115L |
|---|---|---|---|
| Space group: | P212121 | P212121 | P212121 |
| a | 93.42 | 94.05 | 88.69 |
| b | 87.73 | 97.61 | 94.05 |
| c | 109.58 | 110.63 | 113.27 |
| Data analysis: | |||
| Wavelength (Å): | 1.5418 | 1.5418 | 1.0000 |
| Phi step (°): | 1.0 | 1.0 | 0.5 |
| Total Rotation (°): | 360 | 360 | 200 |
| Data processing: | HKL 1.9.1 | HKL 1.9.1 | HKL2000 |
| R-sym: | 0.065 | 0.068 | 0.048 |
| Refinement statistics: | |||
| Program | CNS 1.0 | CNS 1.0 | REFMAC 5.4 |
| Resolution (Å): | 19.90 − 2.20 | 18.08 − 2.20 | 43.44 − 2.35 |
| Completeness* (%): | 96.3 (90.1) | 87.8 (56.9) | 96.36 (97.9) |
| Rcryst* | 0.209 (0.242) | 0.210 (0.222) | 0.195 (0.200) |
| Rfree* | 0.240 (0.272) | 0.245 (0.290) | 0.253 (0.309) |
| R.M.S. Deviations from ideality | |||
| Bond lengths (Å): | 0.005 | 0.006 | 0.016 |
| Bond angles (°): | 1.2 | 1.2 | 1.8 |
| Ramachandran analysis(32)** | |||
| Most favored (%) | 92.0 (99.7) | 92.8 (99.3) | 91.3 (99.7) |
| Disallowed (%) | 0.3 | 0.3 | 0.3 |
| Final model: | |||
| Residues | 65−423 | 65−423 | 65−423 |
| Solvent atoms | 308 | 426 | 342 |
| Detergent molecules | 3 | 3 | 3 |
| Cα Deviations*** (Å) | 0.210 | 0.221 | 0.261 |
| PDB ID | 2PO5 | 2PO7 | 2PNJ |
outer shell values in parentheses
PROCHECK percent in all allowed regions shown in parentheses
R.M.S. Deviations (Cα) between 1HRK structure and variant structure
The structures of the H263C/R115L, H341C/R115L and F337A/R115L variants were solved by difference Fourier analysis since the crystals were isomorphous with those of the R115L variant whose structure (PDB entry 1HRK) has been previously reported (7). Refinement of the variant structures was carried out using CNS (20) for the H263C/R115L and H341C/R115L variants, and REFMAC (21) for the F337A/R115L variant, together with manual model adjustment using XFIT (22) when needed. Refinement statistics for the three structures are collected in Table 1. Overall the structures of the three variants are as expected very similar to each other with r.m.s.d. (Cα) ranging from 0.21 to 0.26Å when the structures (dimers) are superimposed (23) with each other, and with the structure of the R115L variant (the r.m.s.d. (Cα) between the two Fc monomers comprising the 1HRK dimer is 0.16Å). In all cases three cholate molecules (required for solubilization) were found in the putative active site pocket as was observed in the R115L structure. The coordinates for the three ferrochelatase variants described in this work have been deposited in the Protein Data Bank (24) (see Table 1). All graphic representations were created using PYMOL (25) and WinCOOT (26).
Results
Structure Determination for Variants
The crystal structure of a R115L (herein referred to as wild-type) human ferrochelatase variant was initially solved by Wu et al. (7) at 2.0 Å and more recently this has been refined to 1.7 Å by Medlock et al. (14). The enzyme is a homodimer with each subunit possessing a [2Fe-2S] cluster. Additionally the structure of human ferrochelatase with bound protoporphyrin IX substrate has been solved to 2.5 Å (14). In the porphyrin-bound enzyme several active site residues possess altered spatial positions when compared to the porphryin free enzyme. Three of these residues, H263, F337, and H341, were selected as targets for site-directed mutagenesis in the current study. The specific individual mutations made were H263C, F337A, and H341C. While the H263C and H341C variants are purified with less than stoichiometric amounts of porphyrin, the crystallized proteins contained no bound porphyrin or heme. All three variant ferrochelatases crystallized (Table I) in the same space group as the wild-type protein (7) and diffracted to 2.2Å.
The structures of both H263C and H341C variant ferrochelatases are superimposable on the R115L human ferrochelatase variant with the exception of the side chains for N75, M76, R164, H263, F337, H341, and E343 (Figure 1A). In the F337A variant ferrochelatase the spatial orientation of all side chains is identical to the wild-type human ferrochelatase. Interestingly the side chains listed above that are reoriented in the H263C and H341C variants are the same ones that have altered spatial position in human ferrochelatase with bound porphyrin substrate (Figure 1B). None of these side chains makes significant contact with the porphyrin macrocycle.
Figure 1. Reorientation of active site residues.

(A) The orientation of select active site residues in the wild-type and the H263C variant form of human ferrochelatase are pictured. The view into the active site shows the orientation of the side chains of N75, M76, R164, H263, F337, H341 and E343. The wild-type enzyme is shown in cream, red, blue and yellow for carbon, oxygen, nitrogen and sulfur atoms (PDB ID 1HRK), respectively, and H263C variant is shown in orange. (B) The orientation of select active site resides of the substrate bound (E343K variant) and H263C variant forms of human ferrochelatase are illustrated. A view of the active site shows the similarity in orientation of specific active site residues, including N75, M76, R164, H263, H341, E343 and F337. The substrate bound form is shown in green (PDB ID 2HRE) and the H263C variant is shown in orange. The bound protoporphyrin IX is shown in red.
Kinetic Examination
Previously we have produced and kinetically characterized R164L, H263C, H341C, E343Q, Y123F, and Y191F ferrochelatases which are discussed in the current study (15). Additional variants produced for this study are N75A, M76A and F337A. Kinetic data for these variants are listed in Table 2. All of the variants examined possessed intact [2Fe-2S] clusters and were as stable to storage as the wild-type ferrochelatase. Efforts to “rescue” the activity of an H263A mutant by inclusion of 300 mM imidazole in the assay buffer were unsuccessful (15).
Table 2.
Effect of Amino Acid Replacement on the Activity of Human Ferrochelatase
Discussion
The mechanism by which metal is inserted into a porphyrin macrocycle is relatively well understood for solution chemistry, but is poorly understood for enzyme catalyzed reactions. Iron insertion into protoporphyrin to form heme is catalyzed by ferrochelatase and current knowledge of this reaction is based largely upon studies of the human and B. subtilis enzymes. From solution studies it was proposed that macrocycle distortion is a key feature that decreases the energy barrier for metallation. Two additional observations support this hypothesis: i.) small ribozymes can distort porphyrins and facilitate metallation (27, 28) and ii.) antibodies raised against N-alkylated porphyrins (which themselves are bent ∼30°) will bind and distort porphyrins and facilitate metallation (29).
The finding that N-methylprotoporphyrin, which has the alkylated ring bent about 35° out of the plane of the macrocycle, is a tight-binding competitive inhibitor of ferrochelatase fits the distortion model nicely as a putative transition state analog of ferrochelatase. Crystallographic studies on the B. subtilis ferrochelatase with bound N-methylmesoporphyrin led to a number of studies and resulted in proposed models for catalysis that were based upon the assumption that this molecule was bound in the active site in a fashion identical to the position occupied by the substrate protoporphyrin. With the recent publication of human ferrochelatase with bound protoporphyrin it is clear that N-methylmesoporphyrin does not occupy the same site as substrate, and, therefore, cannot be considered a transition state analog (14).
The structure of human ferrochelatase with bound porphyrin substrate differs markedly from the structure of the protein without substrate bound. Most notable is that the mouth of the active site is closed and a set of active site residues occupy different spatial orientations. In the current study we have produced site directed variants for three of these residues, H263, F337, and H341, to see if any single mutation causes either a change in the spatial orientation of other active site residues and/or a closing of the active site mouth. During the course of site-directed mutagenesis studies it was noted that H263A/C/N ferrochelatase preparations have no enzyme activity either in vitro or in vivo and are isolated with bound protoporphyrin (15). Incubation overnight with ferrous iron did not result in the conversion of enzyme-bound porphyrin into heme. Crystallographic studies on H263C were carried out, and it was found that several active site residues were reoriented in the H263C variant relative to the wild type enzyme. In the human ferrochelatase H263C variant, there is an altered orientation of the side chains of N75, M76, R164, F337, H341 and E343 (Figure 1A). No other residues in the active site or outside of the pocket were significantly altered in spatial orientation and the active site mouth is open. Structures of the two additional active site variants, H341C and F337A, were also determined. While the F337A variant appeared identical to wild type except for the replacement of the phenyl side chain, H341C had the same altered orientation of side chains as was found with H263C.
For both H263C and H341C the alteration in orientation of side chains is significant and essentially identical. Interestingly the H341C mutation results in modestly decreased enzymatic activity whereas H263C has no activity. Thus, this reorientation alone does not cause loss of enzyme activity, but these data do suggest that a mutation causing reorientation of these active site residues may have an impact on the catalytic efficiency of the enzyme. The inactivity of the H263C variant may be attributable to the loss of this key residue and its ability to serve as a proton acceptor in the reaction (1). Given that cysteine should be able to serve at some level as a metal ion ligand, it might be expected that H263C should retain some activity if the role of H263 is as a metal ligand for catalysis, but this was not found to be the case.
Examination of the wild-type human structure strongly suggests that a hydrogen bonding network normally exists involving H263, H341 and E343. The altered orientation of H341, E343, and F337 in the variants may be due to disruption of this hydrogen bond network. Disruption of this network results in the side chains of E343 and H341 rotating away from their central pocket position and thereby allowing F337, which is usually inhibited from movement by the imidazole ring of H341, to drop into the pocket.
The crystal structure of the F337A ferrochelatase variant shows that this residue does not play a direct role in alteration of side chain orientation, as might be expected, since it does not actively participate in the hydrogen bond network. Kinetic data from the variant make it clear, however, that F337 plays a key role in catalysis (Table 2). We propose that its role is two-fold. First, movement of the phenyl ring of F337 along with movement of the imidazole ring of H341 reshapes the back of the active site pocket. Second, F337 appears to act as a door that alternatively opens and closes the entrances to two solvent-filled tunnels located at the back of the active site pocket (Figure 2). The F337A variant has a small side chain and so both tunnels are open at all times. We propose that one of these solvent-filled tunnels is the entrance route for the substrate iron on the back side of the ferrochelatase molecule (30) and may also serve as a “rear exit” (31) for active site water molecules which must exit to allow space for the porphyrin macrocycle. Given that kinetic data suggest an ordered reaction with iron binding before porphyrin (11), this tunnel could satisfy both roles by first allowing iron into the active site pocket and then allowing water molecules an exit route as the porphyrin enters the pocket and displaces water.
Figure 2. Tunnels from the protein surface to the active site.
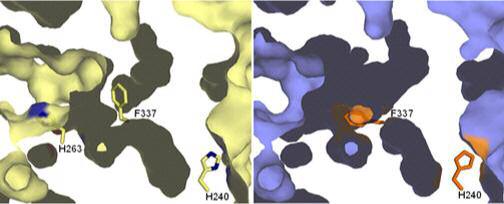
A cross section through the active site of the wild-type and the H263C variant human ferrochelatases is shown. The active site opening is to the left and back. The wild-type surface is colored in cream and the H263C variant surface is in slate. The position of the centrally located and essential H263 is shown in the wild-type structure in cream and blue for carbon and nitrogen, respectively. The figure illustrates that the reorientation of the residues described in the text which result in the closing off of the tunnel opening seen at the bottom of this diagram and the opening of another tunnel entrance at the top of the picture. These alterations result mainly from the movement of the side chain of F337, shown as sticks in the middle of the diagram. The expansion of the pocket seen at the top of the diagram is attributable to the reorientation of the side chain of R164.
Based upon the data above we propose that catalysis involves the following scenario. Following iron binding within the active site, porphyrin enters the active site pocket and at least one pyrrole proton forms a hydrogen bond with the imidazole of H263. This event triggers the catalytic cycle by disrupting the “resting state” hydrogen bond between H263 and the side chain of E343 (3.2 Å). This in turn allows E343 to reorient and shorten the previously existing hydrogen bond with the side chain of H341 from 3.4 Å to 2.9 Å while increasing the distance between H263 and E343 to almost 5 Å. Upon reorientation, the movement of the imidazole side chain of H341 allows the side chain ring of F337 to rotate into the pocket. H341 may now form a hydrogen bond with the hydroxyl of the phenol ring of Y123 (2.9 Å) (Figure 3A). Movement of F337 opens the entrance of a short tunnel and closes the opening of a long amphipathic, solvent-filled tunnel that extends to the surface on the backside of the molecule at H240 (Figure 2).
Figure 3. Hydrogen bonding of active site residues in the resting state and activated state of human ferrochelatase.
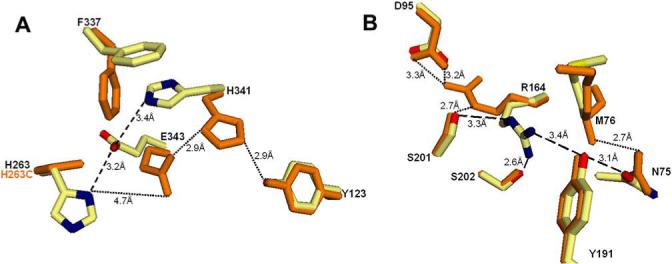
A. Hydrogen bonding of the bottom of the active site pocket involving H263, E343, H341 and Y123 in the resting state (dashed lines) and in the activated state in (dotted lines). The wild-type enzyme is colored in cream, red, blue and yellow for carbon, oxygen, nitrogen and sulfur atoms, respectively, and H263C variant is shown in orange. B. Hydrogen bonding of the top of the active site pocket involving N75, M76, D95, R164, Y191, S201 and S202 in the resting state (dashed lines) and in the activated state in (dotted lines). The wild-type enzyme is colored in cream, red, blue and yellow for carbon, oxygen, nitrogen and sulfur atoms, respectively, and H263C variant is shown in orange.
Reorientation of side chains also occurs on the opposite side of the active site pocket from H263. The impetus for this motion appears to involve N75 which rotates slightly. In the resting state N75 may hydrogen bond with the hydroxyl of Y191 (3.1 Å), which in turn may hydrogen bond to the guanido of R164 (3.4 Å). R164 can also participate in hydrogen bonds with S202 (2.6 Å) and S201 (3.3 Å) and the carbonyl of T198. After reorientation N75 hydrogen bonds to the backbone nitrogen of M76 (2.7 Å) allowing the side chain of M76 to rotate down into the pocket. However, the most dramatic change occurs with R164 which rotates approximately 180° out of the active site pocket. Its new position is stabilized by hydrogen bonds between the guanido of R164, hydroxyl of S201 (2.7 Å) and carboxylate of D95 (3.2 Å and 3.3 Å) (Figure 3B). The movement of R164 creates a pocket over the center of the porphyrin macrocycle adjacent to the side chain of M76. The ultimate impact of these rearrangements is the remodeling of the active site pocket in such a manner that could result in distortion of the porphyrin macrocycle which would facilitate iron insertion (Figure 4). Mutagenesis and characterization of N75, M76, E343, H341, F337, Y191, Y123, and R164 yielded kinetic data that are consistent with this proposal (Table 2) (15).
Figure 4. Remodeling of the active site pocket.
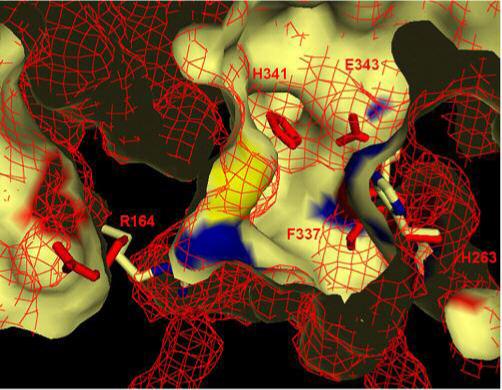
A segment through the active site is shown with a surface representation of the wild-type ferrochelatase in cream. The H263C variant surface is shown as a red mesh. The residue R164 is to the left and reorients outward to the surface in the variant. H341 and E343 of the variant can be seen to reorient into the active site pocket. F337 of the variant moves into the bottom right of the active site as shown in this figure. M76 rotates slightly and its mesh-represented density can be seen near the center of the active site.
The diminished catalytic ability of N75A is of interest since the amide side chain of asparagine has little impact on the shape of the active site pocket and its minor movement from wild-type to H263C or H341C does not spatially alter the active site pocket. It does, however, appear to be key to the hydrogen bond network connecting the H263 side of the pocket with the R164 side. The side chain of M76 protrudes slightly downward into the pocket over one of the pyrrole rings of the macrocycle (Figure 4). An M76A variant eliminates this physical presence and thereby may allow sufficient movement of the porphyrin macrocycle to prevent ring distortion or optimal orientation for metal insertion.
To end the catalytic cycle, metal insertion into the porphyrin nucleus will release H263 to recreate the hydrogen bond network via interaction with E343. Such an event would alter the geometry of the active site allowing release of the heme, which may be facilitated by movement of the guanido of R164 back down over the now metallated product thereby creating charge repulsion between the iron in heme and the guanido of R164. Rotation of F337 reopens the amphipathic tunnel thereby allowing water to move back into the active site pocket as the heme exits.
Since the active site remains open despite significant changes in positions of specific side chains, it is clear that something other than reformation of the the hydrogen bond network must trigger or cause the active site mouth to close. The impetus for this may be hydrophobic interactions between the macrocycle and aromatic active site residues or interactions between the porphyrin propionates and polar residues.
It is of interest that many of the active site residues that are discussed above are not conserved among all ferrochelatases. Indeed, H263, Y123, F337 and E343 are the only conserved residues discussed in the current study and all of these are located on one surface of the active site. Examination of available structures and modeling of other ferrochelatases using threader programs clearly demonstrates that while the overall catalytic function of ferrochelatases may be conserved, the exact residues playing catalytic roles varies considerably. However, the current data support a catalytic model for human ferrochelatase where initial porphyrin substrate binding triggers a cascade of hydrogen bond realignments that result in reshaping of the active site and distortion of the macrocycle thereby facilitating iron insertion.
Acknowledgements
This work was supported by grant DK32303 from the National Institutes of Health to H.A. Dailey. J. Rose and P. Horanyi were supported by grant GM62407.
Footnotes
Conflict of Interest Statement We declare that the authors have no competing interests relating to this work.
References
- 1.Dailey HA, Dailey TA. In: Ferrochelatase, in The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. Academic Press; New York: 2003. pp. 93–121. [Google Scholar]
- 2.Goldberg A, Ashenbrucker H, Cartwright GE, Wintrobe MM. Studies on the Biosynthesis of Heme In Vitro by Avian Erythrocytes. Blood. 1956;11:821–833. [PubMed] [Google Scholar]
- 3.Dailey HA, Dailey TA, Wu CK, Medlock AE, Wang KF, Rose JP, Wang BC. Ferrochelatase at the millennium: structures, mechanisms and [2Fe-2S] clusters. Cell Mol Life Sci. 2000;57:1909–1926. doi: 10.1007/PL00000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dailey TA, Dailey HA. Identification of [2Fe-2S] clusters in microbial ferrochelatases. J Bacteriol. 2002;184:2460–2464. doi: 10.1128/JB.184.9.2460-2464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Karadaghi S, Hansson M, Nikonov S, Jonsson B, Hederstedt L. Crystal structure of ferrochelatase: the terminal enzyme in heme biosynthesis. Structure. 1997;5:1501–1510. doi: 10.1016/s0969-2126(97)00299-2. [DOI] [PubMed] [Google Scholar]
- 6.Karlberg T, Lecerof D, Gora M, Silvegren G, Labbe-Bois R, Hansson M, Al-Karadaghi S. Metal binding to Saccharomyces cerevisiae ferrochelatase. Biochemistry. 2002;41:13499–13506. doi: 10.1021/bi0260785. [DOI] [PubMed] [Google Scholar]
- 7.Wu CK, Dailey HA, Rose JP, Burden A, Sellers VM, Wang BC. The 2.0 A structure of human ferrochelatase, the terminal enzyme of heme biosynthesis. Nat Struct Biol. 2001;8:156–160. doi: 10.1038/84152. [DOI] [PubMed] [Google Scholar]
- 8.Dailey HA, Finnegan MG, Johnson MK. Human ferrochelatase is an iron-sulfur protein. Biochemistry. 1994;33:403–407. doi: 10.1021/bi00168a003. [DOI] [PubMed] [Google Scholar]
- 9.Shepherd M, Dailey TA, Dailey HA. A new class of [2Fe-2S]-cluster-containing protoporphyrin (IX) ferrochelatases. Biochem J. 2006;397:47–52. doi: 10.1042/BJ20051967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lecerof D, Fodje MN, Alvarez Leon R, Olsson U, Hansson A, Sigfridsson E, Ryde U, Hansson M, Al-Karadaghi S. Metal binding to Bacillus subtilis ferrochelatase and interaction between metal sites. J Biol Inorg Chem. 2003;8:452–458. doi: 10.1007/s00775-002-0436-1. [DOI] [PubMed] [Google Scholar]
- 11.Dailey HA, Fleming JE. Bovine ferrochelatase. Kinetic analysis of inhibition by N-methylprotoporphyrin, manganese, and heme. J Biol Chem. 1983;258:11453–11459. [PubMed] [Google Scholar]
- 12.Lecerof D, Fodje M, Hansson A, Hansson M, Al-Karadaghi S. Structural and mechanistic basis of porphyrin metallation by ferrochelatase. J Mol Biol. 2000;297:221–232. doi: 10.1006/jmbi.2000.3569. [DOI] [PubMed] [Google Scholar]
- 13.Hansson MD, Karlberg T, Rahardja MA, Al-Karadaghi S, Hansson M. Amino Acid Residues His183 and Glu264 in Bacillus subtilis Ferrochelatase Direct and Facilitate the Insertion of Metal Ion into Protoporphyrin IX. Biochemistry. 2007;46:87–94. doi: 10.1021/bi061760a. [DOI] [PubMed] [Google Scholar]
- 14.Medlock A, Swartz L, Dailey TA, Dailey HA, Lanzilotta WN. Substrate Interactions with Human Ferrochelatase. Proc Natl Acad Sci USA. 2007;104:1789–1793. doi: 10.1073/pnas.0606144104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sellers VM, Wu CK, Dailey TA, Dailey HA. Human ferrochelatase: characterization of substrate-iron binding and proton-abstracting residues. Biochemistry. 2001;40:9821–9827. doi: 10.1021/bi010012c. [DOI] [PubMed] [Google Scholar]
- 16.Burden AE, Wu C, Dailey TA, Busch JL, Dhawan IK, Rose JP, Wang B, Dailey HA. Human ferrochelatase: crystallization, characterization of the [2Fe-2S] cluster and determination that the enzyme is a homodimer. Biochim Biophys Acta. 1999;1435:191–197. doi: 10.1016/s0167-4838(99)00196-x. [DOI] [PubMed] [Google Scholar]
- 17.Najahi-Missaoui W, Dailey HA. Production and characterization of erythropoietic protoporphyric heterodimeric ferrochelatases. Blood. 2005;106:1098–1104. doi: 10.1182/blood-2004-12-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teng TY. Mounting of crystals for macromolecular crystallography in a freestanding thin-film. J Appl Crystal. 1990;23:387–391. [Google Scholar]
- 19.Otwinowski Z. a. M., W Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymol. 1997;A276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 21.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 22.McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 23.Huang CC, Couch GS, Pettersen EF, Ferrin TE. Chimera: An Extensible Molecular Modeling Application Constructed Using Standard Components. In Pacific Symposium on Biocomputing. 1996;1:724. [Google Scholar]
- 24.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, Feng Z, Gilliland GL, Iype L, Jain S, Fagan P, Marvin J, Padilla D, Ravichandran V, Schneider B, Thanki N, Weissig H, Westbrook JD, Zardecki C. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 25.DeLano WL. The PyMOL Molecular Graphics System. 0.99 ed. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 26.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Sen D. Toward an efficient DNAzyme. Biochemistry. 1997;36:5589–5599. doi: 10.1021/bi962694n. [DOI] [PubMed] [Google Scholar]
- 28.Conn MM, Prudent JR, Schultz PG. Porphyrin Metalation Catalyzed by a Small RNA Molecule. J Am Chem Soc. 1996;118:7012–7013. [Google Scholar]
- 29.Cochran AG, Schultz PG. Antibody-catalyzed porphyrin metallation. Science. 1990;249:781–783. doi: 10.1126/science.2389144. [DOI] [PubMed] [Google Scholar]
- 30.Lange H, Kispal G, Lill R. Mechanism of iron transport to the site of heme synthesis inside yeast mitochondria. The J Biol Chem. 1999;274:18989–18996. doi: 10.1074/jbc.274.27.18989. [DOI] [PubMed] [Google Scholar]
- 31.Meyer E. Internal water molecules and H-bonding in biological macromolecules: a review of structural features with functional implications. Protein Sci. 1992;1:1543–1562. doi: 10.1002/pro.5560011203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]